

Abstract
Cryo-electron tomography (cryoET) allows 3D visualization of cellular structures at molecular resolution in a close-to-physiological state1. However, direct visualization of individual viral complexes in their host cellular environment with cryoET is challenging2, due to the infrequent and dynamic nature of viral entry, particularly in the case of HIV-1. While time-lapse live-cell imaging has yielded a great deal of information about many aspects of the life cycle of HIV-13-7, the resolution afforded by live-cell microscopy is limited (~ 200 nm). Our work was aimed at developing a correlation method that permits direct visualization of early events of HIV-1 infection by combining live-cell fluorescent light microscopy, cryo-fluorescent microscopy, and cryoET. In this manner, live-cell and cryo-fluorescent signals can be used to accurately guide the sampling in cryoET. Furthermore, structural information obtained from cryoET can be complemented with the dynamic functional data gained through live-cell imaging of fluorescent labeled target.
In this video article, we provide detailed methods and protocols for structural investigation of HIV-1 and host-cell interactions using 3D correlative high-speed live-cell imaging and high-resolution cryoET structural analysis. HeLa cells infected with HIV-1 particles were characterized first by confocal live-cell microscopy, and the region containing the same viral particle was then analyzed by cryo-electron tomography for 3D structural details. The correlation between two sets of imaging data, optical imaging and electron imaging, was achieved using a home-built cryo-fluorescence light microscopy stage. The approach detailed here will be valuable, not only for study of virus-host cell interactions, but also for broader applications in cell biology, such as cell signaling, membrane receptor trafficking, and many other dynamic cellular processes.
Keywords: Bioengineering, Issue 76, Molecular Biology, Structural Biology, Virology, Biophysics, Cellular Biology, Physiology, Medicine, Biomedical Engineering, Infection, Microbiology, Technology, Industry, Agriculture, Life Sciences (General), Correlative microscopy, CryoET, Cryo-electron tomography, Confocal live-cell imaging, Cryo-fluorescence light microscopy, HIV-1, capsid, HeLa cell, cell, virus, microscopy, imaging
Introduction
Cryo-electron tomography (cryoET) is a powerful imaging technique that allows three-dimensional (3D) visualization of cells and tissues and provides insights into the organization of native organelles and cellular structures at molecular resolution in a close-to physiological state1. However, the inherently low contrast of unstained frozen-hydrated specimen, combined with their radiation sensitivity, makes it difficult to locate areas of interest inside a cell and subsequently performing the tilt series successfully without damaging the target area. In order to overcome these problems, a correlative approach that combines light and electron microscopy is necessary. Specific features highlighted by fluorescent labeling are identified and located by fluorescence light microscopy, and then their coordinates are transferred to the electron microscope for acquisition of high resolution 3D structural data. This correlative method helps locating the target areas of interest to be addressed. Due to the limitation on sample thickness with cryoEM (<300 nm), currently only the peripheral regions of the cell are suitable for 3D structural analysis by CryoET. Further reducing the thickness of frozen-hydrated specimens by vitreous sectioning8 or by cryo-focused ion beam (FIB) milling9 would expand the capability of correlative imaging.
Previously, correlative methods were primarily used to facilitate cryoET data acquisition for large and static structures10-13. In these studies, cryo-stages have been implemented to accept cryoEM grids and fit onto either an upright microscope or an inverted microscope10,11,14. Although switching grids seems fairly straightforward in their designs, there are additional transferring steps involved for the EM grid, increasing the chance that the grid may be deformed, damaged and contaminated. We recently demonstrated a technical advance in correlative microscopy that allows us to directly visualize dynamic events that are by nature difficult to capture, such as HIV-1 and host cell interactions at early stages of infection15. We accomplished this by designing and implementing a cryo-light microscopy sample stage that adapts a cartridge system to minimize the specimen damage due to grid handling, thus facilitating correlation. Our design includes an integrated specimen cartridge holder, allowing both cryo-light and cryo-electron microscopy to be performed, sequentially, on the same specimen holder, without sample transfer, thus streamlining the correlative process. In addition, we also implemented an accurate and reliable correlation procedure using fluorescent latex beads as fiducial markers.
