

Abstract
Inference of genetic structure and demographic history is fundamental issue in evolutionary biology. We examined the levels and patterns of genetic variation of a widespread mangrove species in the Indo-West Pacific region, Bruguiera gymnorrhiza, using ten nuclear gene regions. Genetic variation of individual populations covering its distribution range was low, but as the entire species it was comparable to other plant species. Genetic differentiation among the investigated populations was high. They could be divided into two genetic clusters: the West and East clusters of the Malay Peninsula. Our results indicated that these two genetic clusters derived from their ancestral population whose effective size of which was much larger compared to the two extant clusters. The point estimate of speciation time between B. gymnorrhiza and Bruguiera sexangula was two times older than that of divergence time between the two clusters. Migration from the West cluster to the East cluster was much higher than the opposite direction but both estimated migration rates were low. The past Sundaland and/or the present Malay Peninsula are likely to prevent gene flow between the West and East clusters and function as a geographical or land barrier.
Keywords: Demographic history, DNA sequence variation, geographical or land barrier, mangrove, population structure
Introduction
Mangrove species constitute a unique ecosystem in coastal zones of tropical and subtropical regions. The distribution of mangrove species is classified into two major regions, Indo-West Pacific (IWP) and Atlantic-East Pacific (AEP) (Tomlinson 1986; Duke et al. 1998). The IWP has about four times greater species diversity than the AEP (Duke et al. 1998). In the last few decades, many mangrove forests are rapidly getting small, fragmented, and lost in both regions (e.g., FAO 2007). Population size reduction and fragmentation cause a loss of genetic diversity and lead to the loss of adaptability for environmental changes. The loss of adaptability increases the extinction risk. For successive preservation and management of this unique ecosystem, the accurate assessment of genetic diversity and population genetic structure of mangrove species is essential. The knowledge of genetic information also provides us a perspective of evolutionary mechanisms that shaped current biodiversity and adaptability, and gives us insight how to restore this unique ecosystem.
Most studies on genetic variability and population structure of mangrove species use simple sequence repeat (SSR) markers and chloroplast DNA (cpDNA) sequences (e.g., Marguire et al. 2000; Liao et al. 2007; Huang et al. 2008; Jian et al. 2010; Pil et al. 2011). Polymorphic SSR loci are “selected” as markers in population genetic studies. This selection leads to the overestimation of the amount of genetic variation in a population. In addition, it is not easy to apply SSR markers for one species to the other species. In contrast, surveys of genetic structure on mangroves using nucleotide sequences are very limited (Zhou et al. 2008; Inomata et al. 2009; Minobe et al. 2010) because nucleotide sequence variation in mangrove species is considered to be low and, therefore, not useful for population genetic studies. However, surveys on gene sequence variation are crucial to find evolutionary forces that shaped genetic structures and to identity genes that are responsible for adaptation. For example, Zhou et al. (2011) reported some candidate genes for local adaptation based on the survey of 71 genes in a mangrove species, Sonneratia alba. To proceed future comparative evolutionary genomics studies, accumulation of gene sequence data is indispensable. Furthermore, for most mangrove species, assessment of genetic structure of populations was mainly focused on limited areas (e.g., Inomata et al. 2009). To draw a general picture of genetic structure and evolutionary history, surveys of populations covering areas of species distribution are necessary.
Mating systems and the ways of pollen and seed dispersal are important factors affecting genetic structure of plant populations. Dispersal distance of pollen and seeds is one of the determinants for geographical distribution range of plant species. Previous studies on mangrove species revealed that, in general, genetic variation within populations is low and genetic differentiation among populations is high (e.g., Goodall and Stoddart 1989; Giang et al. 2006; Liao et al. 2007; Inomata et al. 2009; Minobe et al. 2010; Pil et al. 2011). High genetic differentiation indicates low gene flow between populations, suggesting pollen and seed dispersal are restricted. Indeed, propagules of Rhizophora mucronata and Rhizophora apiculata appear to have short dispersal ability (Drexler 2001) and the average distances of pollen and propagule dispersal are also short in Kandelia candel (Geng et al. 2008).
Plants with sea-drifted seeds and animals living in water cannot migrate through land. In general, this is called a geographical or land barrier hypothesis. In the IWP region, the sea level dropped to 100–120 m below the current level during the Last Glacial Maximum (LGM) (Voris 2000) and thus the Sundaland, which consists of the Malay Peninsula, Sumatra, Java, Borneo islands, and exposed shelves, existed at that time. During the LGM, the Sundaland is considered to have played a role as a land barrier that prevented gene flow between the Pacific and Indian Oceans. At present, the Malay Peninsula is suggested to continue to serve as a barrier to gene flow between the Pacific and Indian Oceans. Because propagules of all mangroves are dispersed only by water (Tomlinson 1986), past and current land barriers are considered to affect genetic structure of the extant mangrove populations (Liao et al. 2007; Huang et al. 2008; Inomata et al. 2009).
Our main objective is to clarify genetic structure and demographic history of typical mangrove species on a global scale. For this purpose, we examined genetic variation of ten nuclear gene regions as well as one cpDNA region using samples from ten Bruguiera gymnorrhiza populations covering its natural distribution range. B. gymnorrhiza (L.) Lamk. (Rhizophoraceae) is a typical mangrove species and one of the predominant mangrove species in the IWP region. It has the broadest distribution in the genus: from eastern coast of Africa in the west through Asia, northward to the Ryukyu Islands of southern Japan and southward in tropical Australia (Tomlinson 1986). It can be found in the middle and upper intertidal zones. Like the other mangrove species of the tribe Rhizophoraceae Blume, B. gymnorrhiza has a unique viviparous propagule (Tomlinson 1986), which is dispersed by water. B. gymnorrhiza is one of the bird-pollinated large-flowered species of Bruguiera (Tomlinson 1986). For interspecies comparison, genetic variation of the identical gene regions of the closely related species, Bruguiera sexangula (Lour.) Poir., was also examined.
We address the following specific questions: (1) What is the extent of nucleotide sequence variation? (2) What is the extent of genetic differentiation between populations? (3) Do the Sundaland and/or the Malay Peninsula play a role of a barrier to prevent gene flow between the Pacific and Indian Oceans? (4) When did the two species diverge?
