

Abstract
Glioblastoma Multiforme (GBM) is recognized as one of the most deadly cancers characterized by cellular atypia, severe necrosis, and high rate of angiogenesis. In this review, we discuss a diversified group of GBM xenograft models and compare them with the genetically engineered mouse (GEM) model systems. Next, we describe common genetic defects observed in GBM and numerous GEM models that recapitulate these abnormalities. Finally, we focus on the clinical value of other vertebrate animal models such as the canine model by examining their contributions to GBM research.
Keywords: brain tumor, animal model, tumorigenesis, cancer therapy
Introduction
Glioma covers various primary tumor types with histological features similar to glia in the central nervous system (CNS). These gliomas are composed predominantly of astrocytes (astrocytomas), oligodendrocytes (oligodendrogliomas), ependymal cells (ependymomas), or mixtures of glial cells. As the most common primary tumors in adult CNS, an estimated of 30,000 patients are diagnosed with glioma in the United States every year [1]. Glioblastoma multiforme (GBM), a grade IV glioma classified by the World Health Organization (WHO), is considered the most malignant and invasive subtype with a median survival of 14.6 months even after radiotherapy and concomitant chemotherapy [2]. GBM is characterized by histopathologic features of cellular atypia, severe necrosis as well as high rate of angiogenesis [3]. There is no cure for this deadly disease and the multimodal treatment for GBM typically consists of surgical resection followed by concomitant chemotherapy and focal radiotherapy. Temozolomide, an oral alkylating agent, is considered the standard care for GBM treatment [2].
GBM itself is divided into two categories. Primary GBM mainly affects older patient population with a mean age of 62 years old and has a predilection to affect men. This type of GBM is referred to as the “de novo” glioblastoma because it is a fully developed tumor resulted from accumulation of multiple genetic aberrations instead of arising from low-grade precursors. In contrast, the secondary GBM results from gradual progression from low-grade astrocytoma (WHO grade II) or anaplastic astrocytoma (WHO grade III) over a period of 5–10 years. Secondary GBM is often diagnosed in younger population with a median age of 45 years old and strikes women with a higher proportion [4]. Primary GBM accounts for up to 95% of GBM cases [5].
Several crucial GBM-contributing genetic aberrations such as EGFR amplification, loss of tumor suppressor genes p53, INK4a/ARF, and PTEN have been well documented (Figure 1). The impact of these mutations in tumor initiation and progression has been investigated both in vitro and in vivo. Like many other cancer types, the genetically engineered mouse (GEM) models allow manipulation of the GBM-contributing genes at the molecular level. The ex vivo xenograft mouse models also offer clinical values in drug screening and gene function investigations. Interestingly, other animal models such as canine models have been used extensively in GBM studies due to their high incidence rate of spontaneous intracranial neoplasia and the resemblance to human tumors.
Figure 1. The key genetic alterations involved in primary and secondary GBM initiation and progression.
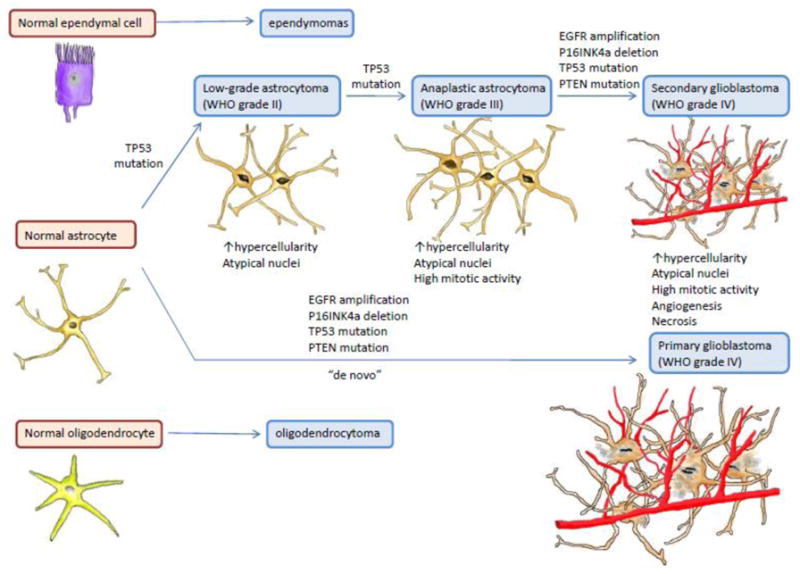
Primary GBM, responsible for up to 95% of GBM cases, resulted from accumulation of multiple genetic aberrations instead of arising from low-grade precursor seen in secondary GBM. Both primary and secondary GBM involves EGFR amplification, TP53 deletion as well as mutations of INK4/ARF and PTEN.