Protocol
1. HeLa Cell Culture on Carbon-coated, Gold EM Finder Grids
Glow discharge the carbon-side of a 200-mesh R2/2 Quantifoil gold EM finder grid under 25 mA for 25 sec.
Coat the EM grid with fibronectin, by floating it, carbon-side down, onto a 40 μl droplet of 50 μg/ml fibronectin solution, then disinfect it under UV light for 2 hr in a tissue culture hood.
Plate HeLa cells onto the grid at a density of 2 x 104 cells/ml (total 2 ml culture) in a glass-bottom culture dish and grow the cells at 37 °C, with 5% CO2, in DMEM supplemented with 4.5 g/L L-glutamine and glucose, 10% heat-inactivated fetal calf serum, 100 units/ml penicillin, and 100 μg/ml streptomycin, for approximately 18 hr. HeLa cells are infected after O/N culture.
2. HIV-1 Infection and Live-cell Imaging
Add a fluorescent cell tracker (Red CMTPX, 1 μl/2 ml) into the glass-bottom culture dish (from step 1.3) and incubate the dish at 37 °C for 10 min, to allow up-take of the fluorescent dye.
Wash the cells with PBS and add 50 μl pre-warmed fresh medium.
Place the dish onto the live-cell chamber, at 37 °C, of a Swept Field Confocal scanner microscope. Select multiple fields (10-15 positions) that contain 1-3 cells/square, with the cells between two mitosis phases when they are most spread-out and flat (see Figures 2c and 2d), since cryoEM requires relatively thin regions of the sample. Store these positions for future imaging (step 2.5).
Infect the cells with VSV-G pseudo-typed HIV-1 particles that contain GFP-Vpr (we use 40 μl of a sample containing 40 ng/ml of p24). When adding the virus particles into the bottom of the dish, be careful to not disturb the EM grid, since the positions for imaging have already been selected and stored.
Immediately after addition of virions, collect time-lapse high-speed 3D confocal images, at the previously selected positions (from step 2.3) for 20-40 min.
Confocal images were acquired using MetaMorph and analyzed by automated 3D particle tracking for single particle dynamics using Imaris software (See Figure 1). Since we are correlating the dynamic behavior of diffraction-limited HIV-1 particles with cellular ultrastructure, time-lapse live-cell imaging and 3D particle tracking are critical to the results. However, for a large and static structure, a simple fluorescence image (without live-cell) would be sufficient for correlative analysis.
3. Frozen-hydrated EM Sample Preparation
To minimize the time delay between collection of the last confocal image and cryo-fixation of the sample, the FEI Vitrobot (or other vitrification device) should be initiated and prepared for plunge-freezing during collection of fluorescence confocal live-cell images.
Turn on the vitrification device and set its climate temperature to 22 °C, the target humidity to 100%, the blotting time to 7 sec, and the wait and drain time to 1 sec.
Place the grid storage box in the plunger dewar and prepare cold liquid ethane. Mount the dewar onto the vitrification device.
Immediately after confocal live-cell imaging (section 2), place the culture dish onto an ice-cooled copper block and transfer it to the cryoEM sample preparation room. Load the EM grid onto the specialized tweezers and quickly blot away any media on the grid. The blotting is done manually, with filter paper, after which 4 μl of 15 nm gold bead solution mixed with 0.2 μm fluorescent microspheres is immediately placed on the grid. The gold beads are used as fiducial markers to aid tomographic data collection and alignment. The fluorescent microspheres are used to aid correlation between fluorescent and cryoEM images (See Figures 2c-2f).
Load the tweezers onto the vitrification device and instruct the device to blot and plunge freeze in liquid ethane16. The blotting parameters optimized to achieve best result are: blot time, 7 sec; blot offset, 0; drain time, 1 sec; temperature, 22 °C; and humidity, 100%.
Transfer the frozen-hydrated grid to a cryoEM holder for immediate cryoET imaging, or to a liquid nitrogen storage dewar for later use.
4. Cryo-fluorescence Light Microscopy
A home-built, cryo-fluorescence sample chamber and stage system (Figure 3) is required to carry out steps 4.1- 4.10. The detailed specifications and description of the stage may be found in Jun et al.15.
Fill a self-pressurized liquid nitrogen dewar with liquid nitrogen at least 2 hr before starting cryo-fluorescence light microscopy (See Figure 3a).
