

Summary
Phosphoinositide 3-kinases (PI3Ks) and Ras and Rho family small GTPases are key regulators of cell polarization, motility, and chemotaxis.They influence each other's activities by direct and indirect feedback processes that are only partially understood. Here, we show that 21 small GTPase homologs activate PI3K. Using a microscopy-based binding assay, we show that K-Ras, H-Ras, and five homologous Ras family small GTPases function upstream of PI3K by directly binding the PI3K catalytic subunit, p110. In contrast, several Rho family small GTPases activated PI3K by an indirect cooperative positive feedback that required a combination of Rac, CDC42, and RhoG small GTPase activities. Thus, a distributed network of Ras and Rho family small GTPases induces and reinforces PI3K activity, explaining past challenges to elucidate the specific relevance of different small GTPases in regulating PI3K and controlling cell polarization and chemotaxis.
Introduction
PI3K as well as Ras family small GTPases are among the most important early signaling components; they mediate cell growth, proliferation, migration, and multiple other cellular processes (Engelman et al., 2006; Etienne-Manneville and Hall, 2002; Heo et al., 2006; Karnoub and Weinberg, 2008; Lee et al., 2007; Rodriguez-Viciana et al., 1997). In addition to regulating downstream components such as Raf, different Ras small GTPases have been shown to directly interact with and activate class I PI3Ks through a Ras binding domain (RBD) that is present in all p110 catalytic subunits (Pacold et al., 2000; Rodriguez-Viciana et al., 1994, 1996). In Drosophila, mutation of the Dp110 RBD impaired interaction with Ras1 (homolog of mammalian H-, K-, and N-Ras) and severely reduced insulin responses, body size, and egg numbers, suggesting that Ras is indispensable for the full activation of class I PI3Ks (Orme et al., 2006). Experiments using mouse neutrophils revealed that a defective p110 RBD significantly reduced activation of PI3K and the migration of neutrophils (Suire et al., 2006). In cells with a homozygous mutation in the p110 RBD, Akt phosphorylation is significantly impaired in response to EGF and completely abrogated in response to FGF2 (Gupta et al., 2007). Nevertheless, most studies to date have focused mostly on the role of the small GTPases H-Ras and K-Ras, and it is not well understood whether other members of the large family of Ras small GTPases directly bind to and activate PI3K.
The role of Rac small GTPases in regulating PI3K is even less understood. Most studies showed that they act downstream of PI3K (Fleming et al., 2000; Han et al., 1998), whereas other studies have suggested that they can also activate PI3K (Servant et al., 2000; Weiner et al., 2002), thereby creating a positive feedback loop. This feedback loop may also involve polarized actin (Peyrollier et al., 2000; Wang et al., 2002) in addition to Rac (Srinivasan et al., 2003). These studies showed that cells expressing constitutively active Rac had elevated levels of PIP3, which could be lowered by inhibiting actin polymerization. In addition, most of the Rho family small GTPases are able to induce or alter actin polymerization (Heo and Meyer, 2003). Nevertheless, the role of different Rho family small GTPases in the positive feedback loop of PI3K activation is not understood. Recent studies have shown that the rapid activation of endogenous Rac triggers effective actin polymerization, but fails to positively activate PI3K (Inoue and Meyer, 2008). This led us to also investigate if and how Rho family small GTPases contribute to a positive feedback activation of PI3K.
Based on these considerations, we performed a systematic analysis of PI3K regulation by the Ras superfamily of small GTPases. Live-cell imaging showed that 21 out of 100 human small GTPases activate PI3K. We employed a microscopy-based interaction assay and found that the seven Ras family small GTPases acted upstream of PI3K and directly interacted with the p110 subunit of PI3K. In contrast, employing chemically inducible versions of different selective and unspecific GEF proteins, we discovered that Rac/CDC42/RhoG small GTPases activate PI3K by an indirect cooperative positive feedback involving not one but multiple endogenous small Rho family GTPases. Mathematical modeling showed that the requirement for multiple small GTPases creates a unique ultrasensitive mechanism for PI3K activation. Thus, PI3K activation is mediated by a systems design that involves multiple direct inputs from Ras family members as well as cooperative parallel positive feedbacks via multiple Rho family members that mediate an ultrasensitive activation of PI3K. This explains previous difficulties in sorting out the roles of different Ras and Rho family small GTPases and PI3K signaling in the establishment of cell polarity.
Results
Identification of 21 Small GTPases as PI3K Activators
We first confirmed that PI3K activity is increased in cells expressing constitutively active versions of either H-Ras or Rac1 (Figure 1A). The pleckstrin homology (PH) domain of AKT1 (PHAKT1) was used to monitor PI3K activity based on the relative increase in plasma membrane translocation of this PIP3 lipid-binding domain. In serum-deprived cells, PHAKT1 was evenly dispersed throughout the cells, whereas cells expressing H-Ras or Rac1 exhibited significant translocation of PHAKT1 to the plasma membrane. Quantitative analysis showed that H-Ras-induced PHAKT1 translocation was 1.5-fold stronger than Rac1-induced translocation (Figure S1).
Figure 1. Identification of Small GTPases that Activate PI3K.
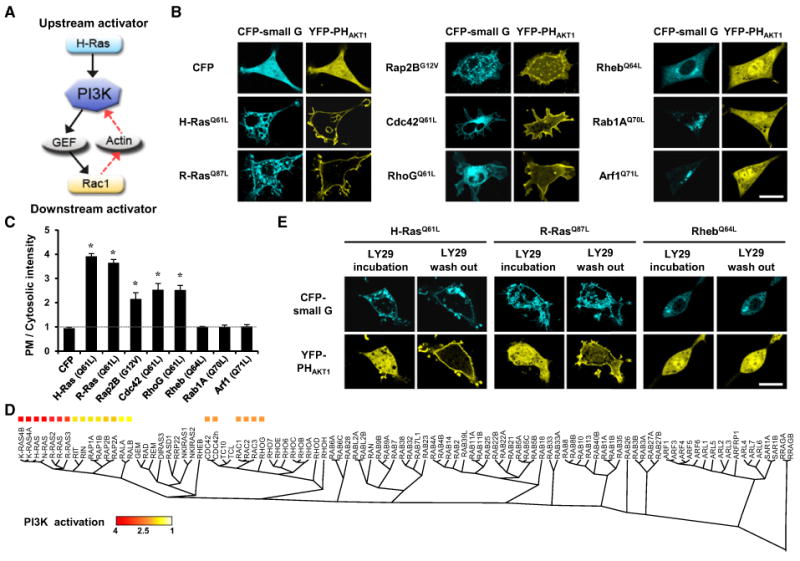
(A) Predicted roles of small GTPases in PI3K activation.
(B) YFP-PHAKT1 localization in NIH 3T3 cells coexpressing constitutively active small GTPases. Scale bar, 20 μm.