Materials and Methods
Plant samples
Leaf materials of B. gymnorrhiza were sampled from different individuals at the following ten locations (Fig. 1); V (Hanoi, Vietnam) located in the east of the Asian Continent, MK (Kuching, Malaysia) located in the northwest of the Borneo Island in the Southeast Asia, ML (Langkawi Island, Malaysia) located in the northwest side of the Malay Peninsula, BMJ (Bali Island, Indonesia) located in the east of the Java Island in the Southeast Asia, AC (Cairns, Australia) located in the northeast coast of Australia, AB (Ballina, Australia) located in the east coast of Australia, K (Kakinada, India) located in the northeast coast of India, G (Goa, India) located in the southwest of India, SL (Tolanaro, Madagascar) located in the southeast coast of Madagascar, and MO (Morondava, Madagascar) located in the west coast of Madagascar. We call each sampling location as a population in the following sections. Leaf materials of a closely related species, B. sexangula, were also sampled at two locations on the western coast of the Malay Peninsula. The details of our samples, including the locations and numbers of individuals, are summarized in Table 1.
Figure 1.
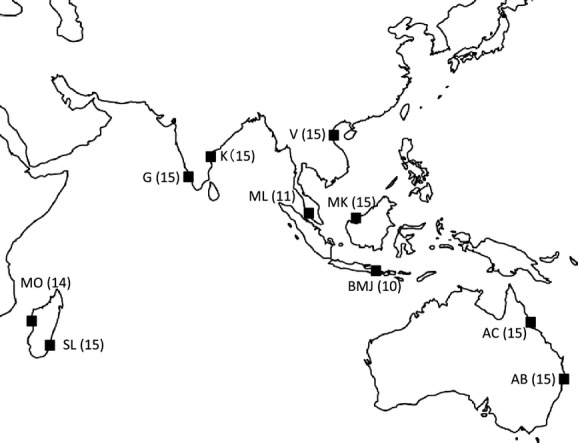
Sampling locations of the 10 populations. Populations are indicated by abbreviations. Number of individuals used in this study is shown in parentheses.
Table 1.
Geographical location and sample size in Bruguiera gymnorrhiza and Bruguiera sexangula
| Sampling location | Abbreviation | Geographical coordinates | Number of individuals |
|---|---|---|---|
| B. gymnorrhiza | |||
| Australia | |||
| Ballina | AB | 28°52′33.26″S, 153°32′47.05″E | 15 (55) |
| Cairns | AC | 16°50′21.47″S, 145°43′25.45″E | 15 (32) |
| Indonesia | |||
| Bali | BMJ | 8°05′45.84″S, 114°28′33.15″E | 10 (10) |
| Vietnam | |||
| Hanoi | V | 21°00′05.62″N, 107°16′35.82″E | 15 (45) |
| Malaysia | |||
| Kuching | MK | 1°38′31.22″N, 110°14′57.25″E | 15 (45) |
| Langkawi | ML | 6°26′02.61″N, 99°50′23.41″E | 11 (11) |
| India | |||
| Goa | G | 15°22′23.85″N, 73°57′42.07″E | 15 (23) |
| Kakinada | K | 16°56′48.14″N, 82°15′23.12″E | 15 (20) |
| Madagascar | |||
| Morondava | MO | 20°16′47.58″S, 44°17′19.81″E | 14 (41) |
| Tolanaro | SL | 25°03′43.35″S, 46°53′42.07″E | 15 (29) |
| B. sexangula | |||
| Malaysia | |||
| Langkawi | 6°26′02.61″N, 99°50′23.41″E | 7 (8) | |
| Thailand | |||
| Ranong | 9°84′43.92″N, 98°55′40.58″E | 2 (2) | |
Number of individuals used in this study is shown. Total number of individuals collected is shown in parentheses.
DNA extraction, PCR, and sequencing
Fifteen individuals were randomly selected from each population. When the number of individuals in a population was smaller than 15, we analyzed all individuals. Genomic DNA was extracted from a piece of a single leaf using modified cetyltrimethylammonium bromide (CTAB) method (Murray and Tompson 1980). In total, genomic DNAs of 140 individuals of B. gymnorrhiza were obtained.
Based on the published sequence information of the expressed sequence tags (ESTs) of B. gymnorrhiza (Miyama et al. 2006), PCR primer sets of eight nuclear genes were designed. Based on the sequence information of the PCR products, we conducted BLAST search against NCBI database (nucleotide collection). According to the blast search results, we tentatively named the eight genes as follows; NAC (BP941102), VVHP (BP944085), PO (BP939063), GM (BP939421), SF (BP939928), EXP2 (BP942725), EPCRF (BP946081), and UNK (BP945250), where the corresponding B. gymnorrhiza ESTs (Miyama et al. 2006) are shown in parentheses. In addition to the aforementioned eight nuclear genes, two nuclear genes, PAL1 and mang-1 (Inomata et al. 2009), and one chloroplast DNA (cpDNA) region, trnS-trnG spacer (Hamilton 1999; Sun and Lo 2011), were also analyzed.
PCR amplification was carried out under the following protocols: an initial denaturation at 95°C for 3 min followed by 30 cycles of denaturation at 95°C for 30 sec, annealing at 50°C, 55°C, or 60°C for 30 sec, polymerization at 72°C for 1.5 min, and a final extension at 72°C for 7 min. The details of the sequence and annealing temperature of PCR primers are listed in Table S1. After the amplification, PCR products were purified. Then we directly determined their sequences using a DNA sequencing kit (BigDye terminator v. 3.1/1.1 cycle sequencing kit, ABI) with the PCR primers and ABI Prism 3100 or 3730 automatic sequencers (Applied Biosystems, Tokyo, Japan).
Two haplotype sequences were determined for each individual. After the direct sequencing when we found no or only one heterozygous site in a sequence of a single individual, we inferred sequences of two haplotypes for each individual. On the other hand, when two or more heterozygous sites or indels were found in a sequence of a single individual, purified PCR product was cloned into the pGEM T-easy vector (Promega KK, Tokyo, Japan). Then, we sequenced each clone using T7 and Reverse primers designed at the promoter sites of the vector (Table S1). Four to sixteen clones were sequenced for each individual. Sequences of two haplotypes of a heterozygous individual were determined when we obtained at least two exactly identical sequences for each haplotype.
