

Abstract
Most GISTs can be recognized by their monotonous cytologic features and overexpression of KIT oncoprotein. Altered morphology and loss of CD117 reactivity has been described previously after chronic imatinib treatment; however, this phenomenon has not been reported in imatinib-naïve tumors. Eight patients with abrupt transition from a classic CD117-positive spindle cell GIST to an anaplastic CD117-negative tumor were investigated for underlying molecular mechanisms of tumor progression. Pathologic and molecular analysis was performed on each of the two components. Genomic DNA PCR for KIT, PDGFRA, BRAF and KRAS hot spot-mutations and FISH for detecting KIT gene copy number alterations were performed. TP53 mutational analysis was performed in 5 cases. There were 7 males and 1 female, with an age range of 23–65 years. Five of the primary tumors were located in the stomach, while one case each originated in small bowel, colon and rectum. In 3 patients, the dedifferentiated component occurred in the setting of imatinib-resistance, while the remaining 5 occurred de novo. The dedifferentiated component had an anaplastic appearance, including one angiosarcomatous phenotype, with high mitotic activity and necrosis, and showed complete loss of CD117 (8/8) and CD34 (5/8) expression, and de novo expression of either cytokeratin (4/8) or desmin (1/8). There was no difference in the KIT genotype between the two components. However, two imatinib-resistant tumors showed co-existence of KIT exon 11 and exon 13 mutations. FISH showed loss of one KIT gene in 3 cases and low level amplification of KIT in 2 other cases in the CD117-negative component, compared to the CD117-positive area. TP53 mutation was identified in 1/5 cases tested, being present in both components. In summary, dedifferentiation in GIST may occur either de novo or after chronic imatinib exposure and can represent a diagnostic pitfall. This phenomenon is not related to additional KIT mutations, but might be secondary to genetic instability, either represented by loss of heterozygosity or low level of KIT amplification.
Keywords: gastrointestinal stromal tumor, dedifferentiation, KIT, imatinib
INTRODUCTION
Gastrointestinal stromal tumors (GISTs) are the most common mesenchymal neoplasms of the digestive tract and are thought to originate from or differentiate toward the interstitial cell of Cajal lineage 1. Nearly all GISTs express KIT protein and the majority show activating mutations in either KIT or PDGFRA proto-oncogenes 2–4. The presence of CD117 immunoreactivity in the overwhelming majority of cases has come to define the pathologic diagnosis of GIST. Only a minority of cases (4%) lack CD117 expression and the diagnosis is usually made on typical histologic features and negative results for desmin, cytokeratin, and S100, which helps to exclude other potential mimics 5.
An altered phenotype secondary to imatinib therapy has been described occasionally and recognized as a potential pitfall 6. These tumors typically lose KIT expression after prolonged treatment with its best known inhibitor, namely imatinib 6, 7. Remarkably CD117 loss is coupled with phenotypic change: i.e. epithelioid morphology and de novo expression of either cytokeratin or desmin or both 8, 9. So far this phenomenon has not been reported in imatinib-naïve tumors and dedifferentiation, although well described in other sarcoma types, has not been well characterized in GIST.
We have identified 8 patients with abrupt transition from a classic CD117-positive spindle cell GIST to anaplastic CD117-negative tumor, in which we sought to investigate the underlying molecular mechanisms of tumor progression. A comparative morphologic, immunohistochemical and molecular investigation was undertaken of both GIST components, in order to better define diagnostic pitfalls, morphologic and molecular features, and possible pathogenesis. We also investigated whether progression from a CD117-positive classic appearance to a CD117-negative anaplastic phenotype might be related to the history of imatinib therapy, perhaps being secondary to the development of imatinib-resistance.
MATERIALS & METHODS
The Pathology files of 5 different institutions (Brigham &Women’s Hospital, Boston, MA, USA, Massachusetts General Hospital, Boston, MA, Chang Gung Memorial Hospital-Kaohsiung Medical Center, Chang Gung University College of Medicine, Kaohsiung, Taiwan, ROC, Treviso General Hospital, Treviso, Italy, and Memorial Sloan-Kettering Cancer Center, New York, NY) were searched for a diagnosis of anaplastic or dedifferentiated GIST. Eight cases were identified in which a CD117-positive GIST component with a classic morphologic appearance was juxtaposed to a CD117-negative anaplastic or pleomorphic type neoplasm.
Morphology and Immunohistochemistry
The two morphologically and immunophenotypically distinct components were analyzed separately. The morphologic appearance of the CD117-positive classic GIST was classified as predominantly spindle, epithelioid, or mixed. The degree of nuclear pleomorphism and presence of giant cells were scored in each component. A mitotic count per 50HPFs was recorded independently in each area. Immunohistochemical studies for the following antibodies were performed: CD117, CD34, desmin, cytokeratin, and p53, and scored independently in each component.
KIT/PDGFRA genotyping for primary and secondary mutations
Mutational analysis was performed using standard protocols. Genomic DNA was isolated separately from each macro-dissected component from paraffin embedded tissue. All 16 samples from the 8 patients were tested for the known sites of KIT (exons 9, 11, 13, 14, and 17), PDGFRA (exons 12 and 18), BRAF exon 15, and KRAS exon 2. One µg of genomic DNA was subjected to PCR using Platinum TaqDNA Polymerase High Fidelity (Life Technologies, Inc Gaithersburg, MD). Primer sequences and annealing temperatures for KIT, PDGFRA and BRAF were as described 7, 10. The KRAS primers used were: KRAS-Ex2Fwd: 5’-GTGTGACATGTTCTAATATAGTCA-3’, KRAS-Ex2Rev: 5’-GTGCAGGACCATTCTTTGATACAG-3’. Direct sequencing of PCR products was performed for all exons tested and each ABI sequence was compared to the NCBI human KIT and PDGFRA gene sequences (NM_000222.2 and NM_006197.1, respectively).
