

Abstract
The objective of this study was to identify quantitative trait loci (QTL) affecting fitness of hybrids between wild soybean (Glycine soja) and cultivated soybean (Glycine max). Seed dormancy and seed number, both of which are important for fitness, were evaluated by testing artificial hybrids of G. soja × G. max in a multiple-site field trial. Generally, the fitness of the F1 hybrids and hybrid derivatives from self-pollination was lower than that of G. soja due to loss of seed dormancy, whereas the fitness of hybrid derivatives with higher proportions of G. soja genetic background was comparable with that of G. soja. These differences were genetically dissected into QTL for each population. Three QTLs for seed dormancy and one QTL for total seed number were detected in the F2 progenies of two diverse cross combinations. At those four QTLs, the G. max alleles reduced seed number and severely reduced seed survival during the winter, suggesting that major genes acquired during soybean adaptation to cultivation have a selective disadvantage in natural habitats. In progenies with a higher proportion of G. soja genetic background, the genetic effects of the G. max alleles were not expressed as phenotypes because the G. soja alleles were dominant over the G. max alleles. Considering the highly inbreeding nature of these species, most hybrid derivatives would disappear quickly in early self-pollinating generations in natural habitats because of the low fitness of plants carrying G. max alleles.
Keywords: Fitness, Glycine soja × G. max, introgression, QTL
Introduction
Many crop species have evolved through recurrent cycles of hybridization with their wild and/or weedy relatives followed by differentiation (Harlan 1992). Gene flow from crops to their wild relatives has been commonly observed in many crop species (Ellstrand et al. 1999; Ellstrand 2003). There is a concern that transgenes in crops will persist in the gene pool of wild relatives and lead to negative environmental effects because of the difficulty in controlling gene flow completely when genetically modified (GM) crops are field planted. Possible concerns related to transgene introgression are the evolution of aggressive weeds from hybrid derivatives (Warwick et al. 2009), the influence on nontarget insects (O'Callaghan et al. 2005), and the changes in genetic diversity of wild populations (Levin et al. 1996; Lu 2008).
The probability of transgene introgression from a crop species into a wild species is largely dependent on the fitness of the F1 hybrid and subsequent generations. Fitness may be defined as the relative ability of an individual to survive and successfully reproduce in a given environment, with the most fit individuals leaving the greatest number of offspring (Jenczewski et al. 2003). Fitness is not only a characteristic of entire genome, it is also a property of individual genes and chromosomal segments (Harrison 1990). The persistence of transgenes from crop plants within the genomes of crop wild relatives is dependent on the fitness conferred by the transgene and by linked genomic regions (Gressel 1999; Jenczewski et al. 2003; Stewart et al. 2003). The fitness of plants carrying domestication-related genes is assumed to be lower than that of their wild relatives when tested in natural habitats (De Wet and Harlan 1975). Transgenes would be expected to disappear in natural populations when linked with domestication-related genes that lead to a selective disadvantage in wild habitats, such as seed dormancy and seed shattering (Gressel 1999; Stewart et al. 2003).
On the other hand, chromosomal blocks can introgress at a higher rate than expected when they contain advantageous gene combinations with positive fitness consequences (Rieseberg et al. 1996). There are some cases in which hybrids between wild relatives and crop plants may be as fit as or even more fit than their parents; examples have been found in Brassica (Snow et al. 1999; Di et al. 2009), Raphanus (Hovick et al. 2012), and Sorghum (Sahoo et al. 2010). Baack et al. (2008) found that some alleles from cultivated sunflower (Helianthus annuus L.) are favored in a noncrop environment and in wild genetic backgrounds. Depending on the effect of the inserted gene itself, transfer of transgenes could lead to a change in allele frequencies through a selective advantage conferred to the recipient (Hails 2000; Gepts and Papa 2003; Jenczewski et al. 2003; Snow et al. 2010; Hartman et al. 2012).
Genetically modified soybean (Glycine max) is economically important and accounted for 81% of the worldwide planting area of soybean in 2012 (81 million ha; James 2012). The annual wild species Glycine soja is found in eastern and northeastern China, Japan, Korea, and far eastern Russia (Carter et al. 2004). In Japan, G. soja is distributed widely in disturbed habitats such as riverbanks, roadsides, and even at the edges of soybean fields (Kaga et al. 2005; Kuroda et al. 2005, 2006b, 2007). Reproductive barriers have not been observed between G. max and G. soja, and the crosses can produce fertile F1 hybrids (Singh and Hymowitz 1989; Carter et al. 2004). The risk of transgene dispersal within Glycine is assumed to be very low in Japan because (1) outcrossing rates between G. max and G. soja are generally less than 1% (Nakayama and Yamaguchi 2002; Kuroda et al. 2008; Mizuguti et al. 2009), (2) natural F1 hybrids between G. max and G. soja are rare in Japan, and (3) plants derived from those hybrids survived only one to a few years in natural habitats (Kaga et al. 2005; Kuroda et al. 2005, 2006b, 2007). However, the genetic and ecological mechanisms for this lack of persistence remain unclear.
During the domestication of soybean, G. max evolved from G. soja to have large and nondormant seeds with a determinate nontwining growing habit that may affect fitness in natural habitats. The seed dormancy of wild soybean is caused by the physical structure of the seed coat, which usually does not imbibe water immediately after immersion (Rolston 1978; Ohara and Shimamoto 1994). In contrast, G. max bears seeds with little to no dormancy because uniform and rapid germination are important for soybean cultivation and food processing. Oka (1983) analyzed reproductive success in seminatural conditions by using hybrid derivatives between G. max and G. soja and found that plants with high seed dormancy and high seed production successfully survived. Thus, knowledge of genomic regions affecting fitness-related traits helps us to understand the reasons why hybrid derivatives between G. max and G. soja are rare in natural habitat. Although domestication-related quantitative trait loci (QTL) such as seed size and growth habit have previously been reported (e.g., Liu et al. 2007), no attempt has been made to identify genetic factors affecting the number of seeds per plant and winter seed survival in the soil.
In this study, artificial F1 hybrids, F2 populations, and backcross populations were made of two combinations of G. soja (W – Wild) and non-GM G. max (D – Domesticated); these combinations had different growth habits and represented northern and southern Japanese germplasm based on the assumption that gene flow from GM G. max to G. soja occurs in both northern and southern Japan. The degree of fitness of the hybrids and their derivatives was compared with their G. soja and non-GM G. max parents in three regions of Japan: north, central, and south. On the basis of the results, we discuss the likelihood of persistence of transgenes from G. max in G. soja populations. This is the first report of the detection of QTLs affecting fitness-related traits such as winter seed survival and seed number per plant of G. soja × G. max hybrids in experimental fields.
Materials and Methods
Plant materials
F1 hybrids between wild and cultivated soybean
F1 hybrids between G. soja and non-GM G. max were produced for two cross combinations. One combination (W1 × D1) was developed from a cross between the G. soja accession “JP036034” (W1) collected in Aomori Prefecture, northern Japan, and non-GM G. max cultivar “Ryuhou” (D1), which is widely grown in the northern Japan. The other combination (W2 × D2) was developed from a cross between G. soja accession “JP110755” (W2) collected in Hiroshima prefecture in southern Japan, and non-GM G. max “Fukuyutaka”(D2), which is widely grown in southern Japan (Table 1). The wild soybean accessions used in these crosses were obtained from the Genebank of the National Institute of Agrobiological Sciences. Non-GM G. max cultivars, D1 and D2, were obtained from the Tohoku Agricultural Research Center and Kyushu Okinawa Agricultural Research Center, respectively.
Table 1.
Artificial hybrids and populations between Glycine soja and G. max evaluated in this study
| Combination (G.soja × G.max) | Generation | Year | Field | Pedigree | Genetic linkage map (no. loci) | Total map length (cM) |
|---|---|---|---|---|---|---|
| W11 × D12 | F1 | 2005 | North5, Central6, South7 | G. soja (W) × G. max (C) | – | – |
| F2 | 2005 | North, south | W × C | 212 | 2720 | |
| BC1F1 | 2006 | Central | W / (W × C) | 214 | 2609 | |
| BC2F1 | 2007 | Central | W / W / (W × C) | 103 | 994 | |
| BC1F2 | 2007 | Central | W / (W × C) | 105 | 931 | |
| W23 × D24 | F1 | 2005 | North, central, south | W × C | – | – |
| F2 | 2005 | North, south | W × C | 208 | 2547 | |
| BC1F1 | 2006 | Central | W / (W × C) | 199 | 2514 | |
| BC2F1 | 2007 | Central | W / W / (W × C) | 72 | 572 | |
| BC1F2 | 2007 | Central | W / (W × C) | 72 | 599 |
W1 (JP036034): G. soja collected from Aomori Prefecture in northern Japan.
D1 (Ryuhou): G. max cultivar commonly planted in northern Japan.
W2 (JP110755): G. soja collected from Hiroshima Prefecture in southern Japan.
D2 (Fukuyutaka): G. max cultivar commonly planted in southern Japan.
North field: Akita prefecture, northern Japan.
Central field: Ibaraki prefecture, central Japan.
South field: Hiroshima prefecture, southern Japan.
F2 populations
F2 populations, which might be expected to grow in natural habitats, were developed for testing because of the highly inbreeding nature of soybean. Two F2 populations, one representing the northern region (W1 × D1: 204 individuals) and the second representing the southern region (W2 × D2: 204 individuals) were developed from seeds by self-pollination of a single F1 hybrid plant per cross (Table 1).
Backcross populations
To confirm the effect of G. max genes in a predominantly G. soja background, backcross (BC) populations were developed for both W1 × D1 and W2 × D2 combinations using G. soja as the recurrent parent (Table 1, Fig. 1). Two BC1F1 populations (W1 × D1: 68 individuals; W2 × D2: 160 individuals) were obtained from crossing each F1 hybrid (donor plant) to the corresponding G. soja accession (recurrent parent). The success of the crossing was confirmed using 60 simple sequence repeat (SSR) markers composed of three markers per linkage group for all 20 linkage groups. Furthermore, two BC2F1 populations (W1 × D1: 60 individuals; W2 × D2: 40 individuals) were developed by crossing selected BC1F1 plants (one plant per population) to G. soja. The selection of the BC1F1 plants, having major G. max QTLs for seed dormancy and total seed number identified in F2 populations, was based on the genotypes of the BC1F1 populations. To investigate the fitness of the populations after an additional generation of self-pollination, the seeds obtained from self-pollination of the selected BC1F1 plants were used to develop two BC1F2 populations (W1 × D1: 150 individuals; W2 × D2: 150 individuals).
Figure 1.

Morphological differences in wild soybean (Glycine soja, W2) with cultivated soybean (G. max, D2); Twining growing habit and small blackish seeds. Photo: Akito Kaga.