Data analyses
Sequences of each gene were aligned using CLUSTAL X 2.1 (Larkin et al. 2007) and further edited by hand. Because, we could not amplify EXP2 gene of one individual sampled from the AB population from the east coast of Australia, we treated it as missing data in the following analysis. A continuous alignment gap was treated as a single indel. All sequences obtained in this study were deposited in the DNA Data Bank of Japan (DDBJ). Accession numbers are AB813916-AB817042.
Molecular population genetic analysis of B. gymnorrhiza was conducted using sequences of B. sexangula as an outgroup. Using inter-simple sequence repeat, ribosomal internal transcribed spacer, and chloroplast DNA markers, Sun and Lo (2011) showed that B. gymnorrhiza and B. sexangula are genetically close relatives. Molecular population genetic parameters, π (the average number of nucleotide differences per site: nucleotide diversity; Nei 1987), Ks (number of nucleotide substitutions per silent site between species), and Ka (number of nucleotide substitutions per replacement site between species), were estimated using the DnaSP ver. 5.10.00 (Librado and Rozas 2009). To assess the deviation from the standard neutral model, Tajima's test, Fu and Li's test, Fay and Fu's test, and MK test were performed using the DnaSP version 5.10.00, and the multilocus HKA test was also performed using HKA program downloaded from http://genfaculty.rutgers.edu/hey/software#HKA.
The extent of genetic differentiation among populations was estimated by analysis of molecular variance (AMOVA) approach (e.g., Weir and Cockerham 1984). The Arlequin software version 3.5.1.3 (Excoffier and Lischer 2010) was used to estimate F-statistics. Three hierarchical levels, (i) among populations, (ii) among individuals/within populations, and (iii) within individuals, were examined for nuclear genes by the locus-by-locus analysis, and two levels, (i) among populations and (ii) within populations, were examined for cpDNA. Each haplotype in a gene was regarded as an allele. Overall values of F-statistics for all the ten nuclear genes were obtained by summing variance components over the genes. In the AMOVA analyses, genotypic data of nuclear genes, where gametic phase of genotype is unknown, and haplotypic data of the cpDNA region were used. A haplotype network of the cpDNA region was constructed using the TCS ver. 1.21 (Clement et al. 2000).
Genetic clusters of populations were inferred using the Bayesian clustering approach, STRUCTURE ver. 2.3.3 (Pritchard et al. 2000; Falush et al. 2003; Hubisz et al. 2009). Under the admixture model, five independent runs were performed for each number of clusters (K: K = 1–10) to confirm the convergence of Markov chain Monte Carlo (MCMC) chains. In each run, 500,000 MCMC iterations were performed after a burn-in period of 1,000,000 iterations. To estimate the number of clusters, we applied the method proposed by Evanno et al. (2005); K is identified when delta K, which is the rate of change in the log probability of data between continuous K values, is maximal.
Population history of genetic clusters identified using the STRUCTURE and evolutionary history of the two species, B. gymnorrhiza and B. sexangula, were inferred. Population parameters were estimated under the isolation-with-migration (IM) model using the IMa2 program (Hey and Nielsen 2007; Hey 2010). In this analysis non-recombinant regions were used. Non-recombinant regions were estimated using the four-gametic test (Hudson and Kaplan 1985) implemented in DnaSp ver. 5.10.00. Population splitting (or speciation) times (T = tu), population sizes (θ = 4Nu), and migration rates (2Nm) between genetic clusters (or species) were estimated, where u is the mutation rate per locus per generation, t is the population splitting (or speciation) time, N is the effective population size, and m is the migration rate. The infinite site model (Kimura 1969) was used except VVHP, EXP2, and mang-1. For the three regions, we applied HKY model (Hasegawa et al. 1985) because there were more than two alleles at some variable sites in these three genes. The upper bounds of the parameters are set to include most part of the posterior distribution of preliminary simulations. We confirmed that the burn-in period was long enough so that the MCMC simulation reached the stationary state. Each simulation run was performed with 40 chains with adjusted heating terms according to the IMa2 manual. To check the convergence of the MCMC simulation to the true stationary state, multiple independent runs were performed. The posterior distributions of parameters were estimated using 100,000 genealogies sampled from multiple independent runs. Peaks of the posterior distribution were defined as estimates of parameters.
Results
Nucleotide variation in B. gymnorrhiza
Sequences of partial regions of the ten nuclear genes, NAC, VVHP, PO, GM, SF, EXP2, UNK, EPCRF, PAL1, and mang-1, were determined for ten populations of B. gymnorrhiza. Sequences of one cpDNA region, trnS-trnG spacer, were also determined. The levels of DNA variation in B. gymnorrhiza are summarized in Table 2. In B. gymnorrhiza total length examined for nuclear genes was 5,295 bp including alignment gaps, where 3,313 bp was coding region. In total 84 segregating sites and five indels were found. In 581 bp of the cpDNA region, there were three segregating sites and five indels.
Table 2.
Summary of nucleotide polymorphism in Bruguiera gymnorrhiza
| Alignment length/Number of silent sites (bp) | n | S | Indel | πt | πs | πa | |
|---|---|---|---|---|---|---|---|
| Nuclear gene | |||||||
| NAC | 370/185.17 | 280 | 0 | 0 | 0.00 | 0.00 | 0.00 |
| VVHP | 575/381.06 | 280 | 26 (22/4) | 2 | 9.06 | 11.89 | 3.29 |
| PO | 536/237.67 | 280 | 0 | 0 | 0.00 | 0.00 | 0.00 |
| GM | 494/282.50 | 280 | 9 (7/2) | 0 | 6.57 | 11.51 | 0.07 |
| SF | 407/100.01 | 280 | 3 (1/2) | 0 | 2.17 | 8.90 | 0.00 |
| EXP2 | 535/433.89 | 278 | 18 (18/0) | 1 | 13.12 | 16.21 | 0.00 |
| UNK | 426/102.33 | 280 | 2 (0/2) | 0 | 1.03 | 0.00 | 1.35 |
| EPCRF | 408/82.75 | 280 | 7 (4/3) | 0 | 5.02 | 25.06 | 0.09 |
| PAL1 | 704/169.66 | 280 | 4 (2/2) | 2 | 1.30 | 3.27 | 0.68 |
| mang-1 | 840/773.67 | 280 | 15 (15/0) | 0 | 2.87 | 3.12 | 0.00 |
| Average | 530/274.87 | 279.8 | 8.4 (6.9/1.5) | 0.5 | 4.11 | 8.00 | 0.55 |
| cpDNA region | |||||||
| trnS-trnG | 581/506.00 | 140 | 3 | 5 | 0.08 | 0.08 | 0.00 |
Alignment length/Number of silent sites, Sequence length includes alignment gaps. Number of silent sites does not include alignment gaps. n, number of sequences; S, Number of segregating sites excluding indels. Numbers of silent/replacement differences are shown in parentheses; Indel, number of indels. Continuous alignment gap was counted as a single indel; πt, number of nucleotide differences per total site (Nucleotide diversity; Nei 1987) with the Jukes and Cantor (1969) correction. Indels are not included. The value was multiplied by 103; πs, number of nucleotide differences per silent site with the Jukes and Cantor (1969) correction. Indels are not included. The value was multiplied by 103; πa, number of nucleotide differences per nonsynonymous site with the Jukes and Cantor (1969) correction. Indels are not included. The value was multiplied by 103.