In this review, we will focus on various genetically engineered mouse models that have assisted unraveling the role of the oncogenes and tumor suppressor genes in GBM pathogenesis (Table 1). We will also discuss xenograft models as well as the clinical significance of other animal models including the canine, zebrafish, and fruit fly models in glioma investigations.
Table 1.
Transgenic and genetically engineered mouse models for glioblastoma multiforme
| Mouse Model | Significant Findings | References |
|---|---|---|
| RCAS-EGFR; tva-INK4a-ARF−/−
RCAS-EGFR; tva-INK4a-ARF+/− |
EGFR mutation alone is insufficient to induce glioma unless the loss of INK4a-ARF occurs concomitantly | Holland, 2000 |
| GFAP-V12Ha-ras; GFAP-EGFRVIII | Aberrant EGFR level is involved in glioma progression rather than initiation and it can give rise to both astrocytoma & oligodendrocytoma | Wei, 2006 |
| S100β-V-erb(EGFR); S100β-INK4a- ARF−/− S100β-V-erb(EGFR); S100β-INK4a- ARF+/− |
Rapid tumor development from low-grade to high-grade with shorter latency and increased penetration | Weiss, 2003 |
| CAGGS-Cre; EGFRVIII; INK4a−/−; PTEN−/− | EGFRVIII alone is insufficient to induce cell transformation and only when concomitant loss of tumor suppresser gene INK4a along with PTEN can promote a fully-penetrant, rapid onset malignant glioma that resembles to human GBM | Zhu, 2009 |
| TgG(ΔZ)T121; PTEN−/− (pRB inactivated through T21 expression) | PTEN inactivation in pRB-inactivated model gives rise to focal hypercellularity, enhancing astrocytoma invasiveness as well as promoting angiogenesis | Xiao, 2005 |
| RCAS-KRas; PTEN−/− | The mice display similar histopathological characteristics such as increased cell density, pseudopalisading necrosis, microvascular proliferation | Hu, 2005 |
| p53−/−; Nf1−/−; PTEN+/− | Heterozygosities of PTEN, Nf1 and p53 cause accelerated glioma formation similar to primary “de novo” GBM | Kwon, 2008 |
| Ntva-INK4a-ARF+/−; RCAS-KRas; RCAS-AKT | The deletion of INK4a-ARF locus in addition to the activation of KRas and AKT permits GBM formation from differentiated astrocytes as well as enhances gliomageneis from neural progenitors. | Uhrbom, 2002 |
| Nf1+/−; p53+/− | The mouse model displays different stages of glioma including GBM. | Reilly, 2000 |
| P53 −/−; Nf1−/−; GFAP-Cre P53+/−; Nf1−/−; GFAP-Cre P53+/−; Nf1+/−; GFAP-Cre |
TP53 loss prior to or concomitant to Nf1 loss is required for malignant astrocytoma formation when early loss of Nf1 fails to induce gliomagenesis | Zhu, 2011 |
Mouse Models
One of the first documented mouse model for cancer study can be traced back to 1916 when Lathrop and Loeb published the effects of hormones on the development of tumors in mice [6]. Since then, mice have remained a favorite animal model to study human diseases due to their evolutionary similarities and fast generation time. Mouse models can help to probe the underlying etiological cause and the detailed mechanism of tumorigenesis. To date, there are four predominant strategies to model malignancies in mice: chemical mutagen-induced, xenograft transplantation, germline genetic modification, and somatic genetic modification mouse models [7]. In this review, we will focus on the most widely employed xenograft model and the genetic engineered mouse model.