Field locations
Among 204 F2 plants in the W1 × D1 population, 104 F2 plants, together with the 10 parents and several F1 hybrid (Table 1), were grown at 1 m × 1 m spacing at the Tohoku Agricultural Research Center (39.5°N, 140.4°E, Akita Prefecture, northern Japan, Appendices A1 and A2), hereafter referred to as the “north field.” The other 100 F2 plants, together with the parents and F1 hybrid, were grown at the same density at the Western Region Agricultural Research Center (34.5°N, 133.4°E, Hiroshima prefecture, southern Japan, Appendices A1 and A2), hereafter referred to as the “south field.” The W2 × D2 population, composed of 204 F2 plants, was grown in the north and south fields in the same manner as the W1 × D1 population (Table 1). As maintenance and evaluation of many climbing plants with a higher G. soja background are difficult, backcross populations (BC1F1, BC2F1, and BC1F2) were only grown at 1 m × 1 m density at the National Institute of Agrobiological Sciences (36.0°N, 140.1°E, Ibaraki Prefecture, central Japan, Appendices A1 and A2), hereafter referred to as the ‘central field’. Seed coats were scratched with a razor blade and germinated in a small pot at the beginning of July in 2005, at the middle of June in 2006, and at the end of May for in 2007. The seedlings were transplanted to the field in the middle of July every year. Three stakes with a net strung between the stakes per plant were used to guide twining stems. During October to November, mature pods with seeds were harvested by hand twice a week. Standard agricultural practices such as applications of fertilizer (650 kg/ha of 3 parts nitrogen, 10 parts phosphate, and 10 parts potassium; 1000 kg/ha of fused magnesium phosphate; 1000 kg/ha of limestone), weeding, insecticides to control stink bug and common cutworm, were conducted.
Trait measurement
Of a total of 11 fitness-related traits (Table 2), 10 were treated as quantitative traits and one (seed coat color) was treated as a qualitative trait. Two seed dormancy–related traits, namely seed winter survival (DORM_1) and seed hardness (DORM_2), were evaluated using the seeds from individual plants. As germination of all the hard seeds from randomly selected lines were confirmed by the mechanical abrasion on the wetted filter paper or soil at room temperature, hard seeds were treated as viable nongerminated seeds. Seed production–related traits, namely total seed number (PROD_1), seed total weight (PROD_2), 100-seed weight (PROD_3), total pod number (PROD_4), stem dry weight (PROD_5), and stem length (PROD_6) were evaluated for each plant (Table 2). Those traits were recorded on a per plant basis after the seeds of each plant had matured. As flowering of the two southern accessions (W2 and D2) as well as F1 plants and most of the W2 × D2 F2 plants were late flowering in the north field, those whole plants (7 of 10 W2, all 5 D2, all 5 of the W2 × D2 F1, and 73 of 104 W2 × D2 F2 plants) were taken from the field before the first snowfall and dried in the greenhouse to obtain mature seeds. The methods of trait evaluation in the backcrossing populations were the same as for the F2 populations. The number of days from sowing to first flowering was recorded as FLOW. The total number of seeds expected to germinate in the following year (SURV) was estimated as PROD_1 multiplied by DORM_1. The mean and standard deviation for each trait and the correlation coefficient between each pair of traits were calculated. Differences in mean values between G. soja, G. max, and F1 hybrids were analyzed separately for each field location in each year with the Mann–Whitney U-test or Kruskal–Wallis test. The median and range instead of mean and standard deviation are reported for segregating populations. All statistical analyses were conducted using R version 2.9.2 (R Development Core Team 2009).
Table 2.
Fitness-related traits evaluated in this study
| General attribute | Trait (unit) | Abbreviation | Evaluation method |
|---|---|---|---|
| Seed dormancy | Seed winter survival (%) | DORM_1 | Percentage of germinated and viable nongerminated seeds after burial of mesh bags containing 20 unscarified seeds 3 cm below the soil surface of each experimental field (north, central, or south) from late December to the following spring (April–May) |
| Seed hardness (%) | DORM_2 | Percentage of nonimbibed seeds after soaking for 4 days in an incubator at 4°C | |
| Seed coat color | DORM_3 | Black or other (buff, green, or brown) | |
| Seed production | Total seed number | PROD_1 | Total number of harvested seeds |
| Total seed weight (g) | PROD_2 | Total weight of harvested seeds | |
| 100-seed weight (g) | PROD_3 | 100-seed weight | |
| Total pod number | PROD_4 | Total number of harvested pods | |
| Stem dry weight (g) | PROD_5 | Stem weight after drying for 7 days at 70°C | |
| Stem length (cm) | PROD_6 | Length from ground to top of stem | |
| Seed dormancy & production | Total number of seeds expected to germinate in the following year | SURV | Total number of seeds (PROD_1) multiplied by the winter seed survival rate (DORM_1) |
| Flowering phenology | Days to first flower (day) | FLOW | Number of days from sowing to flowering of first flower |
Genotyping
Total DNA of each putative F1 seed was extracted from a small piece of cotyledon tissue using an EZ1 DNA Tissue kit (Qiagen, Tokyo, Japan). Total DNA of F2, BC1F1, BC2F1, and BC1F2 individuals was extracted from 100 mg of fresh leaf tissue. DNA concentration was adjusted between 5 and 25 ng/μL by comparing with known concentrations of standard λ DNA on a 1.5% agarose gel. A total of 720 SSR markers from SoyBase (http://soybase.org/) were screened to detect polymorphisms between the parents. Five markers were also included to track the three classical soybean loci, I, T, and Dt1 (Appendix A3). Three markers, dCHS1 (Matsumura et al. 2005), AY262686B, and AY262686Z, were used to track the I locus, which controls seed coat color and might be related to seed winter survival. A single-base indel marker sF3′H1 reported by Toda et al. (2002) was used to detect the T locus, a locus that controls pubescence color and interacts with the I locus. A SSR marker LFsoy3 was designed to track the Dt1 locus, which might be related to stem length and seed total number. These markers were amplified by using KOD-plus polymerase (Toyobo, Osaka, Japan), based on the manufacturer's guide, in a GeneAmp 9700 PCR system (Applied Biosystems, Tokyo, Japan). Polymorphisms were scored by using banding patterns in 12% polyacrylamide gel.
Successful crossing was confirmed by analysis of DNA from putative F1 seeds, based on the genotype of the polymorphic SSR marker Satt207, which has a different allele in each of the four parents (W1, 177 bp; D1, 234 bp; W2, 210 bp; and D2, 231 bp). To genotype F2, BC1F1, BC2F1, and BC1F2 individuals, polymorphic markers were selected at about 20-cM intervals based on the composite map of soybean from SoyBase (http://soybase.org/). Using four types of fluorescent labels (6-FAM, VIC, NED, or PET), multiplex PCR was performed to detect segregation patterns within each population. The PCR reaction mixture consisted of a total volume of 5 μL, containing 1.7 μL of template DNA, 2.5 μL of 2 × Qiagen Multiplex PCR Master Mix, 0.5 μL of a four-primer mix (1.25 μmol/L each), and 0.3 μL of water. PCR amplification was perform in a GeneAmp 9700 (Applied Biosystems) or iCycler (BioRad, Tokyo, Japan) thermal cycler programmed with an initial activation step at 95°C for 15 min; followed by 40 cycles of 30 sec at 94°C for denaturation, 90 sec at 57°C for annealing, and 60 sec at 72°C for extension; followed by 30 min at 60°C for final extension. For analysis, 3 μL of PCR product was denatured at 95°C for 5 min after mixing with 10 μL of Hi-Di formamide (Applied Biosystems) and 15 nL of GeneScan-500LIZ size standard (Applied Biosystems). Denatured samples were analyzed by using a 3100 Genetic Analyser (Applied Biosystems) and the output was analyzed using Gene Mapper 3.0 software (Applied Biosystems).
Linkage map construction
Linkage maps were constructed for F2, BC1F1, BC2F1, and BC1F2 populations by using Joinmap ver. 3.0 software (Van Ooijen and Voorrips 2001) according to the method of Han et al. (2005). The recombination frequencies were converted into map distances using the Kosambi mapping function (Kosambi 1944).
QTL analysis
The QTL analysis for phenotypic data from the BC1F1, BC2F1, and BC1F2 individuals was conducted with MultiQTL ver. 2.6 software according to Peng et al. (2003). For phenotypic data of the F2 individuals from the two field environments (north and south), a single QTL with multiple environment model was fitted to scan the entire genome (Korol et al. 1998, 2001). Statistical significance thresholds (α = 0.05) for putative QTLs were tested by 10,000 runs of a permutation test (Churchill and Doerge 1994). Multiple interval mapping (Kao et al. 1999) was then conducted to reduce the background variation by taking into account QTL effects from other chromosomes. After the permutation test runs, the parameters of significant QTLs (statistical thresholds α = 0.05) were reported as position, additive and dominant effects, and percentage of variance explained (PVE).
Results
Fitness of hybrids and their derivatives
Cultivated and wild soybean
The following domestication-related traits generally differed between the G. soja and G. max parents for both combinations (W1 × D1 and W2 × D2) tested at all three field locations (north, central, and south) in 2005 (Table 3). The means of G. max were higher than those of G. soja for PROD_3, whereas the means of G. soja were generally higher than those of G. max for DORM_1, DORM_2, PROD_1, PROD_4, PROD_6, SURV, and FLOW. In contrast, PROD_2 and PROD_5 were not notably different between G. soja and G. max. Especially, the means of G. max for PROD_2 tended to be similar to or higher than those of G. soja at their recommended regions for growing. Although no G. max data were obtained from the south field in 2005, we confirmed these trends in 2004 (A. Kaga & Y. Kuroda, unpublished data).
Table 3.