Mount a homebuilt cryo-fluorescence sample stage15 (Figure 3) onto an inverted fluorescence light microscope, such as an Olympus IX 71.
Connect the liquid nitrogen inlet of the cryo-sample stage to the self-pressurized dewar and place the liquid nitrogen overflow protection outlet into an appropriate container. Connect a dry nitrogen gas line to a sleeve placed over the objective lens to keep the lens warm and free of frost.
Place a copper block platform (black arrow in Figure 3b) into the cryo-stage chamber for loading the frozen-hydrated sample grid. Cool down and fill the copper chamber of the cryo-stage with liquid nitrogen through the inlet line from the self-pressurized filling dewar.
When the cryo-stage reaches liquid nitrogen temperature (in ~4-6 min), transfer the grid storage box (from step 3.5) to the cryo-stage.
Place the frozen-hydrated sample grid into the EM specimen cartridge on the copper block and clip the grid in place using a C-ring (if using a Polara microscope cartridge), or place a pre-cooled copper ring on top (black arrowhead in Figure 3d), to keep the grid in place and also for easy retrieval of the grid after cryo-fluorescence imaging (if for a non-Polara cryo-electron microscope).
Place the cartridge (from step 4.6) in the inner chamber of the cryo-stage (white arrowhead in Figure 3b).
Search and find the same virus particles from the live-cell imaging data. Since the EM grid has an index, it is straightforward to localize the same particle on the grid in both live-cell images and cryo-florescence images.
Acquire cryo-DIC and GFP images at the identified positions under cryo-condition using a light microscope with a long working (2.7-4 mm) objective lens. During the cryo-fluorescence imaging, periodically check the liquid nitrogen level in the cryo-stage and refill it, as needed, to keep the sample stage below -170 °C.
Upon completion of cryo-fluorescence imaging, carefully return the specimen cartridge to the copper block platform and store the cartridge in a liquid nitrogen dewar for future cryoET analysis with a Polara system. If using a non-Polara cryo-electron microscope, remove the copper ring, retrieve the sample grid, place it in a grid storage box, and store the specimen in a liquid nitrogen dewar until the specimen is examined by cryoET.
For data analysis, overlap the acquired cryo-DIC and GFP images (from step 4.9, see Figure 4b) and correlate the positions of the GFP signals in the cryo-fluorescence image with those obtained in the live-cell imaging series.
5. Cryo-electron Tomography
Load the sample cartridge into the cryo-transfer station of an electron microscope equipped with a field emission gun, such as Polara G2 microscope.
Under low-dose-search mode at a magnification of 140X, identify and save the associated grid squares (from step 4.11) into a stage file.
Under low-dose-search mode at a magnification of 3,500X, insert a 100 μm objective aperture, search and save all the positions correlated with GFP signals into a second stage file.
Under low-dose-exposure mode, find a blank area to adjust beam intensity to a dose of 1 or 2 e-/Å2 and refine the tilt axis, using the stage y function, by burning away the ice in an unimportant carbon region with several gold beads.
Under low-dose-focus mode, adjust the focus distance to ~3 μm and adjust the angle to align the direction of focus to be parallel to the tilt axis.
Under low-dose-search mode, recall saved positions (from step 5.3), adjust the specimen eucentric height and check the tilt range for the area of interest. Acquire a projection image with very low electron-dose (~1 e-/Å2) at 8 to 10 μm defocus, in exposure mode, to confirm the correlated virus particles for cryo-electron tomography.
Switch to the low-dose-focus mode. In TEM auto functions menu, precede the automated eucentric height and focus functions on the focus area.
Set up the parameters for acquiring a tilt-series. For Figure 4 in this paper, tilt angles ranging ±70°, below and above 45°, at 3° and 2° tilt increments, respectively, were used, with a 6 μm defocus and approximately 70 e-/Å2 total dose.
Repeat steps 5.7 and 5.8 for all the saved positions. Batch tomography software can be used for automated data collection at multiple positions to increase the throughput.
6. Three-dimensional Reconstruction using IMOD17
Inspect the raw tilt series to adjust the minimum and maximum density values and determine if there is any view to be excluded due to large image shift.