Xenograft mouse models
Mouse brain tumor models have been employed for research since the mid-1970 [8]. Xenograft mouse models are typically used in preclinical trials to test the efficacy of novel therapeutic agents as well as in studying the gene functions in glioma pathogenesis. In these mouse models, human or mice glioma cell lines are injected under the skin or into the brain of immunocompromised mice. Kaye and colleagues [9] injected 106 cells of rat C6 glioma both subcutaneously and intracranially and found exclusively visible tumor growth in mice with intracranial injection. When the cells are introduced into the frontal lobe of the brain, localized tumor growths were observed in both neonatal and adult mice in a reproducible manner.
One of the most well-known glioma xenograft models is the U251 glioma model established by Ponten et al [10], and over time this cell line has been used both in subcutaneous and intracranial mouse models. Due to the difference in gene expression profile as well as the microenvironment, the intracranial mouse model injected with U251 cell lines better recapitulate the histopathological feature of GBM compared to the subcutaneous model [11]. Not only did it demonstrate infiltrative invasion into brain parenchyma and significant foci of palisading necrosis microscopically, it also reveals striking resemblance to human GBMs under immunohistochemical analysis with positive GFAP, S100B, and Vimentin staining [12]. Moreover, the losses of tumor suppressor genes p53 and PTEN along with the deletion of INK4a/ARF are observed [13]. Another notable cell line established by Ponten and colleagues [10] is the U87 glioma model. Unlike the U251 glioma model, U87 displays a non-diffusely infiltrative growth pattern with a well-demarcated tumor border that is rarely accompanied by necrotic foci [13, 14]. However, the more homogenous tumor vasculature and leakier vessels in the U87 model allows greater entry by systemic therapeutic drugs, making it a superior model for evaluating tumor angiogenesis and anti-angiogenic therapeutic approaches [15].
Although these models display well-defined tumor with predictable proliferation rates, the histopathological feature of the tumors do not always recapitulate what is observed in their human counterpart [16]. Several factors limit the utility of xenograft models, as there are significant differences in selective pressures occurring during cell culture in comparison to the natural brain environment [1]. In addition, these xenograft gliomas in immunocompromised mice grow in the absence of a natural tumor-harboring environment. Other xenograft mouse models include the U1242 MG intracranial model created by Zhao et al [17]. This intracranial xenograft mouse model expresses high level of matrix metalloproteinase (MMP-9) and shows extensive infiltration and hypervascularity, making it a valuable model to study GBM invasion and angiogenesis.
To overcome many shortcomings in recapitulating GBM invasiveness in most established GBM cell lines, Giannini and colleagues [18] injected the tumor cells subcutaneously into the flank of mice, excised these flank xenografts and cultured them before injecting into the brains of the nude mice. They found this heterotopic-to-orthotopic approach for establishing intracranial tumor has a 100% success rate with minimal (<1%) mortality rate. This short-term flank xenograft model retains some of the genetic characterization involved in GBM tumorigenesis such as aberrant level of amplification and overexpression of EGFR gene. The retention of genetic abnormalities along with the highly mitotic and invasive features observed in this model contributes to the versatility of their use in neuro-oncological research. Although this method successfully preserved the invasive nature of tumor cells, necrosis and endothelial proliferation were not observed. Lee and colleagues [19] cultured the GBM cells under NBE growth conditions consisting of serum-free Neurobasal media supplemented with basic FGF and EGF and found that these glioma tumor cells more closely mimic the phenotype and genotype of primary tumors. Due to glioma’s highly proliferative nature and high recurrence rate, Bello et al [20] created xenograft mouse models with human cell lines U87 and D566 in order to assess the post-surgical treatment efficacy for recurring glioma. Twenty days after tumor cell implantation, the primary tumors were surgically resected followed by administration of endogenous inhibitors PEX and a fragment of platelet factor 4 (PF-4/CTF). They found that the systemic administration of PEX or PF-4/CTF after tumor resection significantly improved the survival and delayed glioma reoccurrence in the xenograft mouse models. Although some xenograft models can partially recapitulate human GBM histological features and maintain key molecular characteristics, the differences in tumor microenvironment as well as lack of endogenous spontaneous tumor initiation diminish the clinical value of xenograft models.