Summary of phenotypic values of G. max, G. soja, F1, F2, BC1F1, BC1F2, and BC2F1 populations
| Combination | Field location (year) | No. individual | DORM_ | PROD_ | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 2 | 1 | 2 | 3 | 4 | 5 | 6 | SURV | FLOW | |||||
| W1 × D1 | North (2005) | W1 | 9 | Mean (SD) | 97.5a (3.5) | 95.0a (4.1) | 421ab (203) | 14.0b (7.5) | 3.3c (0.3) | 326ab (193) | 9.5ab (6.1) | 188.2a (28.2) | 410a (197) | 55.8a (2.2) |
| D1 | 6 | Mean (SD) | 0.0b (0.0) | 0.0b (0.0) | 174b (27) | 54.6a (8.3) | 31.4a (1.7) | 75b (11) | 6.6b (0.9) | 23.0b (1.1) | 0b (0) | 37.0c (0.0) | ||
| F1 | 3 | Mean (SD) | 55.0ab (0.0) | 83.3a (5.8) | 688a (241) | 63.6a (21.4) | 9.3ab (0.2) | 676a (73) | 31.9a (10.3) | 176.0a (25.2) | 378a (133) | 53.0ab (2.6) | ||
| F2 | 104 (103)1 | Median (range) | 55.0 (0.0–100.0) | 70.0 (0.0–100.0) | 368 (48–883) | 32.0 (3.1–81.0) | 8.6 (4.4–13.9 | 328 (53–1426) | 14.4 (1.3–48.4) | 109.0 (25.0–272.0) | 187 (0–876) | 49.0 (37.0–63.0) | ||
| Central (2005) | W1 | 7 | Mean (SD) | – | 67.1a (13.8) | 1211a (297) | 41.0a (10.7) | 3.4b (0.3) | 717a (199) | 41.9a (8.4) | 187.0a (36.7) | – | 50.3a (1.0) | |
| D1 | 9 | Mean (SD) | – | 0.0b (0.0) | 50b (21) | 14.6b (6.1) | 29.2a (1.4) | 66b (14) | 25.6b (2.9) | 29.1b (1.4) | – | 34.2b (0.4) | ||
| F1 | 1 | Mean (SD) | – | 40.01ab (–) | 6361ab (–) | 70.71ab (–) | 11.11ab (–) | 3891ab (–) | 45.81ab (–) | 110.01 ab (–) | – | 42.01ab (–) | ||
| South (2005) | W1 | 5 | Mean (SD) | 100.0ns (0.0) | 91.0a (8.9) | 1049ns (561) | 44.9ns (26.3) | 3.8b (0.6) | 575ns (205) | 35.8ns (15.8) | 116.0ns (89.0) | 1049ns (561) | 44.2ns (1.6) | |
| D1 | 0 | Mean (SD) | – | – | – | – | – | – | – | – | – | – | ||
| F1 | 6 | Mean (SD) | 17.5ns (3.5) | 34.2b (17.7) | 796ns (290) | 77.8ns (28.5) | 9.8a (0.4) | 424ns (140) | 23.3ns (6.7) | 134.3ns (26.7) | 139ns (51) | 45.3ns (3.1) | ||
| F2 | 100 (96) 1 | Median (range) | 27.5 (0.0-100.0) | 50.0 (0.0–100.0) | 517 (2–2109) | 54.2 (0.3–217.2) | 9.7 (3.8–13.9) | 302 (31–1369) | 19.0 (2.0–69.0) | 141.5 (24.0–249.0) | 128 (0–1673) | 43.0 (31.0–48.0) | ||
| Central (2006) | W1 | 9 | Mean (SD) | 96.7a (2.9) | 100.0a (0.0) | 2674a (808) | 59.1ns (17.1) | 2.2b (0.2) | 823a (244) | 43.0a (17.1) | 303.8a (55.1) | 2585a (781) | 53.7a (1.8) | |
| D1 | 10 | Mean (SD) | 20.0b (20.0) | 0.0b (0.0) | 111b (9) | 11.4ns (2.3) | 23.5a (3.1) | 29b (5) | 31.7b (8.7) | 27.7b (2.3) | 0b (0) | 40.1b (0.7) | ||
| BC1F1 | 66 | Median (range) | 95.0 (0.0–100.0) | 95.0 (40.0–100.0) | 2588 (617–4550) | 89.8 (9.3–162.7) | 3.5 (1.4–5.4) | 835 (194–1454) | 36.8 (7.2–70.9) | 300.0 (90.0–435.0) | 2166 (0–4416) | 51.0 (45.0–67.0) | ||
| Central (2007) | W1 | 10 | Mean (SD) | 100.0ns (0.0) | 100.0a (0.0) | 5627a (696) | 177.9ns (19.2) | 3.2b (0.1) | 1788a (258) | 334.1a (32.0) | 372.0a (25.8) | 5627a (696) | 79.6a (2.9) | |
| D1 | 9 | Mean (SD) | 0.0ns (0.0) | 5.0b (7.1) | 81b (24) | 22.9ns (6.7) | 28.5a (1.3) | 40b (12) | 30.9b (8.8) | 24.3b (4.1) | 0b (0) | 72.0b (1.8) | ||
| BC2F1 | 150 | Median (range) | 100.0 (60.0–100.0) | 95.0 (45.0–100.0) | 5141 (2682–7297) | 199.7 (112.1–277.6) | 4.0 (3.3–4.8) | 1634 (925–2413) | 314.7 (161.5–465.6) | 375.0 (260.0–460.0) | 5123 (2150–7297) | 82.0 (74.0–89.0) | ||
| BC1F2 | 60 | Median (range) | 80.0 (0.0–100.0) | 85.0 (0.0–100.0) | 3655 (218–5878) | 186.1 (14.4–294.2) | 5.1 (3.4–8.8) | 1216 (81–2027) | 240.8 (16.7–462.9) | 270.0 (45.0–415.0) | 2714 (0–5746) | 80.0 (72.0–92.0) | ||
| W2 × D2 | North (2005) | W2 | 10 | Mean (SD) | 77.5ns (10.6) | 93.3a (2.6) | 1418a (735) | 32.1b (17.7) | 2.2c (0.1) | 1034a (489) | 31.8ns (12.8) | 147.0ns (45.2) | 1099ns (570) | 71.2a (1.1) |
| D2 | 6 (0) 1 | Mean (SD) | – | 0.0b (0.0) | 122b (56) | 21.2b (11.0) | 17.1a (1.6) | 356b (36) | 51.8ns (5.9) | 69.2ns (6.7) | – | 56.0c (0.0) | ||
| F1 | 5 | Mean (SD) | 25.0ns (7.1) | 15.0ab (6.1) | 1076a (320) | 86.6a (27.4) | 8.0ab (0.4) | 796a (295) | 45.8ns (18.8) | 129.0ns (74.1) | 269ns (80) | 65.0b (2.7) | ||
| F2 | 104 (102) 1 | Median (range) | 17.5 (0.0–100.0) | 5.0 (0.0–90.0) | 723(88–1694) | 53.7 (2.9–125.0) | 7.5 (2.5–11.4) | 710 (250–2040) | 41.4 (14.9–106.0) | 111.0 (30.0–298.0) | 144 (0–989) | 63.0 (58.0–85.0) | ||
| Central (2005) | W2 | 6 | Mean (SD) | – | 94.0a (5.5) | 1319a (217) | 42.1b (7.6) | 3.2b (0.2) | 1282a (404) | 50.4ns (6.9) | 117.3a (16.3) | – | 66.7a (0.5) | |
| D2 | 7 | Mean (SD) | – | 0.0c (0.0) | 100b (31) | 23.7c (7.6) | 23.7a (2.1) | 216b (58) | 58.5ns (9.4) | 49.9b (5.5) | – | 53.4b (1.1) | ||
| F1 | 4 | Mean (SD) | – | 42.5ab (15.0) | 1012ab (156) | 99.2a (17.2) | 9.8ab (0.4) | 942ab (419) | 64.5ns (18.5) | 119.5a (13.8) | – | 58.8ab (1.0) | ||
| South (2005) | W2 | 2 | Mean (SD) | 100.0ns (0.0) | 87.5ns (10.6) | 51371ns (–) | 143.31ns (–) | 2.81ns (–) | 17141ns (–) | 54.01ns (–) | 104.01ns (–) | 5137ns(–) | 65.01ns (–) | |
| D2 | 2 | Mean (SD) | – | – | – | – | – | – | – | – | – | – | ||
| F1 | 5 | Mean (SD) | 10.0ns (0.0) | 9.0ns (4.2) | 2744ns (234) | 258.8ns (23.4) | 9.4ns (0.4) | 1354ns (138) | 68.4ns (11.1) | 150.2ns (41.9) | 274ns (23) | 54.8ns (1.6) | ||
| F2 | 100 | Median (range) | 0.0 (0.0–100.0) | 7.5 (0.0–100.0) | 1874 (138–4694) | 186.9 (13.4–468.5) | 9.7 (5.0–18.1) | 1051 (194–2450) | 53.5 (13.0–135.0) | 142.5 (30.0–282.0) | 0 (0–3620) | 53.0 (49.0–62.0) | ||
| Central (2006) | W2 | 10 | Mean (SD) | 98.3a (2.6) | 92.5a (3.5) | 4189a (804) | 104.6ns (24.3) | 2.5b (0.3) | 1354a (274) | 62.6ns (15.4) | 251.9ns (30.1) | 4119a (791) | 83.7a (1.3) | |
| D2 | 10 | Mean (SD) | 10.0b (12.6) | 0.0b (0.0) | 326b (48) | 106.1ns (18.6) | 32.4a (1.5) | 157b (25) | 30.7ns (4.3) | 44.5ns (2.0) | 33b (5) | 63.5b (1.2) | ||
| BC1F1 | 160 | Median (range) | 100.0 (0.0–100.0) | 100.0 (20.0–100.0) | 3973 (328–7574) | 164.1 (14.6–320.0) | 4.1 (2.4–5.3) | 1289 (109–2443) | 75.1 (31.8–145.5) | 280.0 (190.0–380.0) | 3801 (0–7574) | 79.0 (73.0–86.0) | ||
| Central (2007) | W2 | 10 | Mean (SD) | 100.0a (0.0) | 92.5ns (3.5) | 6016a (1764) | 178.6ns (50.0) | 3.0b (0.2) | 2073a (615) | 327.4a (57.4) | 335.5a (33.0) | 6016a (1764) | 105.1a (1.7) | |
| D2 | 10 | Mean (SD) | 0.0b (0.0) | 0.0ns (0.0) | 439b (54.0) | 120.7ns (17.8) | 27.7a (2.1) | 220b (27) | 123.2b (12.3) | 45.9b (6.4) | 0b (0.0) | 88.5b (1.1) | ||
| BC2F1 | 149 | Median (range) | 100.0 (30.0–100.0) | 95.0 (5.0–100.0) | 5863 (4298–9784) | 196.7 (126.0–311.6) | 3.2 (2.4–3.9) | 1999 (1496–3494) | 326.1 (211.4–498.8) | 300.0 (250.0–420.0) | 5673 (1444–9784) | 106.0 (103.0–109.0) | ||
| BC1F2 | 40 | Median (range) | 100.0 (20.0–100.0) | 95.0 (15.0–100.0) | 4244 (1511–7806) | 166.9 (48.3–292.9) | 3.9 (2.9–5.2) | 1429 (521–2692) | 255.1 (75.2–472.8) | 275.0 (80.0–375.0) | 4002 (934–7806) | 105.0 (100.0–109.0) | ||
Different alphabet among parents and populations at each field location indicates significant difference at 5% level by Mann–Whitney's U-test or Kruskal–Wallis test.
Trait abbreviations are as defined in Table 2. –, no data recorded.
Number of seed-producing individuals.
F1 hybrids and F2 populations
The phenotypic values of the F1 and F2 generations in most field locations were intermediate between G. soja and G. max for DORM_1, DORM_2, PROD_1, PROD_3, PROD_4, PROD_6, SURV, and FLOW (Table 3, Fig. 2A and B). However, the means of PROD_2 and PROD_5 in the F1 and F2 generations tended to be similar to or higher than those of G. soja at the recommended regions for growing the G. max parent. Most of the G. soja seeds dug up from the soil in the spring did not imbibe water, whereas the G. max seeds were rotten. Seeds from F1 and F2 plants were of all types: hard seeds that did not absorb water, water-absorbing viable seeds, and rotten seeds. DORM_2 was positively correlated with DORM_1 (P < 0.05) in the F2 generations of W1 × D1 (seeds harvested from the north field, R2 = 0.81; seeds harvested from the south field, R2 = 0.69, Appendix A4) and W2 × D2 (seeds harvested from the north field, R2 = 0.85; seeds harvested from the south field, R2 = 0.60).