TP53 Mutation Analysis
Mutational analysis was performed in 5 cases by DNA PCR using 1µg of genomic DNA extracted from micro-dissected FFPE tissue from each component. The main hot-spots for p53 mutations were screened, including exons 5–8. Primer sequences and annealing temperatures used as were previously reported 7.
Fluorescence in situ hybridization analysis (FISH) for KIT/PDGFRA copy number changes
FISH was performed in 7 cases, using formalin fixed paraffin-embedded sections according to standard procedures. Each of the two components, identified on a matching HE slide, was analyzed and scored separately. Briefly, paraffin sections were de-waxed in xylene, and then micro-waved in 10mM sodium citrate (pH 6~6.5) solution for 5~10 minutes, cooled to room temperature, rinsed and dehydrated. The slides were then denatured in 70% formamide at 68°C for 2 to 4 minutes, quenched, dehydrated, and air-dried. The KIT probes used were two overlapping BAC clones: CTD- 3180G20 and RP11-722F21 (Invitrogen), labeled by nick-translation with Spectrum Green (Vysis, Abbott Laboratories, IL). The PDGFRA probes used included 2 BAC clones that spanned about 290 kb around the gene: RP11-117E8 and RP11-231C18 (Invitrogen). A chromosome 4 centromeric probe labeled with Spectrum Orange (CEP 4, Vysis) was used as reference. The probe mix, 50 to 80 ng of each KIT or PDGFRA BAC and 2 AL Cot-1 DNA (Invitrogen), was ethanol-precipitated, and resuspended in hybridization buffer. The KIT or PDGFRA probe mix was denatured at 70°C for 10 minutes, followed by pre-annealing at 37°C for 30 minutes. The KIT or PDGFRA probe was then combined with the denatured CEP 4 probe on the slide, cover-slipped and incubated overnight at 37°C. After standard post-hybridization washes, the slides were stained with DAPI and mounted in anti-fade (Vectashield, Vector Laboratories). Analysis was done using a Nikon E800 epifluorescence microscope with Isis 3 imaging software (MetaSystems). A minimum of 100 cells was scanned over separate regions for each slide. Z-stack images were captured using a Zeiss Axioplan 2 motorized microscope controlled by Isis 5 software (Metasystems).
RESULTS
Demographics and Imatinib Therapy Information
There were 7 males and 1 female, with a wide age distribution at diagnosis, ranging from 23–65 years old (mean, 50 years-old) (Table 1). The only female patient included in this series was pregnant in her second trimester when the diagnosis was rendered. Primary tumors were located mostly in the stomach, in 5 cases, and one case each originated from the small bowel, colon and rectum, respectively. Primary tumor size ranged from 5.5 to 25 cm. Among the four patients presenting with localized disease at diagnosis, three were classified as high risk for metastasis and one intermediate risk by NCCN criteria 11. Two of the patients with high risk disease recurred in the peritoneum or liver, for which they received imatinib therapy for 40 months and 48 month, respectively, before progressing clinically and being debulked surgically (cases #7,8). Four patients had metastatic implants at diagnosis, three of them being debulked without prior imatinib therapy, while one patient received neoadjuvant imatinib, having being deemed inoperable and subsequently biopsied after progression developed. Thus, all three examples of imatinib therapy-induced dedifferentiation occurred in the clinical setting of imatinib-resistant disease.
Table 1.
Patient demographics, tumor characteristics and history of imatinib therapy
| Age/ Sex |
Location | Size (cm) |
Stage at Presentation |
Metastases | Pre-op imatinib therapy |
|
|---|---|---|---|---|---|---|
| 1 | 23/M | Stomach | NA* | Liver mets | Liver, Peritoneal | Yes |
| 2 | 40/F | Stomach | 8 | Peritoneal mets | Peritoneal | No |
| 3 | 55/M | Stomach | 18 | Primary | No | No |
| 4 | 48/M | Stomach | 5.5 | Peritoneal mets | Liver, Peritoneal | No |
| 5 | 58/M | Rectum | 6 | Primary | No | No |
| 6 | 53/M | Stomach | 7 | Locoregional LN | Peritoneal | No |
| 7 | 60/M | Small bowel | 7.5 | Primary | Liver, Peritoneal | Yes |
| 8 | 65/M | Colon | 25 | Primary | Peritoneal | Yes |
M, male; F, female; NA*, not available, primary not resected, size unknown; LN, lymph nodes.
One additional patient was identified and analyzed separately from the study group since only the CD117-negative anaplastic component was present in the resection specimen, without a classic CD117-positive component. This patient, a 66 year-old man, was diagnosed with chronic myeloid leukemia (CML) in 1998 and treated with imatinib for 10 years until 2010, when he developed cytogenetic relapse. After one year on nilotinib, which was not well tolerated, he was switched to dasatinib for a short period, when he developed massive upper gastrointestinal bleeding and was diagnosed with a small bowel lesion with liver and peritoneal metastases. The palliative surgical resection showed an anaplastic tumor with epithelioid and rhabdoid morphology, with high mitotic count and necrosis. The immunohistochemical studies showed focal cytokeratin positivity, while CD117 and DOG1 were negative. The diagnosis of GIST was confirmed based on molecular demonstration of a KIT exon 11 WK557-8 deletion.