Genetically Engineered Mouse Models
Genetically Engineered Mouse (GEM) models are manipulated at the molecular level by either germline modification or somatic cell gene transfer. In germline-modification models, DNA is incorporated into totipotent cells prior to the developmental stage at which the germline forms and usually involves introducing DNA into gametes such as oocyte, egg or early embryos, or embryonic stem (ES) cells. The first germline GEM models for brain tumors were described by Brinster et al. in 1984 [21] as they microinjected eggs with simian virus SV40 gene containing the metallothionein fusion gene. This random integration of viral oncogene significantly lowers the threshold for other mutation and increases the likelihood of a secondary mutation to occur that is typically seen during tumor development. Germline modifications typically include gain-of function, loss-of-function, and chromosome engineering [22].
Another strategy employed in GEM is the somatic gene transfer, which is not heritable and targets only a specific subset of cell population. Fisher and colleagues [23] expressed the avian retroviral receptor, TVA, under various mammalian promoters in transgenic mice and infected the mice with avian leukosis virus derived gene vectors. This RCAS/tv-a system for oncogene delivery is more specific and allows a single transgenic mouse line to be evaluated for multiple lesions. Unlike germline gene transfer that requires extensive breeding to combine multiple mutations, this system offers a much faster oncogene delivery. However, secondary events are less likely to occur in this strategy and thus tumor formation is the direct result of introduced oncogene without other genetic events in tumorigenesis. Due to the direct injection of oncogene-carrying retrovirus into the brain tissues, local injury and inflammatory response may alter the gene expression.
The glial fibrillary acidic protein (GFAP), an intermediate filament protein found exclusively in astrocytes, is often employed as a promoter in transgenic mice. The very first transgenic mouse model for astrocytomas was reported by Danks et al [24]. They expressed SV40 T large T antigen, which was found to bind and inactivate p53 and Rb under the control of GFAP promoter [25]. These GFAP/T Ag mice displayed rapid transformation of T Ag-expressing astrocytic cells with astrocytoma-like secondary structures; however, all mice died within 30 days postnatally. Weissenberger and colleagues [26] created a transgenic mouse model that constitutively expresses active Src kinase in astrocytes under the control of GFAP gene regulatory elements. Src kinase is a tyrosine kinase that binds and activated EGF receptor, however, integration of the constitutively active v-src gene alone did not result in tumor formation. When the GFAP promoter was used to express an oncogenic mutant form of H-Ras (V12H), the resulting astrocytomas demonstrated histopathological similarities to the human disease including high mitotic activity, infiltration, necrosis, and increased vascularity, and the tumors also exhibited other common mutations associated with astrocytoma [27]. In order to exam the role pRB plays in astrocytoma formation under the GFAP promoter, Xiao et al [28] used a truncated version of SV40 T antigen mutant–T121, and found astrocyte-specific pRB inactivation caused widespread brain abnormalities.
Since xenograft mouse model requires immunocompromised mice to prevent rejection of human tumor cells, the study of GBM tumorigenesis is limited by the modified tumor immunology of these mice. In the contrary, GEM models can be established in immunocompetent animals, allowing a more realistic microenvironment for spontaneous and endogenous tumor growth. The manipulation at the genetic level of GEM models allows thorough investigation of key mutations in a spatial- and temporal-specific fashion. Appropriate interaction between tumor and stromal tissues is also observed in GEM models. With the readily available advanced imaging techniques, non-invasive monitoring of the tumor growth is no longer an issue for the GEM models. However, the GEM model has its own weakness in relatively poor prediction of drug therapeutic response, high cost, time consuming and slow tumor development. The xenograft model still holds significant clinical value in initial drug screening and studying gene functions. In the next section, we discuss the most frequently observed mutations in gliomagenesis and the corresponding GEM models.