Figure 2.
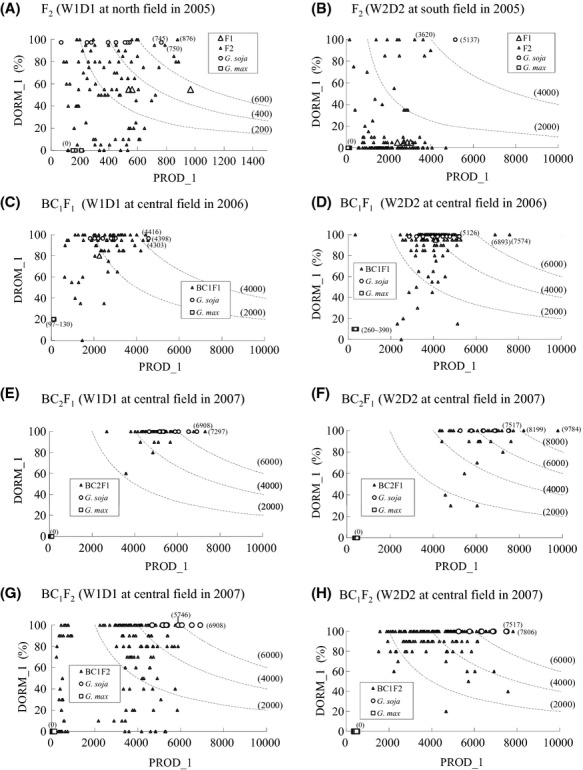
Distribution of SURV, calculated by multiplying DORM_1 and PROD_1 for each combination (W1D1 or W2D2) and generation (F2, BC1F1, BC2F1, or BC1F2). Numbers in brackets indicate SURV values; areas between dotted lines indicate ranges of SURV values.
The extent of DORM_1 was associated with maternal-inherited seed coat color and the pubescence color of the F3 seeds produced on F2 plants (Table 4). G. soja has black seeds and brown pubescence, and G. max has yellow seeds and white pubescence. High DORM_1 was observed for seeds with black or brown seed coat color produced by F2 plants with brown pubescence color, and most of those seeds did not imbibe water when tested in the spring (brown seeds, 75.9%; black seeds, 75.5%). The seeds with other colors of pubescence had relatively low DORM_1. In particular, the seeds with brown seed coat color produced by F2 plants with white pubescence color (22 of 27 F2 plants) were severely cracked or split and could not be found in the following spring (DORM_1, 0.2%).
Table 4.
Relationships between percentage of seed winter survival (DORM_1) and colors of seed coat and pubescence in F2 populations
| Combination | Seed production location | Seed coat color (pubescence color) | ||||||
|---|---|---|---|---|---|---|---|---|
| Black (brown) | Brown (white) | Brown (brown) | Yellow (unclassified) | Green (unclassified) | Unclassified | |||
| [i/i, T/−, R/−]1 | [i/i, t/t, r/r] | [i/i, T/−, R/−] | [I/−2,,] | [I/−2,,] | ||||
| W1 × D1 | North | DORM_1 (%) | 92.7 ± 11.8a | 0.0 ± 0.0b | 81.0 ± 12.4ab | 33.1 ± 25.5b | 51.8 ± 29.2ab | 43.0 ± 35.6b |
| n | 15 | 4 (4)3 | 6 | 19 (1)3 | 54 | 5 | ||
| South | DORM_1 (%) | 83.1 ± 17.4a | 0.0 ± 0.0b | 95.8 ± 4.9a | 12.9 ± 14.2b | 32.0 ± 28.4b | – | |
| n | 17 | 8 (4)3 | 6 | 13 (1)3 | 52 (5)3 | 0 | ||
| W2 × D2 | North | DORM_1 (%) | 51.3 ± 28.7a | 0.0 ± 0.0c | 57.5 ± 32.3ab | 14.0 ± 19.2abc | 15.9 ± 19.3bc | 37.5 ± 23.2abc |
| n | 16 | 6 (6)3 | 4 | 11 (3)3 | 55 (1)3 | 10 | ||
| South | DORM_1 (%) | 74.2 ± 28.4a | 0.6 ± 1.7b | 64.2 ± 44.4a | 4.2 ± 8.4b | 4.2 ± 10.6b | – | |
| n | 13 | 9 (6)3 | 6 | 24 | 48 (4)3 | 0 | ||
| Overall | DORM_1 (%) | 75.5 ± 26.9a | 0.2 ± 1.0c | 75.9 ± 30.4a | 15.6 ± 20.8bc | 26.5 ± 29.6b | 42.1 ± 27.7ab | |
| n | 61 | 27 (22)3 | 22 | 68 (3)3 | 209 (10)3 | 15 | ||
Different alphabet among genotypes at each field location indicates significant difference at 5% level by Kruskal–Wallis test.
Presumed genotypes at the I, T, and R loci.
Genotype at the I locus would be I/ii in the W2 x D2 population.
Number of cracked or split seeds.
The PROD_1 of the F1 plants was generally intermediate between G. soja and G. max for both the W1 × D1 and W2 × D2 combinations (Table 3, Fig. 2A and B). An exception was found in the north field, where PROD_1 of the F1 plants from the W1 × D1 combination (average 688) was similar to or higher than that of the G. soja parent (average 421). The mean values of PROD_2 and PROD_5 in the F1 generation were also higher than those of the parents. In the next generation, PROD_1 of several F2 individuals was similar to or higher than that of the G. soja parent. This transgressive growth of PROD_1 may be explained by heterosis or positional effect within a field for plant size–related traits because of significant (P < 0.05) positive correlations between PROD_1 and plant size–related traits such as PROD_5 and PROD_6 (Appendix A4).
The values for SURV of G. soja and G. max were different within both the W1 × D1 combination and the W2 × D2 combination because G. soja had both high PROD_1 and DORM_1, whereas G. max had low PROD_1 and zero DORM_1 (Table 3, Fig. 2A and B). Average SURV of F1 plants was intermediate between G. soja and G. max for each combination. Greater variation was observed in the F2 progenies than in the F1 plants because of genetic segregation of PROD_1 and DORM_1.
Backcross populations
For the backcross populations (BCs; BC1F1, BC2F1, and BC1F2) from both combinations, plants were grown only in the central field in 2006 and 2007. The phenotypic differentiation between G. soja and G. max in the central field was similar to that seen in the other fields in 2005. All trait values of the BCs were clearly shifted toward those of the G. soja recurrent parents. For both combinations, the medians of the BC1F1 and BC2F1 populations were very close to the means of G. soja for all traits (Table 3). In contrast, the extent of shift in BC1F2 populations for DORM_1, DORM_2, PROD_1, and SURV_1 was not obvious as in the BC1F1 and BC2F1 populations. The plant type of the backcross generations was vigorous in both 2006 and 2007, when mulch sheets were used on the surface of soil; in contrast, the F1 and F2 generations, which were grown without the sheets in 2005, were less vigorous.
Because G. soja had higher PROD_1 and DORM_1 than G. max, SURV of G. soja was higher than that of G. max in both the W1 × D1 and W2 × D2 combinations in all three backcross generations (Table 3, Fig. 2C–H). The medians of SURV in the BC1F1 and BC2F1 generations were very close to G. soja; still, there was variation in both DORM_1 and PROD_1 in the BC1F1 and BC2F1 generations (Fig. 2C–H). Some individuals had the potential to yield large numbers of dormant seed because the number of seeds (PROD_1) was greater than G. soja and the seed dormancy (DORM_1) was similar.
QTL analysis for F2 populations
Of 720 markers screened, 359 and 378 markers revealed clear polymorphisms between G. soja and G. max in the W1 × D1 and W2 × D2 populations, respectively. Of these, 212 and 208 markers were used to develop F2 linkage maps of the W1 × D1 and W2 × D2 populations, respectively (Table 1, Fig. 3). Although gaps of more than 30 cM were observed between Satt285 and Satt414 on LG-J over populations and generations, the SSR markers were otherwise distributed evenly across the soybean genome, and marker orders were conserved between the W1 × D1 and W2 × D2 population maps as well as between those maps and the composite map by Song et al. (2004). The total lengths of the linkage maps developed here were about 2500 cM for the F2 and BC1F1 populations, comparable to the lengths of the SSR-based linkage maps developed by Song et al. (2004) (2524 cM) and Liu et al. (2007) (2383 cM).
Figure 3.

(A) Summary of fitness-related QTLs detected in F2, BC1F1, BC2F1, and BC1F2 generations of both north (W1 × D1) and south (W2 × D2) combinations of Glycine soja × G. max hybrids for linkage group (A1–E). *w and *c indicate G. soja (wild) and G. max (cultivated) homozygote excess, respectively, at the designated SSR locus. Arrows indicate QTLs with allelic effects opposite of those predicted by the parental phenotype for traits differing between G. soja and G. max. (N) or (S) next to an arrow indicates that the effect was observed only in the north field or south field, respectively.
Several markers (1.4% and 3.4% of the markers in the W1 × D1 and W2 × D2 populations, respectively) showed segregation ratios significantly (P < 0.05) deviated from the expected 1:2:1 ratio of G. soja homozygote, heterozygote, and G. max homozygote. Although most markers with segregation distortion were scattered over several linkage groups and were not consistent between the W1 × D1 and W2 × D2 populations, five of the distorted markers were adjacent and located in the upper half of LG-C1 in the W2 × D2 population (Fig. 3A). Paracentric inversions and reciprocal translocations, which can lead to pollen and ovule sterilities and have been found between a specific Chinese accession of G. soja and G. max (Singh and Hymowitz 1988; Palmer et al. 2000), might account for the segregation distortions in these Japanese germplasm sources as well.
In total, 28 and 27 QTLs related to seed dormancy, seed production, and flowering phenology were detected in the F2 generation of W1 × D1 and W2 × D2 populations, respectively (Fig. 3, Appendix A5). Among them, QTLs in three regions (LG-A2, -C2, and -D1b) had large effects on seed dormancy and QTL in one region (LG-L) had a significant effect on seed production.
Seed dormancy
Eight and 6 QTLs associated with seed dormancy were detected in the W1 × D1 and W2 × D2 populations, respectively (Fig. 3, Appendix A5). The G. max alleles at all of those QTLs had additive effects (Add.) of decreasing DORM_1 (Add, –3 to –37%; PVE, 6.4–76.2%) and DORM_2 (Add, –1 to –26%; PVE, 6.2–42.7%). Three major QTLs, which were located on LG-A2, -C2, and -D1b, were associated with DORM_1 and DORM_2 in both populations (Fig. 3A). The G. max allele at the QTL on LG-A2 had larger additive effects in the W2 × D2 population than in the W1 × D1 population. The QTL on LG-A2 was located near the I locus, and the QTL on LG-C2 was close to the T locus. The additive effect of the QTL on LG-A2 detected in seeds harvested from the south field tended to be higher than that for seeds harvested from the north field. In contrast, the additive effect of the QTL on LG-C2 and LG-D1b detected in seeds harvested from the south field tended to be lower than that for seeds harvested from the north field.