(C) Quantitative measurements of PIP3 production by line scan analysis of YFP-PHAKT1 localization. The PI3K activation index is the relative ratio of the fluorescence of the plasma membrane to the fluorescence of the cytosol: (FPM-FBG)/(FCyt-FBG), where F is the fluorescence intensity (FPM, plasma membrane; FBG, background; FCyt, cytosol). Data represent the mean ± SD (n = 5 experiments; * indicates p < 0.001 compared with control [CFP]).
(D) Phylogenic trees of the 100 human small GTPases. Homologies were analyzed with a Clustal WMSA algorithm. The relative amount of PI3K activation is color coded based on the PI3K activation index.
(E) Cells expressing constitutively active small GTPases were preincubated for 1 hr with LY29 (50 μM) to inactivate PI3K. The LY29 was then washed out toobserve the inducible activation of PI3K by small GTPases. Scale bar, 20 μm.
The role of other small GTPases in the activation of PI3K was investigated by selecting and screening 100 CFP-conjugated human small GTPases. YFP-PHAKT1 localization was analyzed in NIH 3T3 cells coexpressing each constitutively active small GTPase. These experiments showed that 21 of these small GTPases significantly activated PI3K, while 79 had no significant effect on PI3K activation (examples in Figures 1B and 1C; see Figure S2 for a full set of images). All of the PI3K-activating small GTPases belonged to the Ras and Rho families (Figure 1D) and could be classified into three groups according to the degree of PI3K activation. H-, K-, N-, and R-Ras induced the strongest PHAKT1 localization to the plasma membrane, with the other Ras family members inducing relatively smaller amounts of translocation. Rho family small GTPases induced intermediate levels of PHAKT1 translocation.
To validate these results, the rate of PI3K activation was measured by testing the kinetics of PHAKT1 translocation. Cells coexpressing constitutively active H-Ras, R-Ras, or Rheb and YFP-PHAKT1 were preincubated with the PI3K inhibitor LY294002 (LY29). After wash-out of LY29, PI3K was reactivated by H- and R-Ras, which led to a rapid translocation of PHAKT1 to the plasma membrane within a few minutes (Figure 1E; Movie S1). Constitutively active Rheb, which had no effect on PI3K activation, did not induce translocation of PHAKT1 in this assay. Thus, this result further supports the data showing that H- or R-Ras triggers PI3K activation and that the process of PI3K activation is very rapid, on a timescale of minutes.
Seven Small GTPases Are Direct Activators of PI3K
To determine the molecular mechanism of PI3K activation by these small GTPases, a recently developed technology called InCell SMART-i (intracellular supramolecular assembly readout trap for interactions) (Lee et al., 2011) was used to determine whether the identified PI3K-activating small GTPases directly interact with the RBD domain of p110 (RBDp110). In InCell SMART-i, ferritin (FT), a genetically encoded nanoparticle, is labeled with fluorescent proteins and engineered to directly or indirectly display bait (B) and prey (P) on its surface. Upon B-P interaction, ferritin nanoparticles (FT-NPs) assemble into nanoclusters, which are visualized as fluorescent dots (Figures S3A and S3B).
Interactions between small GTPases and RBDp110 were assessed using FT-NPs displaying FKBP-rapamycin-binding (FRB) and RBDp110, and fusion proteins comprised of reiterated FK506-binding protein (FKBP) and small GTPases lacking the C-terminal CAAX or polybasic motif necessary for cytoplasmic localization. Small GTPase-RBDp110 interaction and heterodimerization of FKBP and FRB triggered by rapamycin created molecular bridges that assembled the FT-NPs into nanoclusters (Figure 2A). When HeLa cells coexpressing FKBPx2-YFP-H-Ras or R-Ras, RBDp110-CFP-FT, and FRB-mRFP-FT were treated with rapamycin, nanoclusters assembled within 5 min (Figures 2B and 2C; Movie S2). Nanocluster formation was induced by the constitutively active forms of the small GTPases, but not by the dominant-negative forms, demonstrating the specificity of InCell SMART-i (Figure 2C). Interestingly, among the 21 small GTPases that activate PI3K, only 7 members of the Ras family small GTPases (H-Ras, K-Ras4A, K-Ras4B, N-Ras, R-Ras, R-Ras2, and R-Ras3) were able to induce nanocluster formation (Figure 2D; see Figure S3C for a full set of images). The same seven small GTPases were also the most potent PI3K activators. Thus, taken together with the PH domain translocation results, H-, K-, N-, and R-Ras are upstream direct activators of PI3K.
Figure 2. Identification of Small GTPases that Directly Activate PI3K.
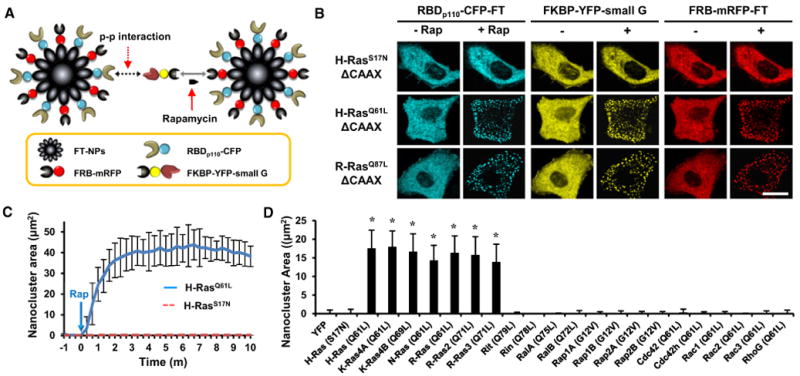
(A) Schematic diagram of the InCell SMART-i technique used to observe protein-protein interactions using ferritin-derived nanoparticles.
(B) Constitutively active H- and R-Ras bound to RBDp110. Scale bar, 20 μm.
(C) Kinetic analysis of the nanoclusters of constitutively active H-Ras (solid line) and dominant-negative H-Ras (dotted line). Data represent the mean ± SD (n> 10).
(D) After rapamycin treatment for 1 hr, the nanoclusters of the 21 constitutively active small GTPases were measured. YFP and dominant-negative H-Ras were used as negative controls. Data represent the mean ± SD (n > 20; * indicates p < 0.001 compared with control [YFP]).
Multiple Rho Family Small GTPases Are Downstream Activators of PI3K
Next, the remaining 14 PI3K-activating small GTPases were analyzed to determine whether they might be part of a positive feedback loop by also acting as downstream effectors of PI3K. It has been suggested that PI3K enhances actin polymerization and membrane protrusion during cell motility through activating the small GTPase Rac1 (Cantley, 2002) and possibly homologous small GTPases. Indeed, we observed that cells expressing each of these 14 small GTPases induced different types of actin-based membrane protrusions (Heo and Meyer, 2003), suggesting that they may have, at least in principle, a role in mediating PI3K-induced morphological changes.