Epidermal Growth Factor Receptor
The epidermal growth factor receptor (EGFR) is a transmembrane glycoprotein that regulates the growth of epithelial cells. Aberrant EGFR expression is the most frequently detected genetic defects observed in GBM and consists of three major mechanisms for oncogenic signaling. The most common mechanism involves overexpression or amplification of the wild-type receptor. Alternatively, the increase in oncogenic signaling may be due to increased autocrine secretion of EGF-family ligands, leading to activation of the wild-type EGF signaling. Lastly, oncogenic signaling is a direct result of constitutive receptor activation in a mutant variant EGFRvIII [29], which is the most common variant of EGFR resulted from deletion of exon 2 to 7 of the EGFR gene. This critical variant is observed in approximately 40% of primary GBM and results in an in-frame deletion of 801 amino acids [30–32]. EGFRvIII’s inability to bind ligand renders it constitutively active, conferring growth advantage for glioma cells [33]. EGFR activation has also been found to contribute to drug resistance in cancer cells [29].
Holland and colleagues [34] initially developed a transgenic system expressing the ALV subgroup A receptor TVA from the Nestin promoter so that the constitutively active mutant form of EGFR can be transferred via a RCAS vector. The resulting transgenic mice with elevated EGFR failed display any glioma-like lesions; however, in conjunction with INK4a/ARF loss, glioma-like lesions with high cell density and vascular proliferation are induced in these mice. This work demonstrates EGFR alone does not lead to gliomagenesis unless another genetic event such as concomitant loss of INK4a/ARF. The presence of EGFR mutation alone is insufficient to induce gliomagenesis was further confirmed by Ding et al [35]. They used ES cell-mediated transgenesis to establish mouse models that expressed either wild-type EGFR or mutant EGFRvIII in astrocytes under the control of GFAP promoter. The authors found both models had an increased number of astrocytes in the brain without glioma formation. They further created a construct that combined both mutant EGFRvIII and oncogenic RAS overexpression, and this GFAP-V12Ha-ras; GFAP-EGFRvIII double transgenic mouse led to accelerated glioma formation with histopathological feature of oligodendrogliomas and mixed oligoastrocytoma. They came into conclusion that aberrant EGFR level is involved in glioma progression rather than initiation, and dysregulated EGFR expression can give rise to both astrocytomas and oligodendrocytomas. In a separate study, Weiss and colleague [36] created a mouse model that expressed v-erbB, a transforming homologue of EGFR, under the control of S100β promoter with a goal to recapitulate high levels of EGFR expression during glioma formation. Most tumors found in this S100-v-erbB model shared histopathological features of low-grade oligodendroglioma with less than 10% displayed resemblance to that of astrocytomas. The authors suggested that overexpression of EGFR occurs early in oligodendrocytoma pathogenesis but late in astrocytoma formation. To further evaluate the significance of EGFR overexpression, they knocked out either tumor suppressor gene INK4a/ARF or p13 in addition to the S100-v-erbB construct and found rapid developed tumors from low-grade to higher-grade with shorter latency and higher penetrance in both models. A more recent work by Zhu et al [37] also demonstrated the constitutively active mutant EGFRvIII alone is not adequate to promote cell transformation in vivo in their Cre-induced conditional transgenic model. However, concomitant loss of the tumor suppressor gene INK4a/ARF along with PTEN resulted in glioma with characteristics resembling to GBM.