Seed production
Sixteen and 17 QTLs related to seed production were detected in W1 × D1 and W2 × D2 populations, respectively (Fig. 3, Appendix A5). Most G. max alleles at those QTLs had additive effects of decreasing PROD_1 (Add, –73 to –496 seeds; PVE, 8.8–33.8%), PROD_2 (Add, –5.1 to –42.5 g; PVE, 6.8–33.5%), PROD_4 (Add, –74 to –229 pods; PVE, 7.5–33%), PROD_5 (Add, –6.7 to –18.6 g; PVE, 10.2–28.8%), and PROD_6 (Add, –4 to –50 cm; PVE, 11.5–40.2%) and increasing PROD_3 (Add, +0.6 to 1.1 g; PVE, 7.8–28.2%).
The G. max alleles at several QTLs had effects opposite of those expected based on the parental phenotypes (Appendix A5), that is, in the W1 × D1 population, toward decreased PROD_3 for the QTL on LG-D1b and LG-M (Add, –0.2 to –0.3 g; PVE, 6.4–8.1%, south field), increased PROD_4 for the QTL on LG-M (Add, +158; PVE, 14.8%), and increased PROD_5 for the QTL on LG-M (Add, +6.6 g; PVE, 15.8%). In the W2 × D2 population, G. max alleles had effects toward decreased PROD_3 for the QTL on LG-C2 (Add, –1.3 g, PVE, 43.5%), increased PROD_5 for the QTL on LG-D2 and LG-M (Add, + 9.5 g; PVE, 22.6%), and increased PROD_6 at the QTL on LG-D2 (Add, +19.0 to +20.1 cm; PVE, 7.7–13.4%).
QTLs with large effect on seed production–related traits such as PROD_1, PROD_2, PROD_3, PROD_4, PROD_5, and PROD_6 were located near a marker LFsoy3 in both the W1 × D1 and W2 × D2 populations (Fig. 3B). The G. max alleles at those QTLs, except for PROD_3, had additive effects of decreasing the phenotypic values for those traits, but the magnitude of effect differed depending on the test location (Appendix A5). For both populations, the additive effects for PROD_1, PROD_2, and PROD_5 were greater than those in the north field. Although the effects of QTLs for PROD_4 would be expected to be consistent, they did not seem to be related in the populations. The frequency of pods with only one or two seeds on plants in the north field was greater than for plants in the south field (data not shown), which may explain this inconsistency.
Flowering phenology
Four QTLs for flowering phenology were detected in each population (Fig. 3, Appendix A5). In general, G. max alleles at these QTLs had additive effects of hastening FLOW (Add, 0 to –3.5 days; PVE, 5.2–54.4%). In contrast, the G. max allele of the QTL on LG-D1b in the W1 × D1 population delayed FLOW (Fig. 3A). The location of the QTL identified on LG-L in the W1 × D1 population (Add, –1.8 to –2.4 days; PVE, 14.1–21.3%) was very near that of a QTL identified in the W2 × D2 population (Add, –0.8 day; PVE, 5.5% [Fig. 3B]). The locations of QTLs with large effects on FLOW were different between the two populations; in the W1 × D1 population, a major QTL was found on LG-O (Add, –2.8 to –3.5 days; PVE, 29.6–54.4%), whereas in the W1 × D1 population, a major QTL was found on LG-H (Add, –1.8 to –3.2 days; PVE, 26.6–36.8%).
QTL analyses for BC1F1 populations
Linkage maps for W1 × D1 and W2 × D2 BC1F1 populations were constructed using 214 and 199 markers, respectively (Table 1, Fig. 3). In total, 8 and 20 QTLs were detected in the W1 × D1 and W2 × D2 populations, respectively (Fig. 3, Appendix A5).
Seed dormancy
Three QTLs for seed dormancy were detected in each population (Fig. 3A, Appendix A5). Although the QTLs for DORM_2 on LG-A2 were detected only in the W2 × D2 population, the QTLs for DORM_1 on LG-A2 were detected across combinations (W1 × D1 and W2 × D2), suggesting that the G. max alleles have a consistent genetic effect even within a high percentage of wild genetic background. The G. max allele at this QTL on LG-A2 had a large negative effect on DORM_1 (Add,–7% to –8%; PVE, 21.2–22.3%).
Seed production
Three and 15 QTLs related to seed production were detected in the W1 × D1 and W2 × D2 BC1F1 populations, respectively (Fig. 3, Appendix A5). No QTL was common between the F2 and BC1F1 generations. For traits PROD_1, PROD_2, PROD_4, and PROD_5, no QTLs were detected in the W1 × D1 population, but several QTLs different from those found in the F2 generation were identified in the W2 × D2 population. As in the F2 generation, most G. max alleles had the effect of decreasing seed production–related traits, that is, at QTLs for PROD_5 (Add, –8.6 to –11.6 g; PVE, 5.1–9.4%) and PROD_6 (Add, –21 to –62 cm; PVE, 7.5–20.2%). However, some G. max alleles at those QTLs from the W2 × D2 population had the effect of increasing PROD_1 on LG-A2 (Add, +354 seeds; PVE, 5.5%), PROD_2 on LG-A2 (Add, +17.9 g; PVE, 7.4%), PROD_4 on LG-E (Add, +113 pods; PVE, 5.1%) and on LG-M (Add, +112 pods; PVE, 4.9%), and PROD_6 on LG-A2 and LG-O (Add, +20 to +21 cm; PVE, 6.9–7.9%). The G. max alleles at all QTLs detected had the effect of increasing PROD_3.
Flowering phenology
Two QTLs for FLOW were detected in each population (Fig. 3B, Appendix A5). Although the QTLs in the two populations were different, the G. max allele at each one had the effect of delayed flowering time (FLOW, –1.8 to –5.0 days; PVE, 11.2–42.5%). There were QTLs in common between F2 and BC1F1 on LG-O in the W1 × D1 population (FLOW, –2.8 to –5.0 days; PVE, 29.6–54.4%) and on LG-H in the W2 × D2 population (FLOW, –1.8 to –3.2 days; PVE, 15.3–36.8%).
QTL analyses for BC2F1 and BC1F2 populations
The linkage maps for W1 × D1 and W2 × D2 BC2F1 populations were constructed by using 103 and 72 markers, respectively (Table 1, Fig. 3). These markers were located in the heterozygous regions in the selected BC1F1 plants. In addition, BC1F2 populations were developed by using seeds from self-pollination of the two selected BC1F1 plants (W1 × D1 and W2 × D2) and partial linkage maps were constructed. The linkage maps for the W1 × D1 and W2 × D2 BC1F2 populations were constructed by using 105 and 72 markers, respectively. The order of markers in each linkage map was well conserved between the W1 × D1 and W2 × D2 populations as well as among the F2, BC1F1, and BC2F1 populations (Fig. 3A and B). Entire linkage groups (LG-A1, -C1, -I, and -M in W1 × D1 and LG-C1, -B2, -D2, -G, -J, and -O in W2 × D2) were found to have been replaced with G. soja genome in the two selected BC1F1 plants, BC2F1 population and BC1F2 population.
In the BC2F1 generation, which had a higher percentage of G. soja genetic background than the BC1F1, but included the selected fitness-related alleles from G. max, 10 QTLs were detected in both the W1 × D1 and W2 × D2 populations (Fig. 3, Appendix A5). Similar to the BC1F1 generation, most QTLs in the BC2F1 generation were different from those detected in the F2 generation. Unlike the situation in the BC1F1 generation, the effects of DORM_1 and DORM_2 QTLs on LG-A2 were not detected in W1 × D1 combination (Fig. 3A).
In the BC1F2 generation, which had a similar percentage of G. soja background to the BC1F1 generation but was homozygous for selected fitness-related alleles from G. max, 19 and 17 QTLs were detected in the W1 × D1 and W2 × D2 populations, respectively (Fig. 3, Appendix A5). The major QTLs for seed dormancy on LG-A2, C2, and D1b (Fig. 3A) and for seed number on LG-L (Fig. 3B) were well conserved between the F2 and BC1F2 generation, except for the DORM_1 QTL on LG-A2 in the W1 × D1 population, which was present in the F2 but not detected in the BC1F2 generation.
Discussion
Life history in relation to hybrid derivatives
In a previous study, hybrid derivatives that had arisen from gene flow between G. soja and G. max were grown in several natural habitats in Japan (Kuroda et al. 2010). Because the hardness of the seed coat, a phenotype related to seed dormancy (Table 4), is largely determined by the phenotype of the maternal G. soja plant, F1 seeds produced by pollen from G. max can survive in the soil several years, and the F1 plants can grow in the wild with G. soja. Here, FLOW of F1 hybrids tended to be similar to that of G. soja parent or intermediate between G. soja and G. max parent (Table 3), indicating that the flowering of natural F1 hybrids and local G. soja could overlap in several parts of Japan where natural hybrids have been identified. Due to genetic segregation in the F2 progenies, the extent of overlapping flowering time with G. soja will be reduced in that generation. However, once secondary gene flow from the F1 hybrid to G. soja has occurred, most of the backcross progenies are expect to have flowering time relatively similar to that of G. soja (Table 3). As the outcrossing rate in wild soybean populations has been reported to be 9.3–19% (Fujita et al. 1997) and 0–6.3% (Kuroda et al. 2008), our results suggest that G. max alleles can persist at some frequency in wild populations as long as gene flow continuously occurs at or near the maximum frequency.
Under the experimental field conditions, the total seed number (PROD_1) of the F1 hybrid was similar to or less than that of the corresponding G. soja parent (Table 3). PROD_1 of most F2 progenies was usually less than that of G. soja, although some F2 individuals revealed a similar or greater PROD_1 than the G. soja parent (Fig. 2A and B). As the proportion of G. soja background increased through backcrossing, the frequency of hybrid derivatives that revealed similar PROD_1 to G. soja also increased (Fig. 2C–F). However, after one round of self-pollination of the BC1F1 progenies, BC1F2 plants with short plant height and low seed production, as was seen in the F2 progenies, appeared again (Fig. 2G and H).
Most G. max seeds died in the soil during the winter, whereas the G. soja seeds survived (DORM_1, Table 3). Although DORM_1 of the F1 hybrids was intermediate between G. max and G. soja, F2 progenies revealed wide variation in DORM_1 (Fig. 2A and B). The extent of DORM_1 of the F2 progenies was related to the seed color (Table 4). As the proportion of G. soja background was increased by backcrossing with G. soja, the seed morphology (i.e., seed coat color and size) became closer to that of G. soja, and DORM_1 of the BC1F1 progenies increased (Fig. 2C–F). However, after one round of self-pollination of the BC1F1 progenies, BC1F2 seed/plants with low DORM_1 appeared (Fig. 2G and H). To understand this further, the phenotypic variation observed in the hybrid progenies was genetically dissected into QTLs by constructing genetic linkage maps.