We first considered that a specific small GTPase is likely downstream of PI3K, if the PI3K-mediated actin polymerization can be reduced or blocked when a dominant-negative mutant of that small GTPase is expressed. To assess this, membrane protrusion was quantitatively analyzed using an inducible PI3K activation strategy. To rapidly induce PI3K activation, we used an inducible protein translocation assay based on an FKBP-rapamycin-FRB interaction. The inter-SH2 (iSH) domain of the p85 regulatory subunit, which is known to interact with the p110 PI3K catalytic subunit, was conjugated to CFP-FKBP (CF). Lyn11-FRB (LDR), a plasma membrane-anchored form of FRB, was then used to recruit CF-iSH to the plasma membrane in response to rapamycin treatment (Suh et al., 2006) (Figure 3A). When cells cotransfected with LDR, CF-iSH, and YFP-PHAKT1 were treated with rapamycin, PIP3 production and membrane protrusion occurred within 5 min (Figures 3B and 3C). We then tested whether PI3K-induced membrane protrusion was diminished by the presence of dominant-negative mutants of the different small GTPases. Dominant-negative Cdc42, RhoG, and Rac1 strongly affected PI3K-induced protrusion, suggesting that these Rho small GTPases are downstream of PI3K. In contrast, dominant-negative mutants of Rit, Rap1A, Rap2B, and RalA did not have any significant effects on PI3K-induced membrane protrusion (Figures 3D and 3E).
Figure 3. Cooperative Positive Feedback from Rho Family Small GTPases to PI3K.
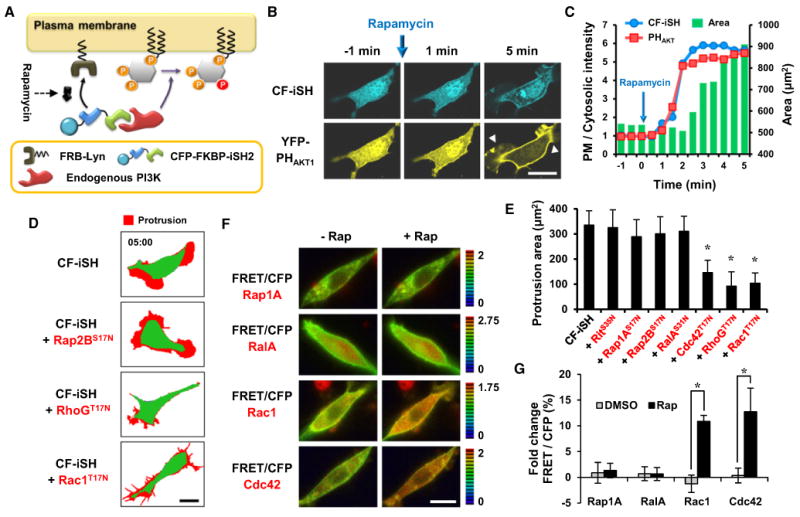
(A) Schematic diagram of the strategy used to activate endogenous PI3K.
(B) Endogenous PI3K activation was sufficient for YFP-PHAKT1 translocation and morphological changes within 5 min. Protrusions are indicated by yellow arrows. Scale bar, 20 μm.
(C) Time series of the translocation of CF-iSH and YFP-PHAKT1 to the plasma membrane. The change in cell area following endogenous PI3K activation is also shown. The y axis shows the ratio of plasma membrane fluorescence intensity to cytosolic fluorescence intensity and the total cell area (μm2).
(D) Protrusion (red) map after activation of endogenous PI3K in cells expressing dominant-negative small GTPases. Scale bar, 20 μm.
(E) Quantitative analysis of the protrusion area. Data represent the mean ± SD (n > 10; * indicates p < 0.001 compared with control [CF-iSH]).
(F) FRET/CFP ratio images show that Rac1 and Cdc42 are activated in response to rapamycin-induced endogenous PI3K activation but that Rap1A and RalA are not. Scale bar, 20 μm.
(G) Kinetic analysis of the percentage of fold change FRET/CFP ratio that was normalized to the initial ratio. Data represent the mean ± SD (n > 10; * indicatesp < 0.001).
To further confirm that these Rho small GTPases are downstream of PI3K, the activities of Rap1A, RalA, Rac1, and Cdc42 were monitored following the activation of PI3K. To assess the activity of these small GTPases, we used small GTPase-specific FRET biosensors (Komatsu et al., 2011). Cells were again cotransfected with the inducible PI3K activation system (mCherry-FKBP-iSH and LDR) together with each FRET biosensor. After serum deprivation to reduce basal small GTPase activities, PI3K was activated by rapamycin-induced translocation of mCherry-FKBP-iSH, and the percentage of the FRET/CFP signal change was measured. Activation of PI3K led to the significant increase in the Rac1- and Cdc42-specific FRET/CFP ratio, while the biosensors for Rap1A and RalA showed no significant changes (Figures 3F and 3G), suggesting that Rac1 and Cdc42 are activated following PI3K activation, whereas Rap1A and RalA are not activated.
These results were further validated using the Cdc42/Rac interactive binding (CRIB) domain of PAK1 (CRIBPAK1) and the RBD of RAF1 (RBDRAF1), which were reported previously to interact with the GTP-bound forms of Rac1 or Rap1A and R-Ras, respectively (Kraynov et al., 2000; Nassar et al., 1995; Spaargaren et al., 1994) (Figures S4A–S4C). In this assay, recruitment of the respective domain to the plasma membrane reflects small GTPase activation. Cells were cotransfected with CF-iSH, LDR, and effector domains, with or without wild-type Rac1, Rap1A, or R-Ras. Consistent with the results in Figure 3, treatment with rapamycin led to the significant translocation of CRIBPAK1 to the plasma membrane in Rac1 cotransfected cells, while RBDRAF1 did not show any translocation even in the presence of Rap1A or R-Ras coexpression (Figures S4D and S4E). This shows that Rac1, but not Ras or Rap, is activated by PI3K. Taken together with the results presented above, this suggests that the Rho small GTPases Rac, CDC42, and likely RhoG, but not the Ras, Rap, or Ral small GTPases, are activated downstream of PI3K.