The level of nucleotide variation was different among the genes. For example, nucleotide diversity (Nei 1987) per silent site, πs, for the total population ranged from 0 (NAC and PO) to 0.02506 (EPCRF) for the nuclear genes (Table 2). Two of the ten genes (NAC and PO) were monomorphic. In the total population, the average πs over the ten nuclear genes was 0.00800. The πs value of the cpDNA region was 0.00008. Nucleotide diversity of the nuclear genes was ∼100 times higher than that of the cpDNA region, indicating that the nuclear genes are more useful markers than the cpDNA region.
The level of nucleotide variation also varied among the ten populations. For example, the average πs value over the ten nuclear genes ranged from 0 (G and K populations from the southwest and northeast India) to 0.00424 (V population from the east of the Asia Continent) (Table S2).
Nucleotide divergence between B. gymnorrhiza and B. sexangula
Nucleotide divergence between B. gymnorrhiza and B. sexangula is summarized in Table 3. No shared polymorphism was found between the two species. The number of fixed nucleotide substitutions ranged from 1 to 11 in the nuclear genes. On average, there were 3.9 fixed nucleotide substitutions between the two species. These results confirm that our sample consists of two distinct species. The number of nucleotide substitutions per silent site between the species (Ks) ranged from 0.00437 to 0.04925 for the nuclear genes and 0.00334 for the cpDNA region. The average Ks value over the ten nuclear genes was 0.02162. Divergence at silent sites of nuclear genes was ∼10 times higher than that of the cpDNA region. The number of nucleotide substitution per replacement site between the species (Ka) ranged from 0.00000 to 0.00485 for the nuclear genes. The highest value of Ka/Ks was 0.32071 for PAL1, indicating that these nuclear genes are not pseudogenes.
Table 3.
Summary of nucleotide divergence between Bruguiera gymnorrhiza and Bruguiera sexangula
| Length (including gaps) | n | Fix | Indel | Ks | Ka | Ka/Ks | |
|---|---|---|---|---|---|---|---|
| Nuclear gene | |||||||
| NAC | 370 | 298 | 4 (2/2) | 0 | 21.92 | 0.00 | 0.00 |
| VVHP | 575 | 298 | 5 (5/0) | 1 | 23.25 | 2.51 | 0.11 |
| PO | 536 | 298 | 1 (1/0) | 1 | 4.37 | 0.00 | 0.00 |
| GM | 495 | 298 | 4 (3/1) | 1 | 22.21 | 4.85 | 0.22 |
| SF | 407 | 298 | 4 (0/4) | 0 | 49.25 | 0.00 | 0.00 |
| EXP2 | 538 | 296 | 4 (4/0) | 1 | 25.70 | 0.00 | 0.00 |
| UNK | 426 | 298 | 3 (2/1) | 0 | 19.81 | 4.04 | 0.20 |
| EPCRF | 408 | 298 | 1 (0/1) | 0 | 24.56 | 3.13 | 0.13 |
| PAL1 | 704 | 298 | 2 (1/1) | 0 | 7.92 | 2.54 | 0.32 |
| mang-1 | 842 | 298 | 11 (11/0) | 2 | 17.23 | 0.00 | 0.00 |
| Average | 530 | 297.8 | 3.9 | 0.6 | 21.62 | 1.71 | 0.10 |
| cpDNA region | |||||||
| trnS-trnG | 581 | 149 | 2 | 0 | 3.34 | 0.00 | 0.00 |
Length, Sequence length includes alignment gaps. n, number of sequences; Fix, number of fixed nucleotide differences between the species excluding indels. Numbers of silent/replacement differences are shown in parentheses; Indel, number of fixed indels between the species. A continuous gap was counted as a single indel; Ks, number of nucleotide substitutions per silent site with the Jukes and Cantor (1969) correction. Indels are not included. The value was multiplied by 103; Ka, number of nucleotide substitution per replacement site with the Jukes and Cantor (1969) correction. Indels are not included. The value was multiplied by 103.
Neutrality tests
Results of neutrality tests are summarized in Table S3. Most results of Tajima's test and Fu and Li's test were not significant. For example, Tajima's D values were not significant except for the VVHP gene of MO population (the west of Madagascar) and the mang-1 gene of the V population (the eastern part of the Asian Continent). No significant results were obtained in Fay and Fu's test (Table S3). The result of the multilocus HKA test was weakly significant (P = 0.048). In MK test no significant result was found (data not shown). These results suggest that gene regions examined could be treated as neutral markers in B. gymnorrhiza.
Genetic structure of populations
F-statistics among the ten populations estimated by the AMOVA approach were shown in Table 4. Two of the ten genes (NAC and PO) had no variation, and therefore they did not have any information for population structure. The estimated FST values were highly significant both for nuclear genes and cpDNA region, indicating that there is genetic differentiation among the ten populations. In nuclear genes most population pairwise FST values were highly significant except for population pairs, AB (eastern Australia) – MK (northwestern Borneo), AB – ML (northwestern part of the Malay Peninsula), AC (northeastern of Australia) – ML, BMJ (eastern Java) – MO (western Madagascar), V (eastern part of Asian Continent) – K (northeastern India), MK – ML, and G (southwestern India) – SL (southeastern Madagascar) (Table S4), indicating that most population pairs are genetically differentiated. Although the pairwise FST values in nuclear genes were not significant for population pairs, AB (eastern Australia) – MK (northwestern Borneo), AB – ML (northwestern part of the Malay Peninsula), and AC (northeastern Australia) – ML, a relationship of cpDNA haplotypes (Figure S2) and geographic distribution of the haplotypes (Figure S3) are likely to indicate genetic differentiation between the Australian populations and others.