Morphology and Immunohistochemistry
Histologically two different components were observed in all tumors: a well-differentiated spindle cell component and an anaplastic component. The well-differentiated spindle cell areas showed classic GIST morphology, being composed of rather uniform fusiform cells, with scant fibrillary, eosinophilic cytoplasm, arranged in intersecting fascicles or a short storiform pattern (figs. 1A, 2A, 3B). The dedifferentiated component had an anaplastic/pleomorphic phenotype, with multinucleated neoplastic giant cells, high mitotic activity (>10MF/50HPFs) and necrosis (figs. 1D, 2E). This component showed consistent loss of CD117 (8/8) and CD34 (6/8) expression (figs. 1B,1C, 2B,2F, 3C,3E), and de-novo expression of either cytokeratin (3/8) or both cytokeratin and desmin (1/8)(figs. 1E,F, Table 2). There was no difference noted in the morphologic appearance of the dedifferentiated component, whether it occurred de novo or secondary to imatinib therapy (Table 2). In one tumor (case#7, fig. 3D) the CD117-negative anaplastic component showed irregular, slit-like vascular spaces, lined by hyperchromatic and anaplastic tumor cells, which were reminiscent of angiosarcoma. This component was positive for CD31 (fig. 3F) and CD34 and was negative for CD117 (fig. 3E). To the best of our knowledge, this is the first example of GIST demonstrating differentiation toward an endothelial lineage.
Figure 1.
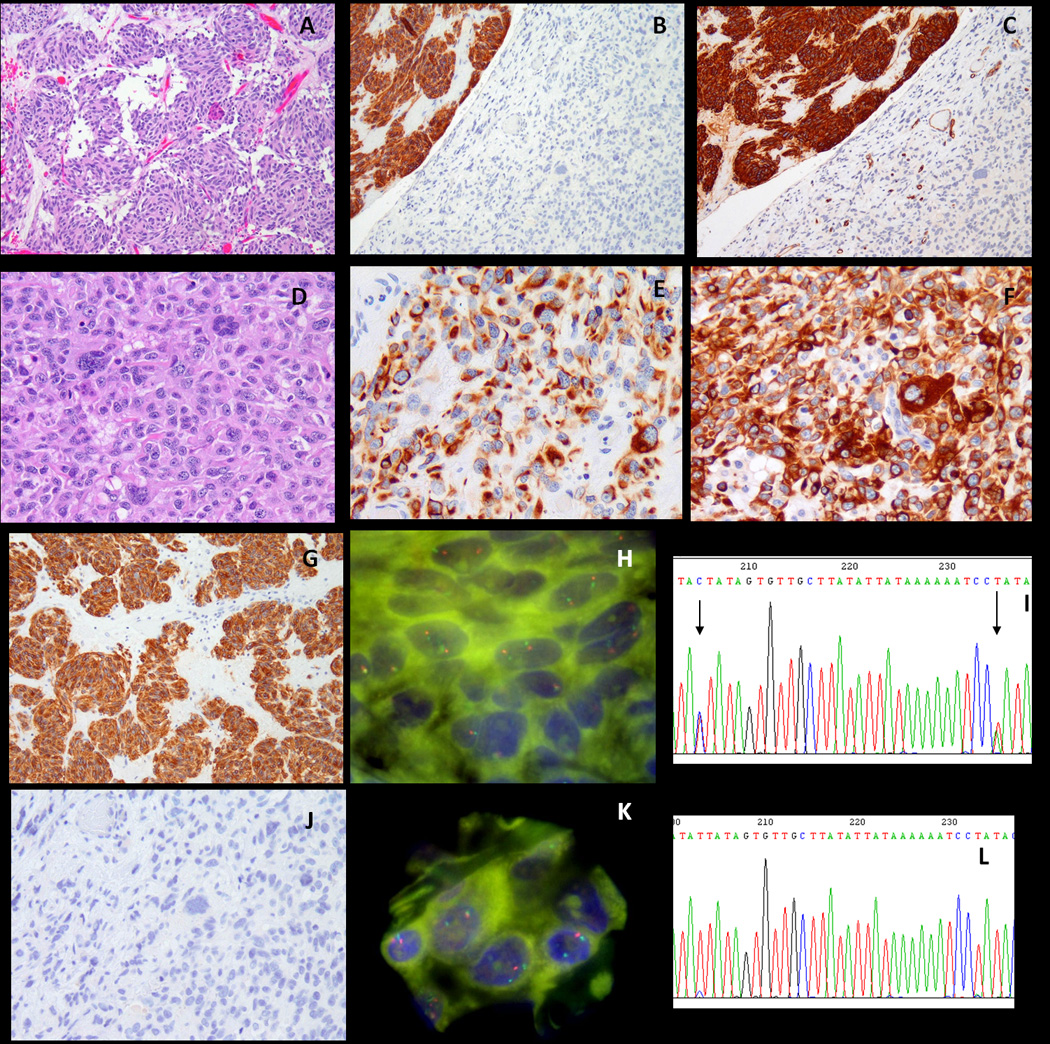
Dedifferentiated GIST (case #1) showing a conventional, spindle cell component (A, 100×), which is strongly positive for CD117 and CD34 (B,C, 100×, upper left corner), in abrupt transition to a pleomorphic, anaplastic area (D, 200×), which is negative for CD117, CD34 (B, C, lower right corner; J), while also showing aberrant expression of cytokeratin AE:AE3 (E, 200×) and desmin (F, 200×). The CD117-positive component (G, 100×) shows two KIT signals by FISH (H) as well as two SNPs in intron 17 (I, ABI seq, arrows). In contrast the KIT-negative dedifferentiated component (J, 200×) shows loss of one KIT signal by FISH, with two reference green CEP4 (K), reflected also by the loss of heterozygosity on the ABI sequence of intron 17 (L).