Multiple Rho Family Small GTPases Are Required for Effective PI3K Activation
We next investigated the mechanism by which downstream Rho family small GTPases activate PI3K. Actin polymerization mediated by small GTPases has been reported to contribute to such a positive feedback loop for PI3K activation (Servant et al., 2000; Srinivasan et al., 2003; Wang et al., 2002; Weiner et al., 2002). In addition, several Rho family small GTPases have been reported to synergistically regulate actin polymerization and cell migration, processes in which PI3K has a major role (Machacek et al., 2009). This made us test the hypothesis that multiple Rho family small GTPases may cooperatively activate PI3K. In order to conditionally activate different Rho family small GTPases at an endogenous level, cells were transfected with FKBP-tagged guanine exchange factors (GEFs), which are activated via rapamycin-induced translocation. Fgd1, Tiam1, and SGEF were used to specifically activate Cdc42, Rac1, and RhoG, respectively, and broad-spectrum Vav2 was used for simultaneous activation of Cdc42, Rac1, and RhoG (Inoue et al., 2005; Rossman et al., 2005). Translocation of each GEF by the addition of rapamycin successfully induced specific morphological changes (Figure 4A). To determine the combinatorial effect of the Rho family small GTPases on PI3K activation, cells were cotransfected with LDR, mCherry-PHAKT1, CF-conjugated GEF, and Lyn-YFP as an internal control and analyzed by total internal reflection fluorescence (TIRF) microscopy. CF and CF-iSH were used as negative and positive controls, respectively. After the induction of CF-iSH translocation to the plasma membrane, PHAKT1 was rapidly recruited to the plasma membrane, whereas CF did not have any notable effect on PHAKT1 translocation. Interestingly, in contrast to the overexpression of constitutively active small GTPases, conditional activation of endogenous levels of individual Rho family small GTPases did not induce significant activation of PI3K. However, simultaneous activation of Cdc42, Rac1, and RhoG by the induced plasma membrane translocation of Vav2 resulted in significant PI3K activation (Figures 4B and 4C). Moreover, the PI3K activation induced by Vav2 occurred in a Grb2 and Ras-independent manner (Figures S5 and S6) and was completely suppressed by siRNA-mediated knockdown of Cdc42 or RhoG (Figure 5A).
Figure 4. Cooperation of Multiple Rho Family Small GTPases Is Required for the Generation of an Effective Positive Feedback Loop at Endogenous Levels.
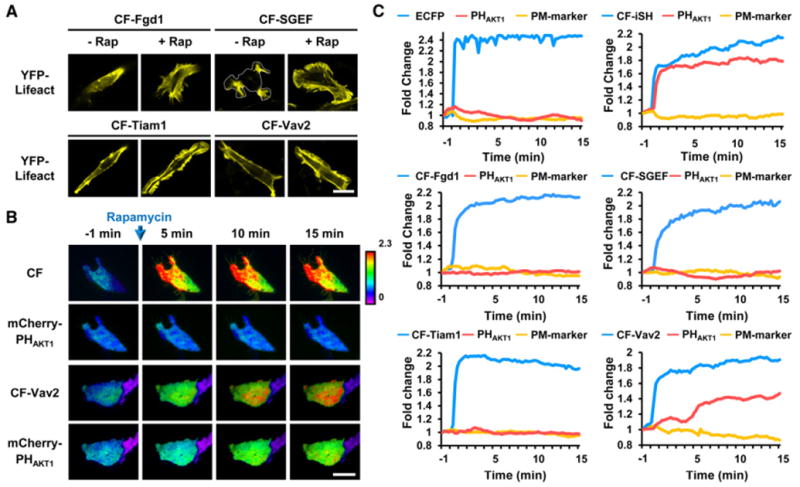
(A) Translocation of different GEFs (DH-PH domains) to the plasma membrane induced marked morphological changes. YFP-LifeAct (an F-actin biosensor) was used to monitor changes in morphology. Scale bar, 20 μm.
(B) Top and bottom rows show the CFP-FKBP-DH-PH domain of each GEF and mCherry-PHAKT1, respectively. Each ratio image was normalized to the plasma membrane marker (YFP-Lyn). CF and CF-iSH were used as negative and positive controls, respectively. Scale bar, 20 μm.
(C) Quantitative measurements of the translocation of CFP-FKBP-GEFs and mCherry-PHAKT1 to the plasma membrane (normalized to the initial value) showed that the induced activation of Vav2 triggered effective PI3K activation through parallel positive feedback loops.
Figure5. Model Showing How PI3K Activity Is Cooperatively Regulated by Multiple Downstream Rho Family Small GTPases through Coupled Positive Feedback Loops.
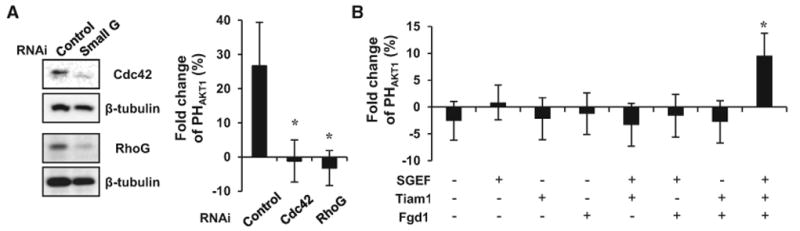
(A) Cdc42 or RhoG knockdown blocked positive feedback activation of PI3K by Vav2. Data represent the mean ± SD (n > 10; * indicates p < 0.001).
(B) Simultaneous activation of combined three GEFs (Fgd1, Tiam1, and SGEF) induced positive feedback activation of PI3K. Data represent the mean ± SD (n > 15; * indicates p < 0.001).
To further confirm the combinatorial effect of GEFs, cells were transfected with a combination of SGEF, Tiam1, and Fgd1. Consistent with Vav2 results, there was significant increase of PI3K activation after recruitment of only three GEFs to the plasma membrane by the rapamycin treatment (Figure 5B). These data argue that Rho family small GTPases cooperate to generate an effective positive feedback loop for PI3K activation.
To analyze the role of multiple positive feedback loops in the regulation of PI3K activity, a mathematical model was constructed comprising three small GTPases (Cdc42, Rac1, and RhoG), PI3K, and two GEFs (Fgd1 and Vav2, Figure 6A). This model focused on the regulation of PI3K activity by Cdc42, Rac1, and RhoG because the expression of Rac2 and Rac3 is limited to hematopoietic cells and neuronal cells (Heasman and Ridley, 2008), respectively, and because Cdc42h shares a high degree of sequence homology with Cdc42. For simplicity, it was assumed that the three feedback loops, which are regulated by Rac1, Cdc42, or RhoG, have similar feedback effects and that they cooperatively activate PI3K. Fgd1 was considered a GEF specific for Cdc42. Simulations using this model showed that PI3K is significantly activated if three positive feedback loops are simultaneously activated by Vav2. However, if only one feedback loop is activated, by Fgd1, PI3K is not significantly activated, consistent with our experimental results (Figure 6B). We also simulated knockout of one feedback loop in the model, demonstrating that all three feedback loops are required for the activation of PI3K based on these plausible model assumptions (Figure 6C). This is also consistent with the experimental data in Figure 5A. For comparison, another mathematical model was constructed in which the PI3K activity is determined by simple summation of the effects of the three feedback loops (rather than cooperation between them). The results of this additive model show that Fgd1 activates PI3K and that two activated feedback loops activate PI3K, which does not agree well with the experimental results (Figure S7). These results indicate that the three coupled positive feedback loops play an important role in increasing the activity of small GTPases (see Kim et al., 2008 for the various roles of coupled feedback loops) and that small GTPases cooperatively regulate PI3K activity.
Figure 6. PI3K Activity Is Cooperatively Regulated by Multiple Downstream Rho Family Small GTPases through Coupled Positive Feedback Loops.