Table 4.
F-statistics among the ten populations
| Nuclear genes (overall 10 loci) | ||
|---|---|---|
| FIS | FST | FIT |
| 0.29192 (≪0.001) | 0.78638 (≪0.001) | 0.84874 (≪0.001) |
| cpDNA region | ||
| FIS | FST | FIT |
| na | 0.88932 (≪0.001) | na |
P-values are showed in parentheses; na, not applicable.
To find the uppermost hierarchical level of the genetic clusters, population structure was estimated using the STRUCTURE program (Pritchard et al. 2000; Hubisz et al. 2009). It assigns individuals into K clusters, each of which is at HWE or not in LD, using multilocus genotype data. According to the method of Evanno et al. (2005), the number of clusters was estimated to be 2 (Figure S1). The result of the STRUCTURE analysis is shown in Figure 2. The first cluster (dark gray) includes two Australian populations (AB and AC; see Figure 1) and three populations located on the east coast of the Malay Peninsula (BMJ, V, and MK; see Fig. 1) (East cluster). The second cluster (light gray) includes five populations located on the west coast of the Malay Peninsula (ML, G, K, MO, and SL; see Fig. 1) (West cluster).
Figure 2.
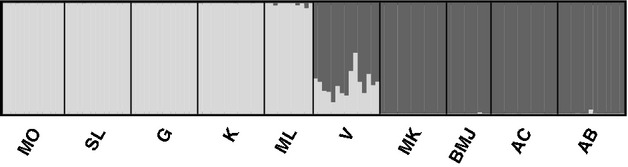
Clustering results of individuals in the 10 popultions of Bruguiera gymnorrhiza were estimated using the STRUCTURE. Each vertical bar in the histogram represents the proportion of cluster memberships in a single individual. Abbreviations of populations ordered from the west to the east are shown under the histogram.
Population history
To infer the population history of the two genetic clusters identified by the STRUCTURE analysis, population parameters were estimated under the IM model using IMa2 program (Hey and Nielsen 2007; Hey 2010). Evolutionary history of the two investigated species was also inferred (Fig. 3). The estimated splitting time between the two population clusters was 0.17 (95% highest posterior density (HPD), lower bound: 0.04, upper bound: 0.54) in units of mutation time scale (time x mutation rate), whereas the estimated speciation time between B. gymnorrhiza and B. sexangula was 0.32 (95% HPD, lower bound: 0.13, upper bound: 0.63). This result indicates the speciation event between B. gymnorrhiza and B. sexangula is roughly two times older than the divergence of the two clusters in B. gymnorrhiza.
Figure 3.
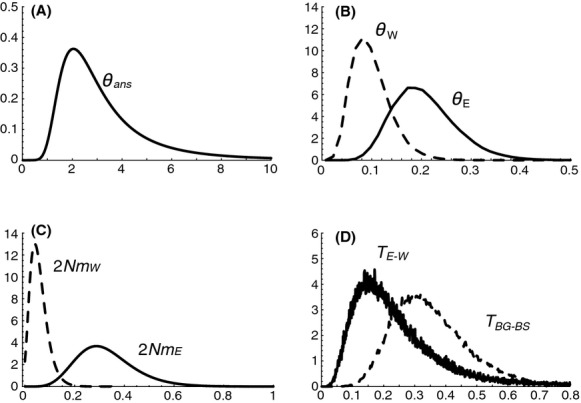
Marginal posterior probability distributions for model parameters estimated using the IMa2. (A) Population size of Ancestor population (θans). (B) Population size of the East cluster (θE) and West cluster (θW). (C) Migration rate from the West cluster (2NmE) to the East cluster and from the East cluster to the West cluster (2NmW). (D) Divergence time of the East cluster and West cluster (TE−W) and speciation time between Bruguiera gymnorrhiza and Bruguiera sexangula (TBG−BS).
The estimate of θ for the East cluster (θE) was 0.17 (95% HPD, lower bound: 0.08, upper bound: 0.38) and θ for the West cluster (θW) was 0.08 (95% HPD, lower bound: 0.04, upper bound: 0.26). θ for the ancestral population was 2.05 (95% HPD, lower bound: 0.82, upper bound: 7.37). This result means that the effective population size of East cluster is larger than that of West cluster, and the ancestral population is much larger than either of the clusters. Larger size in ancestral populations than the extant populations was also reported in the other mangrove species (Sonneratia, Zhou et al. 2007). There was more migration from the West cluster to the East cluster (2NmE = 0.29 [95% HPD, lower bound: 0.12, upper bound: 0.56]) than from the East to West (2NmW = 0.04 [95% HPD, lower bound: 0, upper bound: 0.12]), although in both cases the migration was rare (2Nm < 1). No migration was estimated between B. gymnorrhiza and B. sexangula after the speciation event (data not shown).
Discussion
DNA polymorphism and divergence
To find a general picture of levels and patterns of DNA variation of a broad range species, B. gymnorrhiza, leaf materials were collected from 10 localities covering its distribution range and DNA sequences were determined. General pattern of nucleotide variation of B. gymnorrhiza was as follows: (1) low polymorphism in each local population but (2) heterogeneous variation between the local populations (Table S2). As the entire species, however, the level of polymorphism was comparable to other plant species (e.g., Cryptomeria japonica; Kado et al. 2003; Arabidopsis thaliana; Schmid et al. 2005; Pinus species; Ma et al. 2006; see summarized by Gossmann et al. 2010). Assuming neutral evolution of the investigated gene regions, the simplest interpretation of the result is that the effective population sizes of the local populations are different. Even though the number of individuals is large in nature, effective size of some populations could be quite small.
In each population the level of nucleotide variation was low. For example, in Indian populations, G and K, no polymorphism was found for all the ten genes. Inbreeding is one of the plausible explanations. Other plausible explanation is that originally each local population may have been founded by a small number of individuals and not enough time has elapsed to accumulate new mutations. The bottleneck and/or founder effects caused by the climatic oscillations and environmental changes during the Pleistocene are also possible explanations for the low level of nucleotide variation in local populations. These do not rule out the possibility of local adaptation. In any cases if low genetic variation is general feature in functional (or protein-coding) genes, each population is likely to have little potential for genetic adaptability. This suggests that extinction risk of each population is high. Alternatively, it may be the nature of this species, for example, the species may repeat the following extinction-recolonization cycle; (1) a population starts with a small number of individuals, which adapted (or survived) to a given environment, (2) the population increase in size (number of individuals), and (3) when the environment changes accompanying with climatic and/or geographical changes, the population disappears.