Start eTomo and setup tomogram panel using axis type, pixel size, fiducial diameter, etc. Then create com scripts.
Configure the main window such that several stages of the tomogram computation can be adjusted/followed simultaneously. Modify the necessary parameters and execute the specific programs that are required by each processing step (See eTomo tutorial, http://bio3d.colorado.edu/imod/doc/etomoTutorial.html).
At the final stage, create a full aligned stack (with a mean residual error less than 0.6) and reconstruct tomograms using a weighted back-projection algorithm in IMOD.
Denoise potential areas of interest using a 3D nonlinear anisotropic diffusion edge enhancing program implemented in IMOD with appropriate parameters to enhance the contrast and clarity.
Representative Results
To characterize the dynamic behavior of the virus particles, HeLa cells infected with HIV-1 were imaged by high-speed confocal live-cell microscopy and the particle movements were analyzed by automated 3D particle tracking (Figure 1). To avoid the time lapse of several minutes that can occur between collection of the last confocal live-cell image and plunge-freezing (which may be long enough to lose correlated HIV-1 particles), a cryo-fluorescence light microscopy stage (Figure 3) was designed and constructed for imaging frozen-hydrated samples15, within cryoEM cartridges, on light microscopes. A copper block platform (Figure 3b, black arrow) and copper ring (Figure 3d, arrowhead) was added for use with non-Polara electron microscopes. Correlation between the confocal live-cell, cryo-fluorescence light microscopy, and cryoET was achieved using indexed gold Quantifoil grids and 0.2 μm fluorescent latex beads (Figure 2). Taken together, Figure 4 demonstrates the feasibility of the overall procedure for advanced correlative live-cell microscopy and cryo-electron tomography to visualize fluorescently labeled HIV-1 particles interacting with a host HeLa cell.
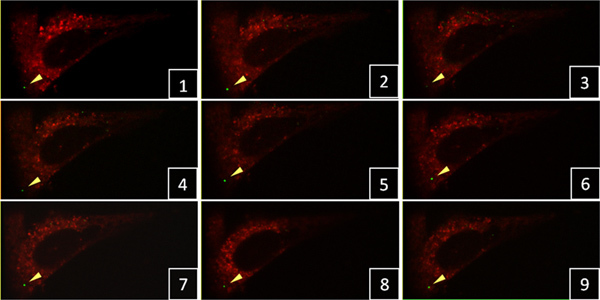 Figure 1. Time-lapse confocal live-cell imaging of HIV-1 particles in HeLa cells. A single green-fluorescent viral particle (yellow arrowhead) was tracked in 3D confocal stacks with a 3 min time interval between frames. Click here to view larger figure.
Figure 1. Time-lapse confocal live-cell imaging of HIV-1 particles in HeLa cells. A single green-fluorescent viral particle (yellow arrowhead) was tracked in 3D confocal stacks with a 3 min time interval between frames. Click here to view larger figure.
 Figure 2. Correlation between fluorescence images and cryoEM images. (a & b) Differential interference contrast (DIC) images of HeLa cells cultured on a Quantifoil gold EM finder grid, recorded with a 2X (a) and 20X (b) objectives. (c) Cryo-fluorescence image of frozen-hydrated HeLa cells (red color for mitochondria) and 0.2 μm fluorescent latex beads (green), recorded with a cryo-stage and a 40X long working distance air objective, overlaid with a DIC image of cells. (d) Low magnification cryoEM image of the corresponding region in (c). (e & f) Correlation between fluorescence and cryoEM images using 0.2 μm fluorescent latex beads. Corresponding beads are circled with the same color. Scale bars, 200 μm in a, 50 μm in b, 25 μm in c, d & e, 5 μm in f.
Figure 2. Correlation between fluorescence images and cryoEM images. (a & b) Differential interference contrast (DIC) images of HeLa cells cultured on a Quantifoil gold EM finder grid, recorded with a 2X (a) and 20X (b) objectives. (c) Cryo-fluorescence image of frozen-hydrated HeLa cells (red color for mitochondria) and 0.2 μm fluorescent latex beads (green), recorded with a cryo-stage and a 40X long working distance air objective, overlaid with a DIC image of cells. (d) Low magnification cryoEM image of the corresponding region in (c). (e & f) Correlation between fluorescence and cryoEM images using 0.2 μm fluorescent latex beads. Corresponding beads are circled with the same color. Scale bars, 200 μm in a, 50 μm in b, 25 μm in c, d & e, 5 μm in f.