Figure 2.
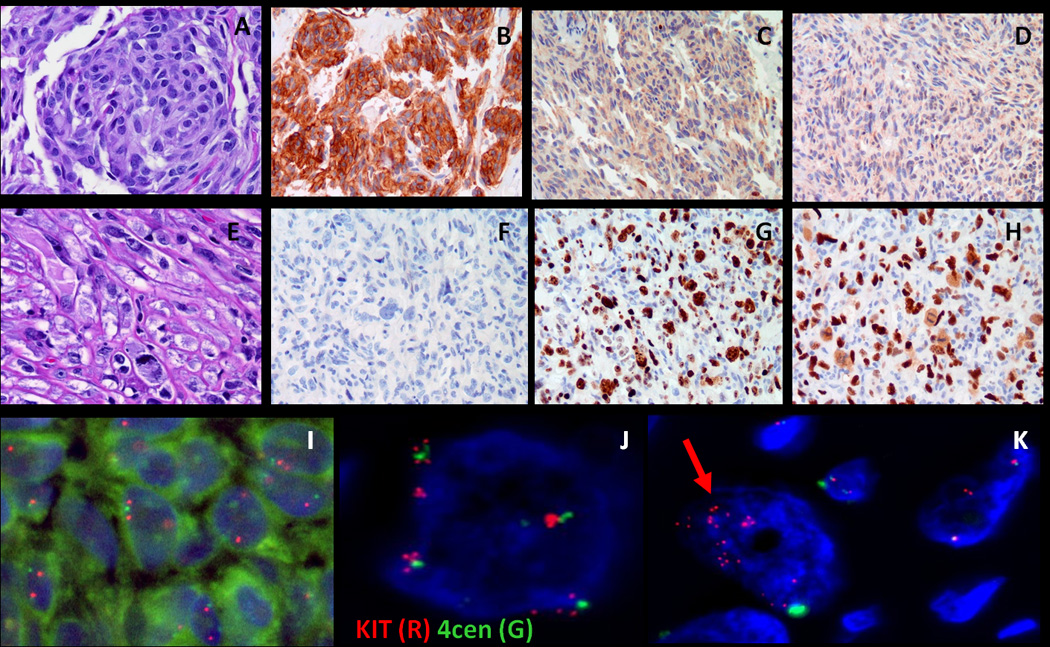
Dedifferentiated GIST (case #2) showing the conventional component (A, 100×) is strongly positive for CD117 (B, 100×), but shows no increased Ki67 labeling (C, 100×) or p53 overexpression (D, 100×). In contrast the CD117-negative anaplastic component (E, F, 200×) shows a high proliferation index (G, 200×) and overexpression of P53 (H, 200×). FISH analysis showed the normal pattern of two red (KIT) and two green (CEP4) signals in the KIT-positive component (I), while a heterogeneous pattern was noted in the KIT-negative component, with either tetraploid cells that have undergone further rounds of replication without centromere separation (4 green (CEP) signals, each green being surrounded by ~4 red (KIT) signals) (J) or occasional cells with numerous, amplified red (KIT) signals (K).
Figure 3.
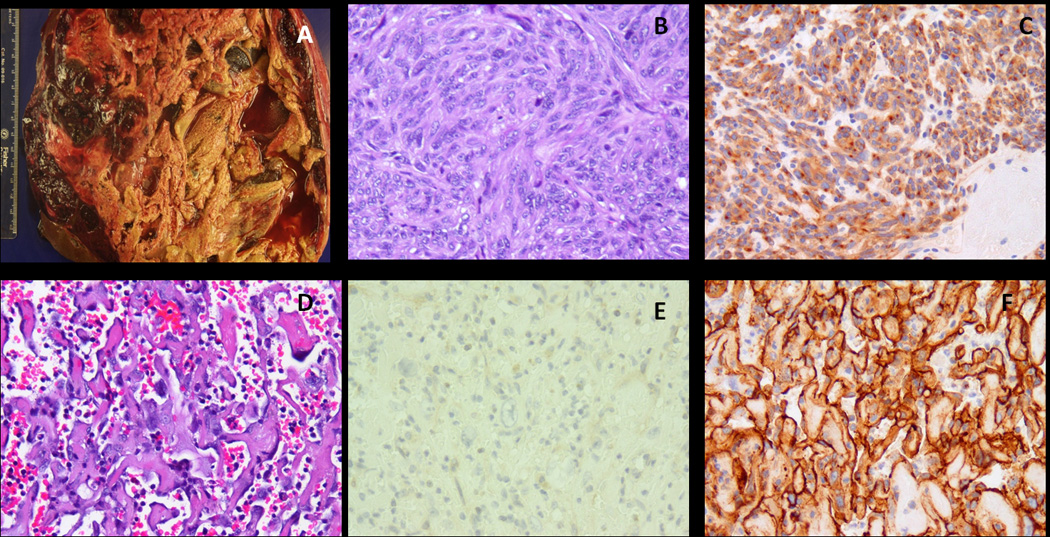
In one of debulking procedure for imatinib-resistant disease (A), in addition to the classic spindle cell GIST component (B, 100×) which was strongly positive for CD117 (C, 100×), there were areas composed of complex, anastomotic vascular spaces, lined by highly atypical cells (D, 200×), which lost CD117 expression (E, 200×) and instead were strongly positive for CD31, in keeping with an angiosarcoma component (case#7).