Seed dormancy–related QTLs
Seedling emergence represents the interface between two demographic events: seed production and seedling recruitment. Because seed dormancy–related traits determine the timing of seedling emergence, the physiology of seed dormancy has a large effect on fitness. Good water permeability is an important trait for uniform and rapid germination in G. max cultivation and food processing. Conversely, rapid water uptake is known to lead to cell damage in the cotyledon (Powell and Matthews 1978) and is disadvantageous to survival of G. soja during winter in natural habitats. The physiological difference has been characterized by many researchers who have measured traits such as seed water imbibition or seed hardness during several days under germinable conditions. However, evaluation of seed dormancy is generally quite different between artificial and natural conditions in terms of time, water, and temperature conditions. Even G. max seed, which imbibes water during winter, could survive winter in 2006 (Table 3), indicating that water imbibition does not always lead to loss of seed viability. In this study, three major QTLs affecting both DORM_1 and DORM_2, which are located on LG-A2, -C2, and -D1b (Fig. 3A), were generally consistent over generations and crossing combinations. A significant high correlation between DORM_1 and DORM_2 was observed (Appendix A4): seeds from hybrid derivatives that had G. max alleles at those QTLs imbibed water easily and appear to have rotted in the soil over the winter. In particular, the G. max allele for DORM_1 on LG-A2 was found to be partially dominant to the G. soja allele because its effect appeared in BC1F1 progenies and it had a large effect of reducing survival rate in the W2 × D2 population (Appendix A5B). Therefore, the effect of such strong G. max alleles may lead to reduced winter survival of the seeds produced by an F1 hybrid plant as well as by later-generation progenies.
Nevertheless, the magnitudes of allele effects at the three major DORM_1/DORM_2 QTLs were slightly different depending on the cross combination. For example, the effect of the QTL on LG-A2 was strongest among the three QTLs in the W2 × D2 population, whereas it was similar to that of the other two QTLs in the W1 × D1 population (Fig. 3A). This explains the different level of seed winter survival between the W1 × D1 and W2 × D2 combinations. All the previously reported QTLs had the effect of causing water imbibition when the alleles at those loci were from G. max. In a G. max × G. soja population, Keim et al. (1990) detected four QTLs on LG-A2, -L, and -D1b by evaluating imbibition of F4 seeds for 7 days at room temperature. In contrast, Sakamoto et al. (2004) and Liu et al. (2007) identified two QTLs, located in LG-C2 and -D1b, by evaluating imbibition of seeds for 12 h and 24 h at room temperature, respectively. Glycine gracilis is an intermediate form between G. max and G. soja that originated in northeastern China (Hymowitz 2004). Three QTLs (on LG-C2, -D1b, and -I) were identified in a G. max × G. gracilis population by testing imbibition of seeds for 24 h at 25°C (Watanabe et al. 2004). These results indicate that QTLs on LG-C2 and -D1b are common among G. max × G. soja populations, but that a QTL on LG-A2 is not consistently detected in such populations. Similarly, in this study, no QTL for seed hardness (DORM_2) was detected on LG-A2 in the W1 × D1 population (Fig. 3A). It is very interesting that QTLs for seed winter survival (DORM_1), which required a long-term evaluation in the field, were successfully identified in the W1 × D1 combination in approximately the same region on LG-A2 where QTLs for DORM_1 and DORM_2 were detected in the W2 × D2 combination. One possible explanation of this finding is that the effect of a QTL on LG-A2 may appear when seeds imbibe water during long-term evaluation if the seed coat of G. max has resistance to water imbibition. The slow imbibition rate seen for D1 parent also supports this explanation and suggests that there is allelic variation within G. max for a seed hardness QTL on LG-A2.
Based on the map locations of gene-derived markers and the magnitude of QTL effects, the DORM_1/DORM_2 QTLs on LG-A2 and LG-C2 are tightly linked to the I locus and T locus, respectively, and the genes responsible for DORM_1 are either I and T themselves or genes closely linked to those loci (Fig. 3A). The I allele, which suppresses seed coat pigmentation, is dominant to the i allele, and the T allele, which confers pigment pubescence, is dominant to the t allele (Bernard and Weiss 1973). Hybrid derivatives without black seed coat (i.e., those with the I allele) showed low seed survival (Table 4), and, thus, the I allele is related to the water imbibition ability of G. max, which might be due to a physical characteristic of the seed coat. Epistatic interaction between the I and T loci has been reported to cause seed coat cracking when the alleles at both I and T locus are recessive and homozygous (Lindstrom and Vodkin 1991). Such cracked F3 seeds produced from several F2 individuals imbibed water quickly and failed to survive during winter (Table 4). Thus, epistatic interactions account for the reduced fitness of progenies derived from self-pollination, in spite of a low proportion of double-recessive individuals in the progenies, through their influence on seed viability or survival.
Seed production–related QTLs
The genes for domestication-related traits, which differentiate between crops and their wild relatives, are not randomly distributed across crop genomes (Ross-Ibarra 2005; Kaga et al. 2008). In this study, QTLs with high contributions to seed production–related traits, representing distinct differences between G. soja and G. max, tended to be concentrated in a particular genomic region on LG-L (Fig. 3B). Those QTLs were common between different cross combinations (W1 × D1 and W2 × D2) as well as across different generations. One possible reason for the positive, high correlation of total number of seed (PROD_1) with traits related to plant size such as stem dry weight (PROD_5) and stem length (PROD_6 [Appendix A4]) would be a gene related to stem elongation. Classically, stem termination in soybean is known to be controlled by two loci, Dt1 and Dt2 (Bernard 1972). The determinate stem type (dt1 allele) shows little growth in stem length after flowering, whereas the indeterminate stem type (Dt1 allele) continues to elongate even after flowering. An intermediate phenotype, called semideterminate, is conditioned by the Dt2 locus (Bernard 1972). Because Dt1 and Dt2 have been mapped on LG-L and LG-G, respectively (Cregan et al. 1999), the QTL with a strong contribution to stem length (PROD_6) on LG-L in this study is likely to be the Dt1 locus (Fig. 3B). Our results indicate that the G. max allele at this locus has the effect of reducing the number of seeds produced by hybrids between G. max and G. soja, as previously reported by Wang et al. (2004). Intriguingly, QTLs for seed weight (PROD_3) as well as other seed production–related traits were closely linked to marker LFsoy3, which was designed to detect a soybean homolog of PsTFL1a, a gene-controlling stem termination in Pisum (Foucher et al. 2003). Further studies are necessary to clarify the pleiotropic effect of soybean TFL1a on these traits. The G. max allele at the QTL for PROD_6 on LG-L was confirmed to have a moderate negative effect in the BC1F1 and BC2F1 populations, but it had no effect on PROD_1 as was found in the progenies from self-pollination (Fig. 3B, Appendix A5). A QTL for both PROD_6 and PROD_1 was identified again on LG-L in the BC1F2 population. These results indicate that the G. max allele is recessive to the G. soja allele because its effects were detected only in progenies generated by self-pollination.
Flowering phenology–related QTL
Photosensitivity is also an important plant response that is heavily involved in the control of flowering as well as in successful seed production. There were clear differences between the W1 × D1 and W2 × D2 populations in terms of both days to first flower (FLOW) (Table 3). The W1 × D1 population, representing northern Japanese germplasm, had shorter FLOW than the W2 × D2 population, representing southern Japanese germplasm. This difference reflects the adaptive strategy of G. soja and G. max in Japan. In northern Japan, the growing season is relatively short; thus, the W1 × D1 population might respond to warm temperatures and start to produce seeds during the short period of moderate climate even if the plants are not large. In contrast, the W2 × D2 population might respond to photoperiod and start to produce seeds only after the plants have grown large because autumn is relatively long in southern Japan.
Based on the location of SSR markers linked to previously reported flowering loci, the FLOW QTLs on LG-O (W1 × D1 population), -L (W1 × D1 and W2 × D2 population), and -I (W1 × D1 population) found in this study (Fig. 3B) are thought to be the classical maturity loci E2 (Bernard 1971), E3 (Buzzell 1971), and E4 (Buzzell and Voldeng 1980). The other FLOW QTLs with a large effect (i.e., that on LG-H) or with a moderate effect (i.e., on LG-E and -F in the W2 × D2 population [Appendix A5B] and on LG-D1b and -K in the W1 × D1 population [Appendix A5A]) have not been previously described and might be new loci for flowering time in soybean. Although a QTL for days to flowering on LG-C2 has been reported in a G. max × G. gracilis population (Yamanaka et al. 2001; Watanabe et al. 2004) and in a G. max × G. soja population (Liu et al. 2007), no flowering time QTL at that location was consistently identified in this study.
Evolutionary aspect of fitness-related QTLs and conclusions
Natural selection is expected to occur on the phenotypes of individuals that constitute G. soja populations, including hybrids between G. soja and G. max. Moreover, the phenotype of the hybrid progenies is influenced by the genetic variability of both G. max and G. soja, in response to a heterogeneous environment such as the natural habitat of G. soja. The results obtained here should be considered as an estimate obtained under conditions of maximum plant growth and seed production because the hybrid derivatives were widely spaced in the field (i.e., at intervals of 1 m); the results might have been different if the plants had been evaluated under conditions favoring high mortality of seedlings and restricted seed production in the competitive native weed population.
Genotype-dependent phenotypic response to different environments is common to quantitative traits and is referred to as phenotypic plasticity (Bradshaw 1965). In particular, the genes for wide adaptability that might have accumulated during human selection of G. max are probably different from those accumulated during ecological adaptation of G. soja, and they are likely to control more than the obvious morphological differences between the two species. For this reason, the effects of G. max genes were examined in this study in two types of hybrids between G. soja and G. max and were tested in two regions of Japan.
A large number of genes and their interactions with environmental changes during plant growth are thought to influence seed production. Nevertheless, the only QTL with a strong effect on PROD_1 between G. soja and G. max across different regions was the one identified on LG-L (Fig. 3B). The limited ability to detect QTLs involved in complex epistatic interactions might have led to underestimation of the number of loci involved in PROD_1 because QTLs for traits such as PROD_4 and FLOW that might be expected to affect PROD_1 were not always detected as QTLs for PROD_1.
Until recently, little has been known about the effect of G. max alleles within a predominantly G. soja genetic background. In this study, the genetic effects of those G. max alleles were not expressed as phenotypes in the BC1F1 and BC2F1 generations, indicating that most G. soja alleles are dominant to G. max alleles; one notable exception was the QTL for seed dormancy on LG-A2 (Fig. 3A). Snow et al. (1999) indicated that after two or three generation of backcrossing, hybrid derivatives in which crop alleles have been introgressed can be just as competitive and successful as wild plants. In this study, PROD_1 and DORM_1 in the BC1F1 and BC2F1 generation approached the values for G. soja as the proportion of G. soja genetic background increased (Table 3). Although QTLs at which G. max alleles had the increasing effect on PROD_1 and DORM_1 were not consistent over generations and crossing combinations (Fig. 3, Appendix A5), these alleles may have the potential to increase the fitness of hybrid derivatives. Individual plants that had higher fitness than G. soja in terms of SURV could be found in most generations of both the W1 × D1 and W2 × D2 populations (Fig. 2).