 Figure 3. Construction of cryo-light microscopy stage.(a) A self-pressurized dewar, filled with liquid nitrogen, used to cool down the copper cryo-sample stage (white arrow). (b) Top, inside view of the cryo-sample stage, showing the inner chamber (white arrowhead). The sample grid is placed onto the EM specimen cartridge, which sits in the center of a copper block (black arrow). Once loaded with the sample grid, the cartridge is transferred to the inner chamber (white arrowhead) for cryo-fluorescence light microscopy. (c & d) The specimen cartridge before (c) and after (d) placing a copper ring (black arrowhead) to keep the grid in place for using with non-Polara cryo-electron microscope.
Figure 3. Construction of cryo-light microscopy stage.(a) A self-pressurized dewar, filled with liquid nitrogen, used to cool down the copper cryo-sample stage (white arrow). (b) Top, inside view of the cryo-sample stage, showing the inner chamber (white arrowhead). The sample grid is placed onto the EM specimen cartridge, which sits in the center of a copper block (black arrow). Once loaded with the sample grid, the cartridge is transferred to the inner chamber (white arrowhead) for cryo-fluorescence light microscopy. (c & d) The specimen cartridge before (c) and after (d) placing a copper ring (black arrowhead) to keep the grid in place for using with non-Polara cryo-electron microscope.
 Figure 4. CryoET of a single fluorescent particle bound to the cellular protrusion of a HeLa cell by correlative microscopy.(a & b) A DIC image recorded with a cryo-light microscopy stage (a) overlaid with a cryo-fluorescence image of GFP-tagged particles (red) (b). (c - e) Low dose cryoEM images of the region containing a fluorescent particle (circled in panel b & c) at 140X (c), 3,500X (d) and 27,500X (e), respectively. Inset, an enlarged view recorded after acquisition of the tomographic tilt series. (f - h) Three 4 nm thick tomographic slices separated by a distance of 21 nm in the z direction. Connections between the particle and HeLa cell membrane are indicated by arrows in both the projection image (inset in panel e) and tomographic slices (panels f & g). Scale bars, 10 μm in a & b, 20 μm in c, 500 nm in d, and 100 nm in e - h.
Figure 4. CryoET of a single fluorescent particle bound to the cellular protrusion of a HeLa cell by correlative microscopy.(a & b) A DIC image recorded with a cryo-light microscopy stage (a) overlaid with a cryo-fluorescence image of GFP-tagged particles (red) (b). (c - e) Low dose cryoEM images of the region containing a fluorescent particle (circled in panel b & c) at 140X (c), 3,500X (d) and 27,500X (e), respectively. Inset, an enlarged view recorded after acquisition of the tomographic tilt series. (f - h) Three 4 nm thick tomographic slices separated by a distance of 21 nm in the z direction. Connections between the particle and HeLa cell membrane are indicated by arrows in both the projection image (inset in panel e) and tomographic slices (panels f & g). Scale bars, 10 μm in a & b, 20 μm in c, 500 nm in d, and 100 nm in e - h.
Discussion
We have presented a straightforward set of protocols to provide an advanced correlative approach to analyze dynamic cellular events using time-lapse confocal, live-cell fluorescence imaging followed by cryoET. Our methodological development to correlate 3D live-cell imaging with high-resolution cryoET is critical to investigate many challenging biological problems, such as visualizing rare, dynamic (not static, as previously reported), and diffraction limited viral particles and their interactions with host cells. The spatial and temporal behavior of specific particles in living cells is characterized by live-cell analysis and the structural details of virus-cell interactions at the defined infection stages are explored. A homemade, cryo-light microscopy stage is used to facilitate direct correlation between fluorescence light images from live cells and images captured by cryoEM.