Table 2.
Morphologic and immunohistochemical features of individual components
| Histologic Type | Pleom | MF/ 50HPF |
Giant Cells |
CD117 | CD34 | Ker | Desmin | |
|---|---|---|---|---|---|---|---|---|
| 1A | Spindle | No | 8 | No | P | P | N | N |
| 1B | Anaplastic | Marked | >10 | Yes | N | N | P | P |
| 2A | Spindle | No | 1 | No | P | N | N | N |
| 2B | Anaplastic | Marked | >10 | Yes | N | N | P | N |
| 3A | Spindle | No | >10 | No | P | P | N | N |
| 3B | Anaplastic | Marked | >10 | Yes | N | N | N | N |
| 4A | Mixed | Moderate | 1 | Yes | P | P | N | N |
| 4B | Anaplastic | Moderate | >10 | Yes | N | N | P | N |
| 5A | Spindle | No | 5 | No | P | P | N | N |
| 5B | Epithelioid/Rhabdoid | Moderate | >10 | Yes | N | N | N | N |
| 6A | Spindle | No | >10 | No | P | P | N | N |
| 6B | Anaplastic | Marked | >10 | Yes | N | N | P | N |
| 7A | Spindle | No | >10 | No | P | P | N | N |
| 7B | Epithelioid/Pleomorphic | Yes | >10 | No | N | P* | N | N |
| 8A | Spindle | No | 8 | No | P | P | N | N |
| 8B | Epithelioid/Pleomorphic | Yes | >10 | No | N | N | N | N |
A, conventional GIST component; B, anaplastic/dedifferentiated component; Pleom, nuclear pleomorphism; MF/HPF, mitotic figures/high power fields; P, positive; N, negative; Ker, cytokeratins AE1:AE3;
CD34 and CD31 co-expression in an angiosarcoma component.
Molecular analyses
There was no difference in the KIT genotype between the two components analyzed. In 4 patients their tumors had a KIT, PDGFRA and BRAF wild-type genotype in both components, for all hot spot exons tested (Table 3). The remaining 4 tumors exhibited a KIT exon 11 mutation: WK557-8 deletion in two cases, a K550-W557 29bp deletion (including deletion of 6 nucleotides from intron 10 and 23 nucleotides from exon 11) in one case, and a V559G substitution in the 4th case. These last two KIT exon 11 genotypes were detected in the dedifferentiated GIST secondary to imatinib-therapy; in both of these cases an additional KIT exon 13 mutation was detected (Table 3). In case #7, the primary tumor (resected in 1998) and the subsequent recurrence (2001), both imatinib-naïve, had only the KIT exon 11 V559G mutation. However, the resistant disease resected after 40 months on therapy (2005), showed an additional secondary KIT exon 13 V654A mutation in both CD117-negative (angiosarcomatous morphology) and CD117-positive components. Compared to the imatinib-naïve heterozygous pattern, both KIT exon 11 and 13 mutations showed a homozygous pattern in the imatinib-resistant samples (in both CD117 positive/negative components), consistent with loss of heterozygosity (LOH) at this locus. This result was further confirmed by FISH, which detected only one KIT signal compared to the normal CEP4 pattern, see below. Case#8 showed one base pair insertion in exon13, V643Sfs6*, resulting in a truncating frame shift mutation, which was detected in both the imatinib-naïve primary tumor (resected from 2008), as well as from the imatinib-resistant tumor (debulked in 2011), but not in the patient’s normal blood DNA. The possibility of this mutation being present at the genomic DNA level only, while being removed from the transcript by nonsense-mediated decay, and therefore not detectable at the cDNA level could not be ruled out, since we were not able to obtain good quality RNA for validating this mutation. The third patient in whom the CD117-negative component was detected after imatinib therapy had a wild-type genotype.
Table 3.