Nucleotide divergence at silent sites between B. gymnorrhiza and B. sexangula was ∼2%. It was similar or smaller that nucleotide divergence between Sonneratia species (2.06 ∼ 3.75%, Zhou et al. 2007) and between R. apiculata and R. mucronata (∼2.7%, Inomata et al. 2009). On the other hand, as the entire species, nucleotide diversity at silent sites in B. gymnorrhiza (0.00800) was comparable to those of Sonneratia alba (0.00432) and S. caseolaris (0.01003) (Zhou et al. 2007), but two order higher than those of R. apiculata (0.00059) and R. mucronata (0.00003) (Inomata et al. 2009). Like in this study, population samples of the two Sonneratia species were collected from across the entire IWP region. However, in the previous study (Inomata et al. 2009), the two Rhizophora species were collected from only three localities in Thailand, which are a part of their distribution range in the IWP region. Wider range of samples in the two Rhizophora species shows higher polymorphism than the previous study (W. L. Ng, N. Inomata, K. M. Teshima, S. Changtragoon, I. Z. Siregar, A. E. Szmidt, unpubl. data). Similar trend was observed in the cpDNA region. In this study, in the trnS-trnG spacer, three and one segregating sites were found in B. gymnorrhiza and B. sexangula, respectively, and one fixed substitution was observed between the two species. Zhou et al. (2008) reported that no polymorphism was observed within the species and the two species differed by a single substitution. No polymorphism in the previous study by Zhou et al. (2008) is probably due to limited range of their samples. Sampling strategy is important for assessing genetic variability of widespread plants.
Two of the ten genes, NAC and PO, were monomorphic in the ten populations investigated in this study. In the NAC gene, nucleotide divergence between the species was comparable to the average divergence of the ten genes, suggesting that the gene is not so conservative. The weakly significant result in the multilocus HKA test probably reflects this inconsistency.
Population structure and demographic history
The FST values among the ten populations were highly significant in both nuclear and cpDNA regions (Table 4), indicating that the ten populations were genetically differentiated. Average distance of pollen and propagule dispersal was short in K. candel (Geng et al. 2008). Like K. candel, B. gymnorrhiza is insect-pollinated and has viviparous propagules. Therefore, pollen and seed dispersal of B. gymnorrhiza appear to be limited. Consistent with this expectation, most pairwise FST values were significant.
The STRUCTURE analysis revealed that there are two possible genetic clusters. One cluster consists of Australian populations and populations located on the eastern side of Malay Peninsula. The other cluster includes populations on the western side of the Malay Peninsula as well as populations of Madagascar and India. The estimated migration rates between two clusters were low (2NmE = 0.29 and 2NmW = 0.04). As is the case of other mangrove species in the South East Asia (e.g., Liao et al. 2007; Inomata et al. 2009), the Sundaland and/or the Malay Peninsula prevented dispersion of B. gymnorrhiza and worked as a land barrier as well.
To obtain the splitting time in the actual demographic unit (in years), the estimated population splitting parameters need to be divided by the mutation rate per locus per year. As it is difficult to estimate mutation rate of Bruguiera species and unfortunately appropriate fossils are not available, the synonymous mutation rate estimated for other tree species was used; 0.7 × 10−9 per site per year in Pinus (Willyard et al. 2007) and 2.6 × 10−9 per site per year in palms (Gaut et al. 1996). We considered these rates as minimum and maximum mutation rates for Bruguiera species. Mutation rate per site per year for nonsynonymous sites was computed by multiplying synonymous mutation rate by the observed Ka/Ks ratio (Table 3). If we assume u = 2.6 × 10−9 per site per year, the geometric mean mutation rate becomes 5.6 × 10−7 per locus per year. Point estimates of the splitting time between the two clusters in B. gymnorrhiza and the speciation time between B. gymnorrhiza and B. sexangula become 0.30 MYA and 0.57 MYA, respectively. If we assume u = 0.7 × 10−9 per site per year, the geometric mean mutation rate becomes 1.5 × 10−7 per locus per year. Point estimates of the splitting time and speciation time become 1.13 MYA and 2.12 MYA, respectively. The estimated splitting time of the two clusters is in the range from 0.30 to 1.13 MYA. The speciation time between B. gymnorrhiza and B. sexangula was in the range from 0.57 to 2.12 MYA. These estimates imply that the speciation of B. gymnorrhiza and B. sexangula happened in the middle Pleistocene, but the dispersion of B. gymnorrhiza did not happen until the late Pleistocene.
During the LGM, the sea level dropped ∼120 m compared with the present geographic area, and the Sundaland was formed in the Southeast Asia. At that time three localities examined in this study (MK, northwestern Borneo; ML, northwestern part of the Malay Peninsula and V, eastern part of Asian Continent) were not coast, while one of them (BMJ, eastern Java) was considered to be the edge of the Sundaland. Extant populations located on the previous Sundaland should be established after the LGM. Consistent with this, the BMJ (eastern Java) population showed higher polymorphism than the ML (northwestern part of the Malay Peninsula) and MK (northwestern Borneo) populations (Table S2). In addition, the BMJ (eastern Java) population harbors five of thirteen haplotypes in the mang-1 and a unique haplotype in the SF, EXP2, and GM (data not shown). Except for comparisons with the V (eastern part of the Asian Continent) population, pairwise FST values were much higher in comparisons with the BMJ (eastern Java) population (Table S4: BMJ – MK (northwestern Borneo): 0.69, BMJ (the east of the Java) – ML (the northwest of the Malay Peninsula): 0.68, MK (northwestern Borneo) – ML (northwestern part of the Malay Peninsula): 0.10). In the Southeast Asia the BMJ (eastern Java) population could be an ancient population compared with others.
Acknowledgments
We thank Ms. M. Kato for her technical assistance. We thank Dr. J. Kusumi for fruitful discussion. We thank Drs. L. Faliniana, A. Rasoamiaramanana, P. Saenger, C. Kusumana, S. Z. Iskandar, T. Yamazaki, A. E. Szmidt, A. Yoshida, and S. Changtragoon for sample collection. We also thank Dr. A. E. Szmidt for English correction. This study was supported by the Global Environment Research Fund (D-0901) of the Ministry of the Environment, Japan to S. M., N. I. and O. K.