There are several important points to keep in mind in order to have optimal data collection and analysis. First, when picking multiple positions for time-lapse confocal live-cell imaging, the selected positions should be within 0.5 mm from the center of the EM grid, since the edge of the grid interferes with the field of view for tilt-series acquisition in cryoET. Further, the cells selected for analysis should be between two mitosis phases, when they are most spread-out and flat. Therefore, the thin regions of cells, available for cryoET analysis, are at their most extended stage. In order to have the majority of cultured cells at the same interphase stage, cell cycle can be synchronized using double thymidine treatment that blocks cell cycle at the early S-phase18. Cells released from thymidine block then progress synchronously through G2- and mitotic phase.
Second, as mentioned earlier, the time delay between the last frame of confocal image and cryo-fixation of the sample should be minimized to allow efficient direct correlation of dynamic objects. In addition, the flatness and integrity of the grid are critical for all procedures and must be maintained throughout the protocol, particularly when bare grids are handled for non-Polara microscopes.
Finally, precise and reliable correlation between the fluorescence image and the electron microscopy image, to find the target, is required for successful subsequent acquisition of cryoET data. In a projection image, at a low magnification, the recognition of the shape and direction of a selected cell and sometimes counting holes on the grid are necessary to locate the correlated region in the EM field. Fluorescent latex beads are added to the sample for a more robust and accurate correlation. Using fluorescent latex beads or Quantum dots, which are both fluorescent and electron-dense, the coordinates of the target in fluorescence image can be directly transferred to EM for a precise localization. It should be noted that the emission wavelength of fluorescent latex beads or Quantum dots is chosen not to have overlap with the emission wavelength of the target.
Our current correlation set up for identifying a target area takes place outside of a cryo-electron microscope, thus involving an additional sample transfer step from an optical microscope to an electron microscope. A more direct and straightforward method has been thought out19, by installing a fluorescent imaging apparatus within the electron microscope. In addition, the resolution afforded by a long working distance air objective with NA of 0.6 is limited (~0.6 μm). Precise and accurate nanoscale single molecule localization in 3D cellular tomograms will further involve super resolution microscopy to correlate fluorescent labeled proteins in the ultrastructure.
Further advances in correlative light microscopy and cryoET are expected by combining additional techniques, such as cryo-focused ion beam (FIB) milling9,20 and vitreous sectioning8 of frozen-hydrated specimens. These techniques overcome the limitations of the specimen thickness, greatly expanding the range of samples amenable for cryoET analysis, thus allowing investigation of viral particles that have traveled deep inside of the cells. In terms of the study of virus particle interactions with host cell factors, further double labeling of specific cellular protein and the HIV-1 cores with a tetra-Cysteine fluorescence probe fused to HIV-1 CA could facilitate the identification of particular early stages of the HIV-1 life cycle and the spatial and functional relationship of host factors and viral particles. We anticipate the correlative 3D live-cell imaging and cryoET methodology to play important roles in study cell signaling, membrane receptor trafficking, and many other dynamic cellular processes in cell biology.
Disclosures
The authors declare no competing financial interests.
Acknowledgments
The authors would like to thank Travis Wheeler and the machine shop at the Department of Cell Biology and Physiology, University of Pittsburgh for construction of the cryo-fluorescence sample stage, Changlu Tao and Cheng Xu at the University of Science and Technology of China for technical assistance, and Dr. Teresa Brosenitsch for critical reading of the manuscript. This work was supported by the National Institutes of Health (GM082251 & GM085043).