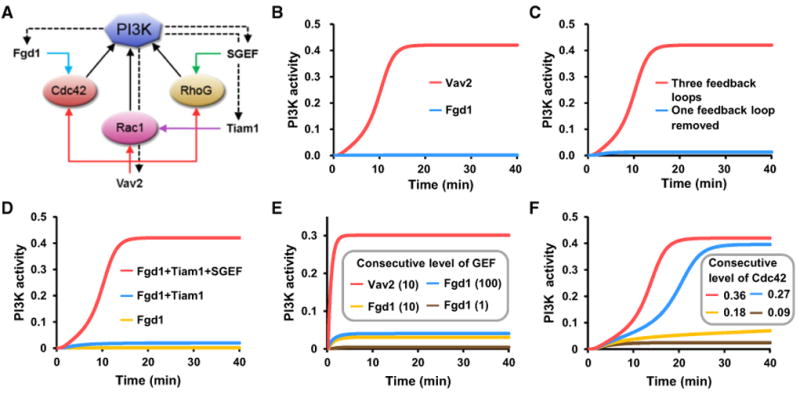
(A) A simplified diagram of the feedback regulation of PI3K by downstream small GTPases used for mathematical modeling.
(B) Temporal profiles of PI3K activity for two different GEFs when endogenous levels of the GTPases are given.
(C) Temporal profiles of PI3K activity when one feedback loop was or was not knocked out.
(D) Temporal profiles of PI3K activity for three combinatorial inputs of three GEFs.
(E) Temporal profiles of PI3K activity for three input level (1, 10, and 100) of Fgd1 and one input level (10) of Vav2 when the endogenous levels of Cdc42, Rac1, and RhoG are low (all are set to 0.1 in this simulation).
(F) Temporal profiles of PI3K activity for four consecutive levels (0.09. 0.18, 0.27, 0.36) of constitutively active Cdc42, which correspond to overexpression experiments of the Cdc42 active form.
We further verified in the model that all of the three GEFs (Fgd1, Tiam1, and SGEF) were required for the activation of PI3K at endogenous levels of Cdc42, Rac1, and RhoG (Figure 6D). Together, these results imply that PI3K activation may require that the concentrations of the active forms of these three GEFs have to exceed a critical threshold and that such a threshold cannot be reached without overexpression. Figure 6E shows that even a very high level of only Fgd1 cannot activate PI3K. Nevertheless, a high level of Vav2 is sufficient for activation even when the endogenous levels of the other three GEFs are low. From these results, we can speculate that an overexpression of constitutively active form of Cdc42 above the normal level can induce activation of PI3K, as shown in Figure 1B. Figure 6F shows the existence of such a threshold in the model. Based on these results, the model explains how PI3K activity can be cooperatively and effectively amplified by downstream activators through coupled positive feedback loops that can be triggered by endogenous levels of small GTPases.
Discussion
Our results reveal a systematic and cooperative mechanism for PI3K activation by a number of small GTPases.A comprehensive screen of 100 small GTPases identified 21 small GTPases that activate PI3K. Interestingly, all of the 21 small GTPases are members of the Ras or Rho families. We used a recently developed platform to measure protein-protein interactions in living cells (InCell SMART-i) and showed that seven small GTPases function as upstream activators by directly interacting with PI3K. These PI3K activators included H-, K-, N-, and R-Ras, and all belonged to the Ras family small GTPases. They also showed the most robust activation of PI3K. Because all these upstream activators localized to the plasma membrane, the interaction between these small GTPases and PI3K likely recruits PI3K to the plasma membrane as part of the activation process. Two experiments utilizing the inducible activation of PI3K for quantifying membrane protrusion and monitoring small GTPase activity showed that Cdc42 and RhoG as well as Rac are downstream, with their activity being activated by PI3K. Moreover, our experimental data and mathematical modeling show that the positive feedback PI3K activation by these downstream small GTPases can only occur in a cooperative manner in the presence of endogenous levels of the small GTPases.
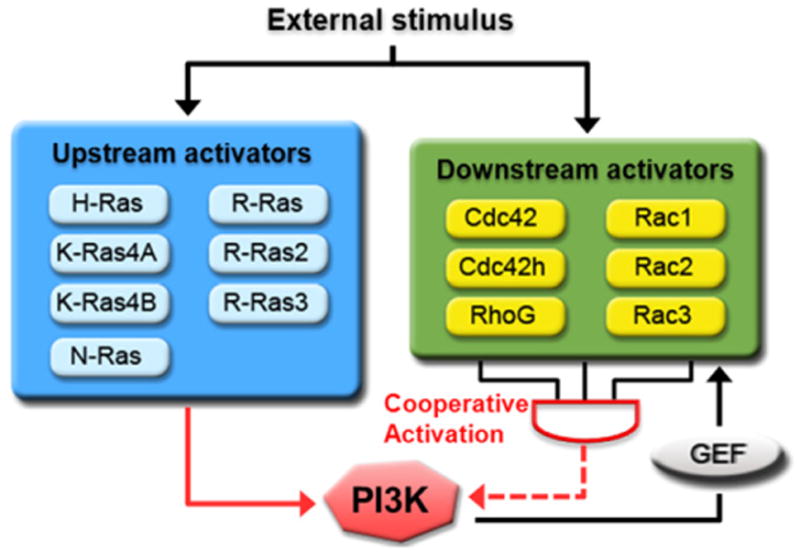
Taken together, our result argues for a cooperative activation of PI3K by a combination of multiple upstream Ras family small GTPase inputs and positive feedback amplification by several Rho family small GTPases (Figure 7). Following external stimulus, GEFs for upstream activators are recruited to the plasma membrane and switch the upstream activators to an active state (Chardin et al., 1993; Mochizuki et al., 2001). The upstream activators then bind to and recruit PI3K to the plasma membrane for the production of PIP3. After PI3K activation, increased PIP3 levels recruit PH domain-containing Rho-GEFs, including Tiam1 and Vav2, to the plasma membrane (Fleming et al., 2000; Han et al., 1998). This results in the activation of downstream GTPases, which induce actin polymerization and generate a positive feedback loop that further activates PI3K in a cooperative manner. During cell migration, several reports have shown that the activity of both PI3K and the PI3K activators identified in this study accumulate and interact at the leading edge of the cell (Li et al., 2005; Machacek et al., 2009; Niggli, 2000; Wang et al., 2002; Weiner et al., 2002; Zhang et al., 2008). This suggests that the systematic activation of PI3K by both upstream and downstream activators is required for cell polarization and proper cell migration. Additionally, it is possible that the simultaneous activation of multiple downstream activators is essential for maintaining the polarity during cell migration.
Figure 7. Coordinated Activation of PI3K by the Ras Superfamily of Small GTPases.