Data Accessibility
DNA sequences: DDBJ accessions AB813916-AB817042. IMa2 and STRUCTURE input files: the Dryad Repository: http://dx.doi.org/10.5061/dryad.bq858
Conflict of Interest
None declared.
Supporting Information
Additional Supporting Information may be found in the online version of this article:
Figure S1. Plot of delta K calculated using the method of Evanno et al. (2005).
Figure S2. Haplotype network of the cpDNA region. Circle size is approximately proportional to the number of individuals of each haplotype. Intermediate black dots indicate putative haplotypes. Solid and dotted lines indicate a single substitution and indels, respectively. Indel length is given above or below each dotted line.
Figure S3. Geographical distribution of the cpDNA haplotypes in Bruguiera gymnorrhiza. Circle size shows the number of individuals used in each population. Colors and patterns correspond to those of haplotype shown in Figure S2.
Table S1. PCR primers and annealing temperature.
Table S2. Nucleotide variation in Bruguiera gymnorrhiza.
Table S3. Summary of neutrality tests.
Table S4. Pairwise FST values (below diagonal) and P-values (above diagonal).
References
- Clement M, Posada D, Crandall K. TCS: a computer program to estimate gene genealogies. Mol. Ecol. 2000;9:1657–1660. doi: 10.1046/j.1365-294x.2000.01020.x. [DOI] [PubMed] [Google Scholar]
- Drexler JZ. Maximum longevities of Rhizophora apiculata and R. mucronata propagules. Pac. Sci. 2001;55:17–22. [Google Scholar]
- Duke NC, Ball MC, Ellison JC. Factors influencing biodiversity and distributional gradients in mangroves. Glob. Ecol. Biogeogr. Lett. 1998;7:27–47. [Google Scholar]
- Evanno G, Regnaut S, Gouget J. Detecting the number of clusters of individuals using the software STRUCTURE: a simulation study. Mol. Ecol. 2005;14:2611–2620. doi: 10.1111/j.1365-294X.2005.02553.x. [DOI] [PubMed] [Google Scholar]
- Excoffier L, Lischer H. Arlequin suite ver 3.5: a new series of programs to perform population genetics analyses under Linux and Windows. Mol. Ecol. Resour. 2010;10:564–567. doi: 10.1111/j.1755-0998.2010.02847.x. [DOI] [PubMed] [Google Scholar]
- Falush D, Stephens M, Prichard JK. Inference of population structure using multilocus genotype data: linked loci and correlated allele Frequencies. Genetics. 2003;164:1567–1587. doi: 10.1093/genetics/164.4.1567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- FAO. The world's mangroves 1980–2005, FAO forestry paper 153. Rome: Forest Resources Division. FAO; 2007. p. 77. [Google Scholar]
- Gaut BS, Morton BR, McCaig BC, Clegg MT. Substitution rate comparisons between grasses and palms: synonymous rate differences at the nuclear gene Adh parallel rate differences at the plastid gene rbcL. Proc. Natl Acad. Sci. USA. 1996;93:10274–10279. doi: 10.1073/pnas.93.19.10274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geng Q, Lian C, Goto S, Tao J, Kimura M, Islam MS, et al. Mating system, pollen and propagule dispersal, and spatial genetic structure in a high-density population of the mangrove tree Kandelia candel. Mol. Ecol. 2008;17:4724–4739. doi: 10.1111/j.1365-294X.2008.03948.x. [DOI] [PubMed] [Google Scholar]
- Giang LH, Geada GL, Hong PN, Tuan MS, Lien TH, Ikeda S, et al. Genetic variation of two mangrove species in Kandelia (Rhizophoraceae) in vietnam and surrounding area revealed by microsatellite markers. Int. J. Plant Sci. 2006;167:291–298. [Google Scholar]
- Goodall JA, Stoddart JA. Techniques for the electrophoresis of mangrove tissue. Aquat. Bot. 1989;35:197–207. [Google Scholar]
- Gossmann TI, Song B-H, Windsor AJ, Mitchell-Olds T, Dixon CJ, Kapralov MV, et al. Genome wide analyses reveal little evidence for adaptive evolution in many plant species. Mol. Biol. Evol. 2010;27:1822–1832. doi: 10.1093/molbev/msq079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamilton MB. Four primer pairs for the amplification of chloroplast intergenic regions with intraspecific variation. Mol. Ecol. 1999;8:521–523. [PubMed] [Google Scholar]
- Hasegawa M, Kishino H, Yano T. Dating of the human-ape splitting by a molecular clock of mitochondrial DNA. J. Mol. Evol. 1985;22:160–174. doi: 10.1007/BF02101694. [DOI] [PubMed] [Google Scholar]
- Hey J. Isolation with migration models for more than two populations. Mol. Biol. Evol. 2010;27:905–920. doi: 10.1093/molbev/msp296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hey J, Nielsen R. Integration within the Felsenstein equation for improved Markov chain Monte Carlo methods in population genetics. Proc. Natl Acad. Sci. USA. 2007;104:2785–2790. doi: 10.1073/pnas.0611164104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang Y, Tan F, Su G, Deng S, He H, Shi S. Population genetic structure of three species in the mangrove genus Ceriops (Rhizophoraceae) from the Indo West Pacific. Genetica. 2008;133:47–56. doi: 10.1007/s10709-007-9182-1. [DOI] [PubMed] [Google Scholar]
- Hubisz MJ, Falush D, Stephens M, Pritchard JK. Inferring weak population structure with the assistance of sample group information. Mol. Ecol. Resour. 2009;9:1322–1332. doi: 10.1111/j.1755-0998.2009.02591.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hudson R, Kaplan N. Statistical properties of the number of recombination events in the history of a sample of DNA sequences. Genetics. 1985;111:147–164. doi: 10.1093/genetics/111.1.147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inomata N, Wang XR, Changtragoon S, Szmidt AE. Levels and patterns of DNA variation in two sympatric mangrove species, Rhizophora apiculata and R. mucronata from Thailand. Genes Genet. Syst. 2009;84:277–286. doi: 10.1266/ggs.84.277. [DOI] [PubMed] [Google Scholar]