References
- Leis A, Rockel B, Andrees L, Baumeister W. Visualizing cells at the nanoscale. Trends in Biochemical Sciences. 2009;34:60–70. doi: 10.1016/j.tibs.2008.10.011. [DOI] [PubMed] [Google Scholar]
- Maurer UE, Sodeik B, Grunewald K. Native 3D intermediates of membrane fusion in herpes simplex virus 1 entry. Proceedings of the National Academy of Sciences of the United States of America. 2008;105:10559–10564. doi: 10.1073/pnas.0801674105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDonald D, et al. Visualization of the intracellular behavior of HIV in living cells. The Journal of Cell Biology. 2002;159:441–452. doi: 10.1083/jcb.200203150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arhel N, et al. Quantitative four-dimensional tracking of cytoplasmic and nuclear HIV-1 complexes. Nature Methods. 2006;3:817–824. doi: 10.1038/nmeth928. [DOI] [PubMed] [Google Scholar]
- Gousset K, et al. Real-time visualization of HIV-1 GAG trafficking in infected macrophages. PLoS Pathogens. 2008;4:e1000015. doi: 10.1371/journal.ppat.1000015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jouvenet N, Bieniasz PD, Simon SM. Imaging the biogenesis of individual HIV-1 virions in live cells. Nature. 2008;454:236–240. doi: 10.1038/nature06998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koch P, et al. Visualizing fusion of pseudotyped HIV-1 particles in real time by live cell microscopy. Retrovirology. 2009;6:84. doi: 10.1186/1742-4690-6-84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang P, et al. Direct visualization of receptor arrays in frozen-hydrated sections and plunge-frozen specimens of E. coli engineered to overproduce the chemotaxis receptor Tsr. Journal of Microscopy. 2004;216:76–83. doi: 10.1111/j.0022-2720.2004.01395.x. [DOI] [PubMed] [Google Scholar]
- Wang K, Strunk K, Zhao G, Gray JL, Zhang P. 3D structure determination of native mammalian cells using cryo-FIB and cryo-electron tomography. Journal of Structural Biology. 2012;180:318–326. doi: 10.1016/j.jsb.2012.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sartori A, et al. Correlative microscopy: bridging the gap between fluorescence light microscopy and cryo-electron tomography. Journal of Structural Biology. 2007;160:135–145. doi: 10.1016/j.jsb.2007.07.011. [DOI] [PubMed] [Google Scholar]
- Schwartz CL, Sarbash VI, Ataullakhanov FI, McIntosh JR, Nicastro D. Cryo-fluorescence microscopy facilitates correlations between light and cryo-electron microscopy and reduces the rate of photobleaching. Journal of Microscopy. 2007;227:98–109. doi: 10.1111/j.1365-2818.2007.01794.x. [DOI] [PubMed] [Google Scholar]
- van Driel LF, Valentijn JA, Valentijn KM, Koning RI, Koster AJ. Tools for correlative cryo-fluorescence microscopy and cryo-electron tomography applied to whole mitochondria in human endothelial cells. European Journal of Cell Biology. 2009;88:669–684. doi: 10.1016/j.ejcb.2009.07.002. [DOI] [PubMed] [Google Scholar]
- Rigort A, et al. Micromachining tools and correlative approaches for cellular cryo-electron tomography. Journal of Structural Biology. 2010;172:169–179. doi: 10.1016/j.jsb.2010.02.011. [DOI] [PubMed] [Google Scholar]
- Gruska M, Medalia O, Baumeister W, Leis A. Electron tomography of vitreous sections from cultured mammalian cells. Journal of Structural Biology. 2008;161:384–392. doi: 10.1016/j.jsb.2007.10.008. [DOI] [PubMed] [Google Scholar]
- Jun S, et al. Direct visualization of HIV-1 with correlative live-cell microscopy and cryo-electron tomography. Structure. 2011;19:1573–1581. doi: 10.1016/j.str.2011.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dubochet J, et al. Cryo-electron microscopy of vitrified specimens. Quarterly Reviews of Biophysics. 1988;21:129–228. doi: 10.1017/s0033583500004297. [DOI] [PubMed] [Google Scholar]
- Kremer JR, Mastronarde DN, McIntosh JR. Computer visualization of three-dimensional image data using IMOD. Journal of Structural Biology. 1996;116:71–76. doi: 10.1006/jsbi.1996.0013. [DOI] [PubMed] [Google Scholar]
- Whitfield ML, et al. Identification of genes periodically expressed in the human cell cycle and their expression in tumors. Molecular Biology of the Cell. 2002;13:1977–2000. doi: 10.1091/mbc.02-02-0030.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Agronskaia AV, et al. Integrated fluorescence and transmission electron microscopy. Journal of Structural Biology. 2008;164:183–189. doi: 10.1016/j.jsb.2008.07.003. [DOI] [PubMed] [Google Scholar]
- Marko M, Hsieh C, Schalek R, Frank J, Mannella C. Focused-ion-beam thinning of frozen-hydrated biological specimens for cryo-electron microscopy. Nature Methods. 2007;4:215–217. doi: 10.1038/nmeth1014. [DOI] [PubMed] [Google Scholar]