Cooperation of Downstream Activators Is Essential for PI3K Activation by a Positive Feedback Loop
Rac small GTPase (Srinivasan et al., 2003) and actin polymerization (Peyrollier et al., 2000; Wang et al., 2002) have been proposed to be key mediators of a positive feedback loop that amplifies PI3K activity. Nevertheless, the rapid activation of endogenous Rac did not result in any significant increase in PI3K activity, despite marked changes in cellular morphology and actin polymerization (Inoue and Meyer, 2008). Interestingly, we now show that Cdc42 and RhoG are also involved in PI3K activation. Moreover, all of the downstream activators induce membrane protrusion by actin polymerization, which is one of the major functions of PI3K in cell migration and polarization (Heo and Meyer, 2003; Rodriguez-Viciana et al., 1997; Wang et al., 2002). Consistently, small GTPases associated with the process of cell shrinkage, such as RhoA, did not activate PI3K. This agrees with previous reports showing that RhoA is an antagonist of PI3K via PTEN activation (Li et al., 2005). The involvement of multiple Rho family small GTPases in a positive feedback loop led us to question how these Rho small GTPases function in this positive feedback loop for the activation of PI3K. To explore this positive feedback mechanism, we utilized the inducible translocation of the GEFs of Tiam1, Fgd1, and SGEF to activate endogenous Rac, Cdc42, and RhoG, respectively. Surprisingly, we found that the activation of individual small GTPases failed to trigger PI3K activation, despite significant actin polymerization. In contrast, PI3K was activated upon the simultaneous activation of endogenous Rac, CDC42, and RhoG by Vav2. The degree of PI3K activation was dependent on the number of downstream activators, although the amount of actin polymerization by these GEFs was similar. Furthermore, siRNA experiments showed that the knockdown of a single downstream activator abolished PI3K activation induced by Vav2. Taken together, these results suggest that a combined activation of small GTPases is critical for effective generation of the postulated positive feedback loop for PI3K activation. In addition, it indicates that PI3K is activated by a synergistic activation mechanism mediated jointly by active Rac, Cdc42, and RhoG.
Indirect Activation of PI3K by Ras Family Small GTPases
We also identified eight Ras family small GTPases, including Rit, Rap1A, Rap2B, and RalA, which do not directly interact with PI3K and are also not downstream of PI3K. These small GTPases have been reported previously to regulate PI3K activity (Rodriguez-Viciana et al., 2004). Although the detailed mechanism of PI3K activation by these small GTPases was not elucidated in this study, it is possible that they may indirectly induce PI3K activation through regulating upstream or downstream activators (Arthur et al., 2004; Hoshino and Nakamura, 2003; Schwamborn and Püschel, 2004). Consistent with this, the levels of PI3K activation induced by these small GTPases were less robust than those induced by upstream or downstream activators. In addition, both indirect and downstream activator small GTPases induce actin polymerization and morphological changes. An example of this is shown by Rap2B, which induces the formation of “eyelash-like” morphological structures that are likely a combination of lamellipodia and filopodia induced by Rac and Cdc42, respectively (Heo and Meyer, 2003). The mechanism of PI3K activation by these indirect regulators will be an interesting topic for further investigation.
Conclusion
Our data present a comprehensive analysis of the interrelationships between Ras superfamily small GTPases and PI3K signaling. We determined that 21 out of 100 human small GTPases examined activate PI3K. All of the PI3K-activating small GTPases belong to the Ras and Rho families of small GTPases and are classified as upstream or downstream activators. To initiate effective activation of PI3K by positive feedback, multiple downstream activators must be simultaneously upregulated and cooperate with each other in coupled positive feedback loops. We propose that the cooperative regulation of PI3K by Ras and Rho family small GTPases is essential to generate and maintain effective PI3K activation.
Experimental Procedures
Mammalian DNA Constructs
The 100 human small GTPases analyzed (wild-type, constitutively active, and dominant-negative forms) have been described previously (Heo et al., 2006; Heo and Meyer, 2003). CFP- and YFP-PHAKT1 have been described previously (Shin et al., 2011). FRB-mRFP-FT, FKBP-GFP-FT, LDR, and CF-iSH, which contain the iSH domain (420–615) of p85β, have also been described previously (Inoue et al., 2005; Suh et al., 2006). Raichu-Cdc42 and Rac1 have been described previously (Komatsu et al., 2011). Raichu-Rap1A and RalA were kindly provided by Michiyuki Matsuda. Twenty-one small GTPases were produced by cloning the CAAX- or poly basic-deleted small GTPase ORFs into the FKBPx2-YFP backbone vector. RBDp110 from p110α (133– 332) was cloned into CFP-FT. All GEF constructs containing DH and PH domains, CF-Fgd1 (346–720), CF-SGEF (413–811), Tiam1 (1012–1592), and CF-Vav2 (164–536) were generated using RT-PCR cloning with gene-specific primers containing attB sequences to create entry clones for the gateway system (Invitrogen). The coding region of each entry clone was transferred into YFP, mCherry, and CFP-FKBP expression vectors using LR Clonase (Invitrogen). For TIRF imaging, CRIBPAK1 (69–108), RBDRAF1 (51–131), and PHAKT1 (2–147) were cloned into the mCherry-C1 (Clontech) vector.
Cell Culture and Transfection
NIH 3T3 and HeLa cells were purchased from the American Type Culture Collection (ATCC). Cells were cultured in Dulbecco's Modified Eagle's Medium (DMEM) supplemented with 10% FBS in 10% CO2 at 37°C and were passaged every 3 days. Transfection was performed with a Neon (Invitrogen) or Lipofectamine 2000 (Invitrogen) according to the manufacturer's instructions.
InCell SMART-i Assay
For the screening of small GTPase-RBDp110 interactions, HeLa cells were incubated at 24 hr after transfection with rapamycin (500 nM, Calbiochem)for 1 hr. The cells were then fixed in 4% paraformaldehyde for 10 min. Images were randomly captured with A1R confocal microscope.
Live-Cell Imaging and Data Acquisition
NIH 3T3 cells were incubated in serum-free medium (DMEM) for 6 hr. Just prior to imaging, the medium was replaced with DPBS that included glucose (Invitrogen). For dual color measurements, CFP and YFP were used for confocal microscopy, and CFP and mCherry were used for TIRF microscopy. CFP, YFP, and mCherry were used for triple color measurements. All live images were captured using A1R confocal (Nikon) and Ti TIRF (Nikon) microscopes (60× magnification). The images were analyzed with Nikon imaging software (NIS-element AR 64-bit version 3.00, Laboratory Imaging) and MetaMorph software (version 7.6.0.0, MDS Analytical Technologies).
siRNA and Western Blotting
A universal negative control, Cdc42 (5′-UUU GGG UCC CAA CAA GCA AGA AAG G-3′) and RhoG (5′-AUG ACG AAG ACG UUG GUC UGA GGG U-3′) specific siRNAs were obtained from Invitrogen. At 48 hr after siRNA transfection (10 pM), whole-cell extracts were prepared and resolved by 4%–10% gradient SDS-PAGE. The proteins were transferred to a PVDF membrane, which was then incubated overnight at 4°C with anti-Cdc42 (Millipore) and anti-RhoG(Millipore) antibodies. Bound antibodies were visualized with horse-radish peroxidase-conjugated secondary antibodies (GE Healthcare) and the ECL system (Millipore).