KIT, PDGFRA and p53 sequencing and KIT copy number by FISH analyzed separately in the KIT-positive and KIT negative components
| KIT/PDGFRA genotype | FISH | P53 mutation |
|
|---|---|---|---|
| 1A | WT | Normal | ND |
| 1B | WT | Loss of one KIT copy | ND |
| 2A | WT | Normal | WT |
| 2B | WT | low level KIT amplification | WT |
| 3A | KIT exon 11 557-8WKdel | Normal | WT |
| 3B | KIT exon 11 557-8WKdel | Normal | WT |
| 4A | WT | Normal | Ex 8 del |
| 4B | WT | low level KIT amplification | Ex 8 del |
| 5A | KIT exon 11 557-8WKdel | Normal | WT |
| 5B | KIT exon 11 557-8WKdel | Normal | WT |
| 6A | WT | ND | ND |
| 6B | WT | ND | ND |
| 7A |
KIT exon 11 V559G KIT exon13 V654A |
Normal | ND |
| 7B |
KIT exon 11 V559G / KIT exon13 V654A |
Loss of one KIT copy | ND |
| 8A |
KIT exon 11 29bp DEL KIT exon 13: V643Sfs6* |
Normal | WT |
| 8B |
KIT exon 11 29bp DEL KIT exon 13: V643Sfs6* |
Loss of one KIT copy | WT |
A, conventional GIST component; B, anaplastic/dedifferentiated component; del, deletion; ex, exon; ND, not done
The anaplastic KIT-negative GIST occurring in the patient with long-term imatinib therapy for CML showed a concomitant KRAS G12V mutation to the KIT exon 11. Since there was no associated CD117-positive, morphologically bland, spindle cell component or tissue available from the specimen prior to imatinib therapy, we cannot establish if the presence of the KRAS G12V mutation is secondary to imatinib-therapy or related to the CD117-negative dedifferentiation phenotype. As a consequence of this finding we have studied all the other cases for mutations at this hot spot; however, no other dedifferentiated GIST showed this abnormality. Also, no mutations were identified in BRAF exon 15 in any of the cases tested. TP53 mutation was identified in only one of the five cases tested (case# 4). The same mutation was present in both components.
FISH showed loss of one KIT gene copy in the dedifferentiated component in all 3 cases that occurred secondary to imatinib therapy (fig. 1K), while the CD117-positive component of the imatinib-resistant GIST showed two normal copies, detected by 2 CEP4 signals (fig. 1H). This result was also confirmed by comparing the ABI sequencing of intron 17 of the 2 components in these three cases, showing LOH in the dedifferentiated component, while the CD117-positive areas genotype retained the heterozygous pattern of common SNP present in this region (figs. 1I,L). Additionally a low level of KIT amplification was detected in two of three de novo dedifferentiated GIST tumors, compared to the CD117-positive components with classic morphology (figs. 2I–K).
DISCUSSION
Dedifferentiation was first described in chondrosarcoma12, wherein a low grade chondrosarcoma component is juxtaposed to an anaplastic, non-chondroid, spindle and often pleomorphic sarcomatous component. Subsequent examples of dedifferentiation have been described in liposarcoma, leiomyosarcoma, solitary fibrous tumor, etc 13–15. However, all these examples have been described de novo, unrelated to the use of therapeutic agents (chemotherapy, radiation, etc), occurring most likely due to acquisition of secondary genetic events, which trigger tumor progression and induce phenotypic changes. In the setting of GIST, the concept of dedifferentiation has initially been used by Pauwels et al. to illustrate the dramatic morphologic change induced by KIT-independent mechanisms of resistance to tyrosine kinase inhibitors 6. In their study, GIST dedifferentiation was defined as tumor progression from a conventional CD117-positive GIST to an anaplastic/pleomorphic CD117-negative tumor, and recognized as a potential diagnostic pitfall to the practicing pathologist, due to the change in both morphology and immunoprofile from its conventional GIST counterpart.
A somewhat similar example of morphologic and immunohistochemical switch occurring as an escape mechanism to chronic inhibition of KIT-signaling during imatinib therapy is the development of divergent rhabdomyoblastic differentiation, defined by a CD117-negative component resembling embryonal rhabdomyosarcoma and acquiring immunophenotypic evidence of skeletal muscle differentiation (desmin and myogenin reactivity)9. In at least a subset of these cases, secondary BRAF mutations have been implicated in the mechanism of inducing imatinib-resistance 10. However, no BRAF mutations were detected in any of the dedifferentiated GIST cases tested in our study.
One of the pitfalls encountered when confronted with two very different morphologies is to suggest the possibility of a collision between a conventional GIST and a different neoplasm. That was the case in our case#1, where the referring pathologist questioned the possibility of a carcinoma next to the peritoneal GIST recurrence. It is likely that the case recently reported as a collision between a giant cell-rich leiomyosarcoma and GIST also represents an example of dedifferentiated GIST 16. Another potential diagnostic pitfall is the non-random association of GIST with desmoid-type fibromatosis17. As these lesions may express weak/focal CD117 reactivity they can be confused with recurrent/progressing GIST. Conversely, based on their negative CD117 expression, these tumors, when growing on imatinib therapy, could potentially be misinterpreted as CD117-negative imatinib-resistant GIST. However, the characteristic uniform fascicular spindle cell appearance of desmoid tumors, with distinctive myofibroblastic differentiation is very different from the anaplastic and pleomorphic areas of dedifferentiation described here.
KRAS mutations have been implicated recently as a mechanism of primary resistance, being reported in 3 of 60 (5%) GIST patients and co-existing with imatinib-sensitive KIT or PDGFRA mutations in naïve GIST18. Although none of these patients have in fact been treated with imatinib to explore the impact of concomitant KRAS mutations on clinical response, in vitro studies suggested that imatinib treatment of KITΔ559/KRAS double-mutants only partially reduced the phosphorylation levels of ERK1/2 and AKT18. Interestingly, a KRAS G12V mutation was identified in one of the dedifferentiated GIST tumors; however, its significance in inducing this phenotype remains unclear, since it was not present in the other cases.
Although TP53 mutations have frequently been implicated in driving tumor progression and leading to genomic instability 19, it does not seem to play an important role in KIT-independent dedifferentiation, since it was detected in only one of the clinically advanced cases in our series and was present in both components.