- Jian S, Ban J, Ren H, Yan H. Low genetic variation detected within the widespread mangrove species Nypa fruticans (Palmae) from Southeast Asia. Aquat. Bot. 2010;92:23–27. [Google Scholar]
- Jukes TH, Cantor CR. Evolution of protein molecules. In: Munro HN, editor. Mammalian protein metabolism. New York: Academic Press; 1969. pp. 21–131. [Google Scholar]
- Kado T, Yoshimaru H, Tsumura Y, Tachida H. DNA variation in a conifer, Cryptomeria japonica (Cupressaceae sensu lato) Genetics. 2003;164:1547–1559. doi: 10.1093/genetics/164.4.1547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimura M. The number of heterozygous nucleotide sites maintained in a finite population due to steady flux of mutations. Genetics. 1969;61:893–903. doi: 10.1093/genetics/61.4.893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larkin MA, Blackshields G, Brown NP, Chenna R, McGettigan PA, McWilliam H, et al. Clustal W and Clustal X version 2.0. Bioinformatics. 2007;23:2947–2948. doi: 10.1093/bioinformatics/btm404. [DOI] [PubMed] [Google Scholar]
- Liao PC, Havanond S, Huang S. Phylogeography of Ceriops tagal (Rhizophoraceae) in Southeast Asia: the land barrier of the Malay Peninsula has caused population differentiation between the Indian Ocean and South China Sea. Conserv. Genet. 2007;8:89–98. [Google Scholar]
- Librado P, Rozas J. DnaSP v5: a software for comprehensive analysis of DNA polymorphism data. Bioinformatics. 2009;25:1451–1452. doi: 10.1093/bioinformatics/btp187. [DOI] [PubMed] [Google Scholar]
- Ma XF, Szmidt AE, Wang XR. Genetic structure and evolutionary history of a diploid pine Pinus densata inferred from the nucleotide variation at seven gene loci. Mol. Biol. Evol. 2006;23:807–816. doi: 10.1093/molbev/msj100. [DOI] [PubMed] [Google Scholar]
- Marguire TL, Saenger P, Baverstock P, Henry R. Microsatellite analysis of genetic structure in the mangrove species Avicennia marina (Forsk.) Vierh. (Avicenniaceae) Mol. Ecol. 2000;9:1853–1862. doi: 10.1046/j.1365-294x.2000.01089.x. [DOI] [PubMed] [Google Scholar]
- Minobe S, Fukui S, Sasaki R, Kajita T, Changtragoon S, Shukor NAA, et al. Highly differentiated population structure of a mangrove species, Bruguiera gymnorrhiza (Rhizophoraceae) revealed by one nuclear GapCp and one chloroplast intergenic spacer trnFtrnL. Conserv. Genet. 2010;11:301–310. [Google Scholar]
- Miyama M, Shimizu H, Sugiyama M, Hanagata N. Sequencing and analysis of 14,842 expressed sequence tags of burma mangrove, Bruguiera gymnorrhiza. Plant Sci. 2006;171:234–241. [Google Scholar]
- Murray MG, Tompson WF. Rapid isolation of high molecular weight plant DNA. Nucleic Acid Res. 1980;8:4321–4325. doi: 10.1093/nar/8.19.4321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nei M. Molecular evolutionary genetics. New York, NY: Columbia Univ. Press; 1987. [Google Scholar]
- Pil WP, Boeger MRT, Muschner VC, Pie MR, Ostrensky A, Boeger WA. Postglacial north-south expansion of populations of Rhizophora mangle (Rhizophoraceae) along the Brazilian coast revealed by microsatellite analysis. Am. J. Bot. 2011;98:1031–1039. doi: 10.3732/ajb.1000392. [DOI] [PubMed] [Google Scholar]
- Pritchard JK, Stephens M, Donnelly P. Inference of population structure using multilocus genotype data. Genetics. 2000;155:945–959. doi: 10.1093/genetics/155.2.945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmid KJ, Ramos-Onsins S, Ringys-Beckstein H, Weisshaar B, Mitchell-Olds T. A multilocus sequence survey in Arabidopsis thaliana reveals a genome-wide departure from a neutral model of DNA sequence polymorphism. Genetics. 2005;169:1601–1615. doi: 10.1534/genetics.104.033795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun M, Lo EYY. Genomic markers reveal introgressive hybridization in the Indo-West Pacific mangroves: a case study. PLoS ONE. 2011;6:e19671. doi: 10.1371/journal.pone.0019671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomlinson PB. The botany of mangroves. Cambridge: Cambridge Univ. Press; 1986. [Google Scholar]
- Voris HK. Maps of Pleistocene sea levels in Southeast Asia: shorelines, river systems and time durations. J. Biogeogr. 2000;27:1153–1167. [Google Scholar]
- Weir BS, Cockerham CC. Estimating F-statistics for the analysis of population structure. Evolution. 1984;38:1358–1370. doi: 10.1111/j.1558-5646.1984.tb05657.x. [DOI] [PubMed] [Google Scholar]
- Willyard A, Syring J, Gernandt DS, Liston A, Cronn R. Fossil calibration of molecular divergence infers a moderate mutation rate and recent radiations for Pinus. Mol. Biol. Evol. 2007;24:90–101. doi: 10.1093/molbev/msl131. [DOI] [PubMed] [Google Scholar]
- Zhou R, Zeng K, Wu W, Chen X, Yang Z, Shi S, et al. Population genetics of speciation in nonmodel organisms: i. Ancestral polymorphism in mangroves. Mol. Biol. Evol. 2007;24:2746–2754. doi: 10.1093/molbev/msm209. [DOI] [PubMed] [Google Scholar]
- Zhou R, Gong X, Boufford D, Wu CI, Shi S. Testing a hypothesis of unidirectional hybridization in plants: observations on Sonneratia, Bruguiera and Lingularia. BMC Evol. Biol. 2008;8:149. doi: 10.1186/1471-2148-8-149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou R, Ling S, Zhao W, Osada N, Chen S, Meng Z, et al. Population genetics in nonmodel organisms: II. Natural selection in marginal habitats revealed by deep sequencing on dual platforms. Mol. Biol. Evol. 2011;28:2833–2842. doi: 10.1093/molbev/msr102. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
DNA sequences: DDBJ accessions AB813916-AB817042. IMa2 and STRUCTURE input files: the Dryad Repository: http://dx.doi.org/10.5061/dryad.bq858