Mathematical Modeling
To investigate the cooperative regulatory mechanisms of three small GTPases for PI3K, we developed a mathematical model using Hill-type functions (Kim et al., 2007, 2008) as follows:
where the parameter values were H = 3, k1 = 1, k2 = 0.3, ki = 1 (3≤i≤18), PI3Ktotal = 1, and Cdc42total = Ractotal = RhoGtotal = 0.5.
Statistical Methods
Statistical significance was evaluated by two-tailed unpaired Student's t test using SigmaPlot (version 9.0). Significance was established at p < 0.001.
Supplementary Material
Acknowledgments
This work was supported by the National Research Foundation of Korea (NRF), grants funded by the Korea Ministry of Education, Science & Technology (MEST; 2009-0086964, 2010-0017662, 2011-0000840), KAIST Future Systems Healthcare Project, the Korea Science and Engineering Foundation (KOSEF) grant funded by the Korean government (MEST; 2011-0019510), WCU (World Class University) Program through the National Research Foundation of Korea funded by the Ministry of Education, Science & Technology (R31-2011-000-10071-0), and the Korea Research Foundation through the Basic Research Program (2008-05943). The work was further supported by NIH grants GM063702 and MH064801 to T.M.
Footnotes
Supplemental Information: Supplemental Information includes four figures, three tables, and Supplemental Experimental Procedures and can be found with this article online at doi:10.1016/j.molcel.2012.05.007.
References
- Arthur WT, Quilliam LA, Cooper JA. Rap1 promotes cell spreading by localizing Rac guanine nucleotide exchange factors. J Cell Biol. 2004;167:111–122. doi: 10.1083/jcb.200404068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cantley LC. The phosphoinositide 3-kinase pathway. Science. 2002;296:1655–1657. doi: 10.1126/science.296.5573.1655. [DOI] [PubMed] [Google Scholar]
- Chardin P, Camonis JH, Gale NW, van Aelst L, Schlessinger J, Wigler MH, Bar-Sagi D. Human Sos1: a guanine nucleotide exchange factor for Ras that binds to GRB2. Science. 1993;260:1338–1343. doi: 10.1126/science.8493579. [DOI] [PubMed] [Google Scholar]
- Engelman JA, Luo J, Cantley LC. The evolution of phosphati-dylinositol 3-kinases as regulators of growth and metabolism. Nat Rev Genet. 2006;7:606–619. doi: 10.1038/nrg1879. [DOI] [PubMed] [Google Scholar]
- Etienne-Manneville S, Hall A. Rho GTPases in cell biology. Nature. 2002;420:629–635. doi: 10.1038/nature01148. [DOI] [PubMed] [Google Scholar]
- Fleming IN, Gray A, Downes CP. Regulation of the Rac1-specific exchange factor Tiam1 involves both phosphoinositide 3-kinase-dependent and -independent components. Biochem J. 2000;351:173–182. doi: 10.1042/0264-6021:3510173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta S, Ramjaun AR, Haiko P, Wang Y, Warne PH, Nicke B, Nye E, Stamp G, Alitalo K, Downward J. Binding of ras to phosphoinositide 3-kinase p110alpha is required for ras-driven tumorigenesis in mice. Cell. 2007;129:957–968. doi: 10.1016/j.cell.2007.03.051. [DOI] [PubMed] [Google Scholar]
- Han J, Luby-Phelps K, Das B, Shu X, Xia Y, Mosteller RD, Krishna UM, Falck JR, White MA, Broek D. Role of substrates and products of PI 3-kinase in regulating activation of Rac-related guanosine triphosphatases by Vav. Science. 1998;279:558–560. doi: 10.1126/science.279.5350.558. [DOI] [PubMed] [Google Scholar]
- Heasman SJ, Ridley AJ. Mammalian Rho GTPases: new insights into their functions from in vivo studies. Nat Rev Mol Cell Biol. 2008;9:690–701. doi: 10.1038/nrm2476. [DOI] [PubMed] [Google Scholar]
- Heo WD, Meyer T. Switch-of-function mutants based on morphology classification of Ras superfamily small GTPases. Cell. 2003;113:315–328. doi: 10.1016/s0092-8674(03)00315-5. [DOI] [PubMed] [Google Scholar]
- Heo WD, Inoue T, Park WS, Kim ML, Park BO, Wandless TJ, Meyer T. PI(3,4,5)P3 and PI(4,5)P2 lipids target proteins with polybasic clusters to the plasma membrane. Science. 2006;314:1458–1461. doi: 10.1126/science.1134389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoshino M, Nakamura S. Small GTPase Rin induces neurite outgrowth through Rac/Cdc42 and calmodulin in PC12 cells. J Cell Biol. 2003;163:1067–1076. doi: 10.1083/jcb.200308070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inoue T, Meyer T. Synthetic activation of endogenous PI3K and Rac identifies an AND-gate switch for cell polarization and migration. PLoS ONE. 2008;3:e3068. doi: 10.1371/journal.pone.0003068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inoue T, Heo WD, Grimley JS, Wandless TJ, Meyer T. An inducible translocation strategy to rapidly activate and inhibit small GTPase signaling pathways. Nat Methods. 2005;2:415–418. doi: 10.1038/nmeth763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karnoub AE, Weinberg RA. Ras oncogenes: split personalities. Nat Rev Mol Cell Biol. 2008;9:517–531. doi: 10.1038/nrm2438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim JR, Bae WS, Yoon Y, Cho KH. Topological difference of core regulatory networks induces different entrainment characteristics of plant and animal circadian clocks. Biophys J. 2007;93:L01–L03. doi: 10.1529/biophysj.107.106658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim JR, Yoon Y, Cho KH. Coupled feedback loops form dynamic motifs of cellular networks. Biophys J. 2008;94:359–365. doi: 10.1529/biophysj.107.105106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Komatsu N, Aoki K, Yamada M, Yukinaga H, Fujita Y, Kamioka Y, Matsuda M. Development of an optimized backbone of FRET biosensors for kinases and GTPases. Mol Biol Cell. 2011;22:4647–4656. doi: 10.1091/mbc.E11-01-0072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kraynov VS, Chamberlain C, Bokoch GM, Schwartz MA, Slabaugh S, Hahn KM. Localized Rac activation dynamics visualized in living cells. Science. 2000;290:333–337. doi: 10.1126/science.290.5490.333. [DOI] [PubMed] [Google Scholar]
- Lee JY, Engelman JA, Cantley LC. Biochemistry. PI3K charges ahead. Science. 2007;317:206–207. doi: 10.1126/science.1146073. [DOI] [PubMed] [Google Scholar]