Most mechanisms of drug resistance in GIST relieve the imatinib-induced apoptotic effect by reactivating the original driving oncogene, KIT, through either secondary mutations or yet unidentified pathways (so-called ‘functional resistance’). Thus, most imatinib-resistant GIST remain addicted to KIT for cell proliferation and show hyperactivation of the KIT receptor. The acquisition of secondary KIT mutations on the same allele as the primary mutation is the prevailing mechanism of failure, which typically occurs after a prolonged interval of KIT-inhibition (of at least a year)20, 21. In contrast, acquired resistance in tumors lacking detectable 2nd site mutations occur in patients who have been on the drug for a shorter period of time20. Only occasionally, imatinib resistance occurs through KIT-independent mechanisms, in which KIT expression is lost in progressing tumors, often being associated with phenotypic changes 6, 9. This phenomenon of receptor tyrosine kinase switch, which results in KIT being removed from driving cell growth, remains to be further elucidated. Although, one in vitro study implicated AXL receptor tyrosine kinase in substituting KIT to induce imatinib-resistance in GIST882 cells22, others have not been able to validate these findings.
As the presence of second site mutations in KIT has been associated with KIT-hyperactivation-driven mechanisms of resistance, it is not surprising that KIT-negative dedifferentiation is not triggered through similar pathways. Interestingly, half of the cases included in this study showed a wild-type genotype, suggesting preferential escape from KIT-addiction in tumors which exhibit oncogenic KIT signaling through mechanisms other than activating mutations. Furthermore, it has been previously shown that in contrast to KIT-mutant GIST, the wild-type tumors develop imatinib resistance in the absence of acquired KIT mutations, through yet undefined mechanisms 20, 23. The results of the present study show that loss of heterozygosity, with common loss of one KIT gene copy, was the prevalent finding in KIT-independent tumor progression on imatinib therapy. Thus, loss of KIT expression due to haploinsufficiency was found in all three cases of CD117-negative dedifferentiated GIST detected in imatinib-resistant patients. Remarkably, in two patients with de novo dedifferentiation, a low level of KIT amplification was noted, suggesting that oncogene expression is not necessarily regulated by gene dosage, and potential other mechanisms, such as KIT promoter methylation or miRNA, may play a role in this phenomenon.
In summary, we provide a detailed molecular characterization of CD117-negative dedifferentiated GIST, compared to the CD117-positive conventional GIST component. In addition to dedifferentiation in GIST occurring secondary to chronic KIT-inhibition and imatinib resistance, we provide evidence for this phenomenon developing as de-novo progression. Furthermore, we expand the morphologic spectrum of these tumors to report, for the first time, a phenotypic shift to angiosarcomatous histology. We also report the presence of a KRAS G12V mutation which co-existed with a KIT exon 11 mutation in one of the dedifferentiated GIST, in the setting of prolonged chronic inhibition of KIT signaling due to therapy for CML. However, overall the process of dedifferentiation is not associated with acquisition of additional mutations in the original driver oncogene. Instead, our findings suggest that genetic instability, represented either by loss of heterozygosity or low level amplification of KIT, is a common abnormality in the CD117-negative dedifferentiated component. The remarkable morphologic and immunophenotypic changes present in the CD117-negative anaplastic component can pose significant challenges to the practicing pathologist, eventually resulting in misdiagnoses. The previous history of GIST and/or tyrosine kinase inhibitor therapy should be a hint in raising the suspicion of dedifferentiation in GIST. It is tempting to speculate that KIT silencing may induce loss of the conventional GIST phenotype as well as abrogation of the common signalling pathway.
Acknowledgments
Supported in part by: ACS MRSG CCE-106841 (CRA), P01CA47179 (CRA), P50 CA 140146-01 (CRA), Life Raft Group (CRA), GIST Cancer Research Fund (CRA), Shuman Family Fund for GIST Research (CRA).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
The authors have no conflicts of interest or funding to disclose.
REFERENCES
- 1.Antonescu CR. The GIST paradigm: lessons for other kinase-driven cancers. J Pathol. 2011;223:251–261. doi: 10.1002/path.2798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hirota S, Isozaki K, Moriyama Y, et al. Gain-of-function mutations of c-kit in human gastrointestinal stromal tumors. Science. 1998;279:577–580. doi: 10.1126/science.279.5350.577. [DOI] [PubMed] [Google Scholar]
- 3.Rubin BP, Singer S, Tsao C, et al. KIT activation is a ubiquitous feature of gastrointestinal stromal tumors. Cancer Res. 2001;61:8118–8121. [PubMed] [Google Scholar]