In contrast, QTLs at which G. max alleles had negative effects on fitness were consistently detected in both cross combinations and in different generations. In particular, QTLs for DORM_1 on LG-A2, -C2, and -D1b (Fig. 3A) and for PROD_1 on LG-L (Fig. 3B) were found in both cross combinations. This is one reason why hybrid derivatives do not survive in natural habitats (Kaga et al. 2005; Kuroda et al. 2005, 2006b, 2007), and why genetic differentiation is maintained between G. soja and G. max (Maughan et al. 1996; Powell et al. 1996; Xu and Gai 2003; Kuroda et al. 2006a). Previously, it was reported that hybrids between wild and crop species should be less fit than their wild parents due to the burden that crop traits would introduce into wild plants (De Wet and Harlan 1975). Current knowledge of the genetic basis of domestication traits suggests that few genomic regions are usually involved in domestication (White and Doebley 1998; Gross and Olsen 2010); thus, these regions could be purged quite rapidly with no long-term impact on fitness within the first few generations after hybridization. Our results support these studies and suggest that the risk of transgene dispersal into the wild soybean gene pool is generally low in Japan. The simulation studies as to what extent G. max alleles persist under a mixed mating system (i.e., considering the relative proportions of progenies both from self-fertilization and from outcrossing events) is required to improve the assessment of environmental transgene dispersal from GM soybeans.
Acknowledgments
We thank the staff of the experimental farms at NIAS, National Agricultural Research Center for Tohoku Region, and National Agricultural Research Center for Western Region for their assistance with the field experiments. We would also like to thank the National Agricultural Research Center for Kyushu Okinawa Region for the supply of “Fukuyutaka” seed. We are grateful to Yutaka Tabei, Hikaru Saji, and Ryo Osawa for technical assistance. This study was supported in part by the Global Environment Research Fund of the Japanese Ministry of the Environment (FY2003-FY2005) and principally by a grant (Assurance of Safe Use of Genetically Modified Organisms Project) from the Ministry of Agriculture, Forestry, and Fisheries of Japan.
Conflict of Interest
None declared.
Supporting Information
Additional Supporting Information may be found in the online version of this article:
Appendix A1. Geographical locations of experimental fields.
Appendix A2. Meteorological data for the north (Akita), central (Ibaraki), and south (Hiroshima) fields for 2005, 2006, and 2007, and mean values for the years 1979–2000.
Appendix A3. Primer sequences for five markers.
Appendix A4. Correlations among fitness-related traits for both W1D1 and W2D2 hybrids.
Appendix A5A. QTLs detected in F2, BC1F1, BC1F2, and BC2F1 generations of the W1 × D1 population.
Appendix A5B. QTLs detected in F2, BC1F1, BC1F2, and BC2F1 generations of the W2 × D2 population.
References
- Baack EJ, Sapir Y, Chapman MA, Burke JM, Rieseberg LH. Selection on domestication traits and quantitative trait loci in crop-wild sunflower hybrids. Mol. Ecol. 2008;17:666–677. doi: 10.1111/j.1365-294X.2007.03596.x. [DOI] [PubMed] [Google Scholar]
- Bernard RL. Two major genes for time of flowering and maturity in soybeans. Crop Sci. 1971;11:242–244. [Google Scholar]
- Bernard RL. Two genes affecting stem termination in soybeans. Crop Sci. 1972;12:235–239. [Google Scholar]
- Bernard RL, Weiss MG. Qualitative genetics. In: Caldwell BE, editor. Soybean: improvement, production and uses. Madison, WI: American Society of Agronomy; 1973. pp. 117–149. [Google Scholar]
- Bradshaw AD. Evolutionary significance of phenotypic plasticity in plants. Adv. Genet. 1965;13:115–156. [Google Scholar]
- Buzzell RI. Inheritance of a soybean flowering response to fluorescent-daylength conditions. Can. J. Genet. Cytol. 1971;13:703–707. [Google Scholar]
- Buzzell RI, Voldeng HD. Inheritance of insensitivity to long daylength. Soyb. Genet. Newsl. 1980;7:26–29. [Google Scholar]
- Carter TE, Hymowitz T, Nelson RL. Biogeography, local adaptation, Vavilov and genetic diversity in soybean. In: Werner D, editor. Biological resources and migration. Berlin: Springer; 2004. pp. 47–59. [Google Scholar]
- Churchill GA, Doerge RW. Empirical threshold values for quantitative trait mapping. Genetics. 1994;138:963–971. doi: 10.1093/genetics/138.3.963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cregan PB, Jarvik T, Bush AL, Shoemaker RC, Lark KG, Kahler AL, et al. An integrated genetic linkage map of the soybean genome. Crop Sci. 1999;39:1464–1490. [Google Scholar]
- De Wet JMS, Harlan JR. Weeds and domesticates: evolution in the man-made habitat. Econ. Bot. 1975;29:99–108. [Google Scholar]
- Di K, Stewart CN, Wei W, Shen BC, Tang ZX, Ma KP. Fitness and maternal effects in hybrids formed between transgenic oilseed rape (Brassica napus L.) and wild brown mustard [B. juncea (L.) Czern et Coss.] in the field. Pest Manag. Sci. 2009;65:753–760. doi: 10.1002/ps.1749. [DOI] [PubMed] [Google Scholar]
- Ellstrand NC. Current knowledge of gene flow in plants: implications for transgene flow. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2003;358:1163–1170. doi: 10.1098/rstb.2003.1299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellstrand NC, Prentice HC, Hancock JF. Gene flow and introgression from cultivated plants into their wild relatives. Annu. Rev. Ecol. Evol. Syst. 1999;30:539–563. [Google Scholar]
- Foucher F, Morin J, Courtiade J, Cadioux S, Ellis N, Banfield M, et al. DETERMINATE and LATE FLOWERING are two TERMINAL FLOWER1/CENTRORADIALIS homologs that control two distinct phases of flowering initiation and development in pea. Plant Cell. 2003;15:2742–2754. doi: 10.1105/tpc.015701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujita R, Ohara M, Okazaki K, Shimamoto Y. The extent of natural cross-pollination in wild soybean (Glycine soja. J. Hered. 1997;88:124–128. [Google Scholar]
- Gepts P, Papa R. Possible effects of (trans) gene flow from crops on the genetic diversity from landraces and wild relatives. Environ. Biosaf. Res. 2003;2:361–366. doi: 10.1051/ebr:2003009. [DOI] [PubMed] [Google Scholar]
- Gressel J. Tandem constructs: preventing the rise of superweeds. Trends Biotechnol. 1999;17:361–366. doi: 10.1016/s0167-7799(99)01340-2. [DOI] [PubMed] [Google Scholar]
- Gross BL, Olsen KM. Genetic perspectives on crop domestication. Trends Plant Sci. 2010;15:529–537. doi: 10.1016/j.tplants.2010.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hails RS. Genetically modified plants – the debate continues. Trends Ecol. Evol. 2000;15:14–18. doi: 10.1016/s0169-5347(99)01751-6. [DOI] [PubMed] [Google Scholar]
- Han OK, Kaga A, Isemura T, Wang, et al. A genetic linkage map for azuki bean. [Vigna angularis (Willd.) Ohwi & Ohashi] Theor. Appl. Genet. 2005;111:1278–1287. doi: 10.1007/s00122-005-0046-8. [DOI] [PubMed] [Google Scholar]
- Harlan JR. Crops and man. Crop Science Society of America, Madison, WI: American Society of Agronomy; 1992. The dynamics of domestication; pp. 115–133. [Google Scholar]
- Harrison RG. Hybrid zones: windows on evolutionary process. Vol. 7. Oxford, UK: Oxford Surveys in Evolutionary Biology; 1990. pp. 69–128. [Google Scholar]
- Hartman Y, Hooftman DAP, Uwimana B, Smulders CCM, van de Wiel MJM, Visser RGF, et al. Genomic regions in crop–wild hybrids of lettuce are affected differently in different environments: implications for crop breeding. Evol. Appl. 2012;5:629–640. doi: 10.1111/j.1752-4571.2012.00240.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hovick SM, Campbell LG, Snow AA, Whitney KD. Hybridization alters early life-history traits and increases plant colonization success in a novel region. Am. Nat. 2012;179:192–203. doi: 10.1086/663684. [DOI] [PubMed] [Google Scholar]
- Hymowitz T. Speciation and cytogenetics. In: Boerma HR, Specht JE, editors. Soybeans: improvement, production and uses. Madison, WI: American Society of Agronomy–Crop Science Society of America–Soil Science Society of America; 2004. pp. 97–136. Agronomy Monograph No. 16. [Google Scholar]
- James C. Global status of commercialized biotech/GM crops: 2012. Ithaca, NY: ISAAA; 2012. ISAAA Brief No. 44. [Google Scholar]
- Jenczewski E, Ronfort J, Chevre AM. Crop-to-wild gene flow, introgression and possible fitness effect of transgenes. Environ. Biosaf. Res. 2003;2:9–24. doi: 10.1051/ebr:2003001. [DOI] [PubMed] [Google Scholar]
- Kaga A, Tomooka N, Phuntsho U, Kuroda Y, Kobayshi N, Isemura T, et al. Exploration and collection for hybrid derivatives between wild and cultivated soybean: preliminary survey in Akita and Hiroshima Prefectures, Japan. Annual Report on Exploration and Introduction of Plant Genetic Resources. 2005;21:59–71. Available at http://www.gene.affrc.go.jp/plant/pdf/report/parts/2004_1-8.pdf (accessed 20 May 2013) [Google Scholar]
- Kaga A, Isemura T, Tomooka N, Vaughan DA. The genetics of domestication of the azuki bean (Vigna angularis. Genetics. 2008;178:1013–1036. doi: 10.1534/genetics.107.078451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kao CH, Zeng ZB, Teasdale RD. Multiple interval mapping for quantitative trait loci. Genetics. 1999;152:1203–1216. doi: 10.1093/genetics/152.3.1203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keim P, Diers BW, Shoemarker RC. Genetic analysis of soybean hard seedness with molecular markers. Theor. Appl. Genet. 1990;79:465–469. doi: 10.1007/BF00226154. [DOI] [PubMed] [Google Scholar]