- Lee S, Lee KH, Ha JS, Lee SG, Kim TK. Small-moleculebased nanoassemblies as inducible nanoprobes for monitoring dynamic molecular interactions inside live cells. Angew Chem Int Ed Engl. 2011;50:8709–8713. doi: 10.1002/anie.201101467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Z, Dong X, Wang Z, Liu W, Deng N, Ding Y, Tang L, Hla T, Zeng R, Li L, Wu D. Regulation of PTEN by Rho small GTPases. Nat Cell Biol. 2005;7:399–404. doi: 10.1038/ncb1236. [DOI] [PubMed] [Google Scholar]
- Machacek M, Hodgson L, Welch C, Elliott H, Pertz O, Nalbant P, Abell A, Johnson GL, Hahn KM, Danuser G. Coordination of Rho GTPase activities during cell protrusion. Nature. 2009;461:99–103. doi: 10.1038/nature08242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mochizuki N, Yamashita S, Kurokawa K, Ohba Y, Nagai T, Miyawaki A, Matsuda M. Spatio-temporal images of growth-factor-induced activation of Ras and Rap1. Nature. 2001;411:1065–1068. doi: 10.1038/35082594. [DOI] [PubMed] [Google Scholar]
- Nassar N, Horn G, Herrmann C, Scherer A, McCormick F, Wittinghofer A. The 2.2 A crystal structure of the Ras-binding domain of the serine/threonine kinase c-Raf1 in complex with Rap1A and a GTP analogue. Nature. 1995;375:554–560. doi: 10.1038/375554a0. [DOI] [PubMed] [Google Scholar]
- Niggli V. A membrane-permeant ester of phosphatidylinositol 3,4, 5-trisphosphate (PIP(3)) is an activator of human neutrophil migration. FEBS Lett. 2000;473:217–221. doi: 10.1016/s0014-5793(00)01534-9. [DOI] [PubMed] [Google Scholar]
- Orme MH, Alrubaie S, Bradley GL, Walker CD, Leevers SJ. Input from Ras is required for maximal PI(3)K signalling in Drosophila. Nat Cell Biol. 2006;8:1298–1302. doi: 10.1038/ncb1493. [DOI] [PubMed] [Google Scholar]
- Pacold ME, Suire S, Perisic O, Lara-Gonzalez S, Davis CT, Walker EH, Hawkins PT, Stephens L, Eccleston JF, Williams RL. Crystal structure and functional analysis of Ras binding to its effector phos-phoinositide 3-kinase gamma. Cell. 2000;103:931–943. doi: 10.1016/s0092-8674(00)00196-3. [DOI] [PubMed] [Google Scholar]
- Peyrollier K, Hajduch E, Gray A, Litherland GJ, Prescott AR, Leslie NR, Hundal HS. A role for the actin cytoskeleton in the hormonal and growth-factor-mediated activation of protein kinase B. Biochem J. 2000;352:617–622. [PMC free article] [PubMed] [Google Scholar]
- Rodriguez-Viciana P, Warne PH, Dhand R, Vanhaesebroeck B, Gout I, Fry MJ, Waterfield MD, Downward J. Phosphatidylinositol-3-OH kinase as a direct target of Ras. Nature. 1994;370:527–532. doi: 10.1038/370527a0. [DOI] [PubMed] [Google Scholar]
- Rodriguez-Viciana P, Warne PH, Vanhaesebroeck B, Waterfield MD, Downward J. Activation of phosphoinositide 3-kinase by interaction with Ras and by point mutation. EMBO J. 1996;15:2442–2451. [PMC free article] [PubMed] [Google Scholar]
- Rodriguez-Viciana P, Warne PH, Khwaja A, Marte BM, Pappin D, Das P, Waterfield MD, Ridley A, Downward J. Role of phosphoinositide 3-OH kinase in cell transformation and control of the actin cytoskeleton by Ras. Cell. 1997;89:457–467. doi: 10.1016/s0092-8674(00)80226-3. [DOI] [PubMed] [Google Scholar]
- Rodriguez-Viciana P, Sabatier C, McCormick F. Signaling specificity by Ras family GTPases is determined by the full spectrum of effectors they regulate. Mol Cell Biol. 2004;24:4943–4954. doi: 10.1128/MCB.24.11.4943-4954.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rossman KL, Der CJ, Sondek J. GEF means go: turning on RHO GTPases with guanine nucleotide-exchange factors. Nat Rev Mol Cell Biol. 2005;6:167–180. doi: 10.1038/nrm1587. [DOI] [PubMed] [Google Scholar]
- Schwamborn JC, Püschel AW. The sequential activity of the GTPases Rap1B and Cdc42 determines neuronal polarity. Nat Neurosci. 2004;7:923–929. doi: 10.1038/nn1295. [DOI] [PubMed] [Google Scholar]
- Servant G, Weiner OD, Herzmark P, Balla T, Sedat JW, Bourne HR. Polarization of chemoattractant receptor signaling during neutrophil chemotaxis. Science. 2000;287:1037–1040. doi: 10.1126/science.287.5455.1037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin SY, Yang HW, Kim JR, Do Heo W, Cho KH. A hidden incoherent switch regulates RCAN1 in the calcineurin-NFAT signaling network. J Cell Sci. 2011;124:82–90. doi: 10.1242/jcs.076034. [DOI] [PubMed] [Google Scholar]
- Spaargaren M, Martin GA, McCormick F, Fernandez-Sarabia MJ, Bischoff JR. The Ras-related protein R-ras interacts directly with Raf-1 in a GTP-dependent manner. Biochem J. 1994;300:303–307. doi: 10.1042/bj3000303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Srinivasan S, Wang F, Glavas S, Ott A, Hofmann F, Aktories K, Kalman D, Bourne HR. Rac and Cdc42 play distinct roles in regulating PI(3,4,5)P3 and polarity during neutrophil chemotaxis. J Cell Biol. 2003;160:375–385. doi: 10.1083/jcb.200208179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suh BC, Inoue T, Meyer T, Hille B. Rapid chemically induced changes of PtdIns(4,5)P2 gate KCNQ ion channels. Science. 2006;314:1454–1457. doi: 10.1126/science.1131163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suire S, Condliffe AM, Ferguson GJ, Ellson CD, Guillou H, Davidson K, Welch H, Coadwell J, Turner M, Chilvers ER, et al. Gbetagammas and the Ras binding domain of p110gamma are both important regulators of PI(3)Kgamma signalling in neutrophils. Nat Cell Biol. 2006;8:1303–1309. doi: 10.1038/ncb1494. [DOI] [PubMed] [Google Scholar]
- Wang F, Herzmark P, Weiner OD, Srinivasan S, Servant G, Bourne HR. Lipid products of PI(3)Ks maintain persistent cell polarity and directed motility in neutrophils. Nat Cell Biol. 2002;4:513–518. doi: 10.1038/ncb810. [DOI] [PubMed] [Google Scholar]
- Weiner OD, Neilsen PO, Prestwich GD, Kirschner MW, Cantley LC, Bourne HR. A PtdInsP(3)- and Rho GTPase-mediated positive feedback loop regulates neutrophil polarity. Nat Cell Biol. 2002;4:509–513. doi: 10.1038/ncb811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang S, Charest PG, Firtel RA. Spatiotemporal regulation of Ras activity provides directional sensing. Curr Biol. 2008;18:1587–1593. doi: 10.1016/j.cub.2008.08.069. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.