- 4.Heinrich MC, Corless CL, Duensing A, et al. PDGFRA activating mutations in gastrointestinal stromal tumors. Science. 2003;299:708–710. doi: 10.1126/science.1079666. [DOI] [PubMed] [Google Scholar]
- 5.Medeiros F, Corless CL, Duensing A, et al. KIT-negative gastrointestinal stromal tumors: proof of concept and therapeutic implications. Am J Surg Pathol. 2004;28:889–894. doi: 10.1097/00000478-200407000-00007. [DOI] [PubMed] [Google Scholar]
- 6.Pauwels P, Debiec-Rychter M, Stul M, et al. Changing phenotype of gastrointestinal stromal tumours under imatinib mesylate treatment: a potential diagnostic pitfall. Histopathology. 2005;47:41–47. doi: 10.1111/j.1365-2559.2005.02179.x. [DOI] [PubMed] [Google Scholar]
- 7.Agaram NP, Besmer P, Wong GC, et al. Pathologic and molecular heterogeneity in imatinib-stable or imatinib-responsive gastrointestinal stromal tumors. Clin Cancer Res. 2007;13:170–181. doi: 10.1158/1078-0432.CCR-06-1508. [DOI] [PubMed] [Google Scholar]
- 8.Diaz Delgado M, Hernandez Amate A, Pereira Gallardo S, et al. Gastrointestinal stromal tumors: morphological, immunohistochemical and molecular changes associated with kinase inhibitor therapy. Pathology oncology research : POR. 2011;17:455–461. doi: 10.1007/s12253-011-9362-2. [DOI] [PubMed] [Google Scholar]
- 9.Liegl B, Hornick JL, Corless CL, et al. Monoclonal antibody DOG1.1 shows higher sensitivity than KIT in the diagnosis of gastrointestinal stromal tumors, including unusual subtypes. Am J Surg Pathol. 2009;33:437–446. doi: 10.1097/PAS.0b013e318186b158. [DOI] [PubMed] [Google Scholar]
- 10.Agaram NP, Wong GC, Guo T, et al. Novel V600E BRAF mutations in imatinibnaive and imatinib-resistant gastrointestinal stromal tumors. Genes Chromosomes Cancer. 2008;47:853–859. doi: 10.1002/gcc.20589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Demetri GD, von Mehren M, Antonescu CR, et al. NCCN Task Force report: update on the management of patients with gastrointestinal stromal tumors. Journal of the National Comprehensive Cancer Network : JNCCN. 2010;8(Suppl 2):S1–S41. doi: 10.6004/jnccn.2010.0116. quiz S42–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dahlin DC, Beabout JW. Dedifferentiation of low-grade chondrosarcomas. Cancer. 1971;28:461–466. doi: 10.1002/1097-0142(197108)28:2<461::aid-cncr2820280227>3.0.co;2-u. [DOI] [PubMed] [Google Scholar]
- 13.Evans HL. Liposarcoma: a study of 55 cases with a reassessment of its classification. Am J Surg Pathol. 1979;3:507–523. doi: 10.1097/00000478-197912000-00004. [DOI] [PubMed] [Google Scholar]
- 14.Chen E, O'Connell F, Fletcher CD. Dedifferentiated leiomyosarcoma: clinicopathological analysis of 18 cases. Histopathology. 2011;59:1135–1143. doi: 10.1111/j.1365-2559.2011.04070.x. [DOI] [PubMed] [Google Scholar]
- 15.Mosquera JM, Fletcher CD. Expanding the spectrum of malignant progression in solitary fibrous tumors: a study of 8 cases with a discrete anaplastic component--is this dedifferentiated SFT? Am J Surg Pathol. 2009;33:1314–1321. doi: 10.1097/pas.0b013e3181a6cd33. [DOI] [PubMed] [Google Scholar]
- 16.Insabato L, Masone S, Campione S, et al. Coexistence of primary gastric giant cell-rich leiomyosarcoma and gastrointestinal stromal tumor: report of a very rare combination and review of the literature. International journal of surgical pathology. 2012;20:74–78. doi: 10.1177/1066896911414018. [DOI] [PubMed] [Google Scholar]
- 17.Dumont AG, Rink L, Godwin AK, et al. A nonrandom association of gastrointestinal stromal tumor (GIST) and desmoid tumor (deep fibromatosis): case series of 28 patients. Ann Oncol. 2012;23:1335–1340. doi: 10.1093/annonc/mdr442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Miranda C, Nucifora M, Molinari F, et al. KRAS and BRAF mutations predict primary resistance to imatinib in gastrointestinal stromal tumors. Clin Cancer Res. 2012;18:1769–1776. doi: 10.1158/1078-0432.CCR-11-2230. [DOI] [PubMed] [Google Scholar]
- 19.Romeo S, Debiec-Rychter M, Van Glabbeke M, et al. Cell cycle/apoptosis molecule expression correlates with imatinib response in patients with advanced gastrointestinal stromal tumors. Clin Cancer Res. 2009;15:4191–4198. doi: 10.1158/1078-0432.CCR-08-3297. [DOI] [PubMed] [Google Scholar]
- 20.Antonescu CR, Besmer P, Guo T, et al. Acquired resistance to imatinib in gastrointestinal stromal tumor occurs through secondary gene mutation. Clin Cancer Res. 2005;11:4182–4190. doi: 10.1158/1078-0432.CCR-04-2245. [DOI] [PubMed] [Google Scholar]
- 21.Debiec-Rychter M, Cools J, Dumez H, et al. Mechanisms of resistance to imatinib mesylate in gastrointestinal stromal tumors and activity of the PKC412 inhibitor against imatinib-resistant mutants. Gastroenterology. 2005;128:270–279. doi: 10.1053/j.gastro.2004.11.020. [DOI] [PubMed] [Google Scholar]
- 22.Mahadevan D, Cooke L, Riley C, et al. A novel tyrosine kinase switch is a mechanism of imatinib resistance in gastrointestinal stromal tumors. Oncogene. 2007;26:3909–3919. doi: 10.1038/sj.onc.1210173. [DOI] [PubMed] [Google Scholar]
- 23.Liegl B, Kepten I, Le C, et al. Heterogeneity of kinase inhibitor resistance mechanisms in GIST. J Pathol. 2008;216:64–74. doi: 10.1002/path. [DOI] [PMC free article] [PubMed] [Google Scholar]