- Korol AB, Ronin YI, Nevo E. Approximate analysis of QTL-environment interaction with no limits on the number of environments. Genetics. 1998;148:2015–2028. doi: 10.1093/genetics/148.4.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korol AB, Ronin YI, Itskovich AM, Peng J, Nevo E. Enhanced efficiency of quantitative trait loci mapping analysis based on multivariate complexes of quantitative traits. Genetics. 2001;157:1789–1803. doi: 10.1093/genetics/157.4.1789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kosambi DD. The estimation of map distance from recombination values. Ann. Eugen. 1944;12:172–175. [Google Scholar]
- Kuroda Y, Kaga A, Apa A, Vaughan, et al. Exploration, collection and monitoring of wild soybean and hybrid derivatives between wild soybean and cultivated soybean: based on field surveys at Akita, Ibaraki, Aichi, Hiroshima and Saga Prefectures. Annual Report on Exploration and Introduction of Plant Genetic Resources. 2005;21:73–95. Available at http://www.gene.affrc.go.jp/plant/pdf/report/parts/2004_1-9.pdf (accessed 20 May 2013) [Google Scholar]
- Kuroda Y, Kaga A, Tomooka N, Vaughan DA. Population genetic structure of Japanese wild soybean (Glycine soja) based on microsatellite variation. Mol. Ecol. 2006a;15:959–974. doi: 10.1111/j.1365-294X.2006.02854.x. [DOI] [PubMed] [Google Scholar]
- Kuroda Y, Kaga A, Guaf J, Vaughan DA, Tomooka N. Exploration, collection and monitoring of wild soybean, cultivated soybean and hybrid derivatives between wild soybean and cultivated soybean: based on field surveys at Akita, Ibaraki, Kochi and Saga Prefectures. Annual Report on Exploration and Introduction of Plant Genetic Resources. 2006b;22:1–12. Available at http://www.gene.affrc.go.jp/plant/pdf/report/parts/2005_1-1.pdf (accessed 20 May 2013) [Google Scholar]
- Kuroda Y, Kaga A, Poafa J, Vaughan DA, Tomooka N, Yano H. Exploration, collection and monitoring of wild soybean, cultivated soybean and hybrid derivatives between wild soybean and cultivated soybean: based on field surveys at Akita, Hyogo and Saga Prefectures. Annual Report on Exploration and Introduction of Plant Genetic Resources. 2007;23:9–27. Available at http://www.gene.affrc.go.jp/plant/pdf/report/parts/2006_1-2.pdf (accessed 20 May 2013) [Google Scholar]
- Kuroda Y, Kaga A, Tomooka N, Vaughan DA. Gene flow and genetic structure of wild soybean (Glycine soja) in Japan. Crop Sci. 2008;48:1071–1078. [Google Scholar]
- Kuroda Y, Kaga A, Tomooka N, Vaughan DA. The origin and fate of morphological intermediates between wild and cultivated soybeans in their natural habitats in Japan. Mol. Ecol. 2010;19:2346–2360. doi: 10.1111/j.1365-294X.2010.04636.x. [DOI] [PubMed] [Google Scholar]
- Levin DA, Francisco-Ortega J, Jansen RK. Hybridization and the extinction of rare plant species. Conserv. Biol. 1996;10:10–16. [Google Scholar]
- Lindstrom JT, Vodkin LO. A soybean cell wall protein is affected by seed color genotype. Plant Cell. 1991;3:561–571. doi: 10.1105/tpc.3.6.561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu B, Fujita T, Yan ZH, Sakamoto S, Xu D, Abe J. QTL mapping of domestication-related traits in soybean (Glycine max. Ann. Bot. 2007;100:1027–1038. doi: 10.1093/aob/mcm149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu BR. Transgene escape from GM crops and potential biosafety consequences: an environmental perspective. International Centre for Genetic Engineering and Biotechnology, Collection of Biosafety Reviews. 2008;4:66–141. [Google Scholar]
- Matsumura H, Watanabe S, Harada K, Senda M, Akada S, Kawasaki S, et al. Molecular linkage mapping and phylogeny of the chalcone synthase multigene family in soybean. Theor. Appl. Genet. 2005;110:1203–1209. doi: 10.1007/s00122-005-1950-7. [DOI] [PubMed] [Google Scholar]
- Maughan PJ, Saghai Maroof MA, Buss GR, Huestis GM. Amplified fragment length polymorphism (AFLP) in soybean: species diversity, inheritance, and near-isogenic line analysis. Theor. Appl. Genet. 1996;93:392–401. doi: 10.1007/BF00223181. [DOI] [PubMed] [Google Scholar]
- Mizuguti A, Yoshimura Y, Matsuo K. Flowering phonologies and natural hybridization of genetically modified and wild soybeans under field condition. Weed Biol. Manag. 2009;9:93–96. [Google Scholar]
- Nakayama Y, Yamaguchi H. Natural hybridization in wild soybean (Glycine max ssp. soja) by pollen flow from cultivated soybean (Glycine max ssp. max) in a designed population. Weed Biol. Manag. 2002;2:25–30. [Google Scholar]
- O'Callaghan M, Glare TR, Burgess EPJ, Malone LA. Effects of plants genetically modified for insect resistance on nontarget organisms. Annu. Rev. Entomol. 2005;50:271–292. doi: 10.1146/annurev.ento.50.071803.130352. [DOI] [PubMed] [Google Scholar]
- Ohara M, Shimamoto Y. Some ecological and demographic characteristics of two growth forms of wild soybean (Glycine soja. Can. J. Bot. 1994;72:486–492. [Google Scholar]
- Oka H. Genetic control of regenerating success in semi-natural conditions observed among lines derived from a cultivated x wild soybean hybrid. J. Appl. Ecol. 1983;20:937–949. [Google Scholar]
- Palmer RG, Sun H, Zhao LM. Genetics and cytology of chromosome inversions in soybean germplasm. Crop Sci. 2000;40:683–687. [Google Scholar]
- Peng J, Ronin Y, Fahima T, Röder MS, Li Y, Nevo E, et al. Domestication quantitative trait loci in Triticum dicoccoides, the progenitor of wheat. Proc. Natl Acad. Sci. USA. 2003;100:2489–2494. doi: 10.1073/pnas.252763199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Powell AA, Matthews S. The damaging effect of water on dry pea embryos during imbibition. J. Exp. Bot. 1978;29:1215–1229. [Google Scholar]
- Powell W, Morgante M, Doyle JJ, McNicol JW, Tingey SV, Rafalski JA. Genepool variation in genus Glycine subgenus Soja revealed by polymorphic nuclear and chloroplast microsatellites. Genetics. 1996;144:793–803. doi: 10.1093/genetics/144.2.793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- R Development Core Team. R: a language and environment for statistical computing. Vienna, Austria: R Foundation for Statistical Computing; 2009. Available at http://www.r-project.org (accessed 20 May 2013) [Google Scholar]
- Rieseberg LH, Linder CR, Seiler GJ, Ungerer MC, Arias DM. Role of gene interactions in hybrid speciation: evidence from ancient and experimental hybrids. Science. 1996;272:741–745. doi: 10.1126/science.272.5262.741. [DOI] [PubMed] [Google Scholar]
- Rolston MP. Water impermeable seed dormancy. Bot. Rev. 1978;44:365–396. [Google Scholar]
- Ross-Ibarra J. Quantitative trait loci and the study of plant domestication. Genetica. 2005;123:197–204. doi: 10.1007/s10709-004-2744-6. [DOI] [PubMed] [Google Scholar]
- Sahoo L, Schmidt JJ, Pedersen JF, Lee DJ, Lindquist JL. Growth and fitness components of wild × cultivated Sorghum bicolor (Poaceae) hybrids in Nebraska. Am. J. Bot. 2010;97:1610–1617. doi: 10.3732/ajb.0900170. [DOI] [PubMed] [Google Scholar]
- Sakamoto S, Abe J, Kanazawa A, Shimamoto Y. Marker-assisted analysis for soybean hard seededness with isozyme and simple sequence repeat loci. Breed. Sci. 2004;54:133–139. [Google Scholar]
- Singh RJ, Hymowitz T. The genomic relationship between Glycine max (L.) Merr. and G. soja Sieb. and Zucc. as revealed by pachytene chromosome analysis. Theor. Appl. Genet. 1988;76:705–711. doi: 10.1007/BF00303516. [DOI] [PubMed] [Google Scholar]
- Singh RJ, Hymowitz T. The genomic relationships among Glycine soja Sieb. and Zucc., G. max (L.) Merr., and ‘G. gracilis’ Skvortz. Plant Breeding. 1989;103:171–173. [Google Scholar]
- Snow AA, Andersen B, Jorgensen RB. Costs of transgenic herbicide resistance introgressed from Brassica napus into weedy B. rapa. Mol. Ecol. 1999;8:605–615. [Google Scholar]
- Snow AA, Culley TM, Campbell LG, Sweeney PM, Hegde SG, Ellstrand NC. Long-term persistence of crop alleles in weedy populations of wild radish (Raphanus raphanistrum. New Phytol. 2010;186:537–548. doi: 10.1111/j.1469-8137.2009.03172.x. [DOI] [PubMed] [Google Scholar]
- Song QJ, Marek LF, Shoemaker RC, Lark KG, Specht JE, Concibido VC, et al. A new integrated genetic linkage map of the soybean. Theor. Appl. Genet. 2004;109:122–128. doi: 10.1007/s00122-004-1602-3. [DOI] [PubMed] [Google Scholar]
- Stewart CN, Halfhill MD, Warwick SI. Transgene introgression from genetically modified crops to their wild relatives. Nat. Rev. Genet. 2003;4:806–817. doi: 10.1038/nrg1179. [DOI] [PubMed] [Google Scholar]
- Toda K, Yang D, Yamanaka N, Watanabe S, Harada K, Takahashi R. A single-base deletion in soybean flavonoid 3′-hydroxylase gene is associated with gray pubescence color. Plant Mol. Biol. 2002;50:187–196. doi: 10.1023/a:1016087221334. [DOI] [PubMed] [Google Scholar]
- Van Ooijen JW, Voorrips RE. JoinMap 3.0 software for the calculation of genetic linkage maps. Wageningen, The Netherlands: Plant Research International; 2001. [Google Scholar]
- Wang D, Greaf GL, Procopiuk AM, Diers BW. Identification of putative QTL trait that underlie yield in interspecific soybean backcross populations. Theor. Appl. Genet. 2004;108:458–467. doi: 10.1007/s00122-003-1449-z. [DOI] [PubMed] [Google Scholar]
- Warwick SI, Beckie HJ, Hall LM. Gene flow, invasiveness, and ecological impact of genetically modified crops. Ann. N. Y. Acad. Sci. 2009;1168:72–99. doi: 10.1111/j.1749-6632.2009.04576.x. [DOI] [PubMed] [Google Scholar]
- Watanabe S, Tajuddin T, Yamanaka N, Haysshi M, Harada K. Analysis of QTLs for reproductive development and seed quality traits in soybean using recombinant inbred lines. Breed. Sci. 2004;54:399–407. [Google Scholar]
- White S, Doebley J. Of genes and genomes and the origin of maize. Trends Genet. 1998;14:327–332. doi: 10.1016/s0168-9525(98)01524-8. [DOI] [PubMed] [Google Scholar]
- Xu DH, Gai JY. Genetic diversity of wild and cultivated soybeans growing in China revealed by RAPD analysis. Plant Breeding. 2003;122:503–506. [Google Scholar]
- Yamanaka N, Ninomiya S, Hoshi M, Tsubokura Y, Yamamoto K, Yano M, et al. An informative linkage map of soybean reveals QTLs for flowering time, leaflet morphology and regions of segregation distortion. DNA Res. 2001;8:61–72. doi: 10.1093/dnares/8.2.61. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.