PTEN
PTEN (Phosphatase and tensin homologue) acts as a tumor suppressor gene through its phosphatase activity keeping cell proliferation in check [38, 39]. PTEN is thought to antagonize the function of PI3K and induce cell cycle arrest and apoptosis in glioma cells [40]. In addition to its role as a cell proliferation inhibitor, inactivation of PTEN is found to promote an undifferentiated state with high self-renewal and tumorigenic potential [41]. PTEN mutation is found in approximately 30% primary GBMs, usually by complete loss of its locus on chromosome 10q, however, this mutation is rarely detected in secondary GBMs [42, 43]. PTEN mutation in patients with Cowden disease also showed higher incidence of gliomas development compared to normal individuals [44].
It has been shown that PTEN-deficient human glioma cell lines are highly invasive and when PTEN activity is restored, the cell invasion is significantly decreased [45]. To study the astrocytoma suppression mechanisms of PTEN, Xiao et al [46] conditionally inactivated PTEN with localized somatic retroviral (MSCV)-Cre in a GEM astrocytoma model with inactivated pRB. Inactivation of pRB family proteins induced aberrant proliferation and apoptosis in astrocytes. The localized PTEN inactivation in these mice alleviated apoptosis induced from pRB inactivation and gave rise to focal hypercellularity, enhancing astrocytoma invasiveness and a more pronounced angiogenesis.
The protein kinase AKT typically promotes cell survival and proliferation by activating a downstream effector mammalian Target of Rapamycin (mTOR) that has been found to be overexpressed without identified mutation in most GBMs. To investigate the relationship between AKT and PTEN, Hu and coworkers [47] injected a combination of cells producing RCAS-KRas and RCAS-Cre intracranially to 37 PTEN −/− mice and these mice displayed similar histopathological characteristics such as increased cell density, pseudopalisading necrosis and microvascular proliferation that resemble to the KRas+AKT-induced GBM in their previous study [48, 49]. These findings suggested the association of an oncogenic effect of AKT activity in the context of PTEN loss. They also found loss of PTEN alone was insufficient to induce GBMs as none of the 32 PTEN-deficient mice injected with RCAS-Cre displayed tumor formation. Although PTEN mutation or deletion is frequently found in GBM, it requires other genetic event to give rise to GBM formation. Wei and colleagues generated a conditional PTEN−/− model by crossing mice with null PTEN alleles to mice that express Cre recombinase under the control of the GFAP promoter [50]. The conditional PTEN knockout mice showed characteristics of generalized seizures and all died by 6 weeks of age. The brains of these mice weighed approximately 30% more than that of control with a 3-fold increase in the number of astrocytes, demonstrating an increase in brain mass correlating with increased astroglial cell proliferation. However, no glioma formation was observed in these conditional PTEN mice possibly due to their early lethality before glioma formation could take place. This suggests PTEN inactivation by itself only contributes to glioma progression instead of initiating gliomagenesis. The role of PTEN in tumor progression rather than tumor initiation was further confirmed by Kwon et al [51]. The authors introduced a loxP-PTEN allele in the Nf1- and p53-deficient astrocytoma model that was previously found to develop malignant astrocytoma. The presence of somatic heterozygosities of PTEN, Nf1 and p53 cause accelerated glioma formation similar to primary “de novo” GBM and significantly shortens the survival of mice. All GEM mouse models mentioned above consistently demonstrate the role of PTEN in promoting tumorigenesis rather than glioma initiation.
INK4a/ARF
The INK4a/ARF locus on chromosome 9p21 by differential splicing and alternate reading frame encodes two distinct proteins, p16INK4a and P19ARF, that ultimately links the retinoblastoma and p53 tumor suppressor pathways, respectively [52]. P16INK4a is a cyclin-dependent kinase inhibitor that induces cell cycle arrest in the G1 phase by binding to and inhibiting CDK4/6, as sequential phosphorylation of Rb by CDK4/6 is required for cell cycle progression. Homozygous deletions at the INK4a/ARF locus are identified in 57% of GBMs [53], while loss of heterozygosity (LOH) on chromosome 13q where Rb gene is located is observed in 45% of glioblastomas [54, 55]. In contrast to P16INK4a, ARF does not bind to CDKs, instead, it inhibits MDM2, an ubiquitin ligase that targets p53 for degradation.
The first transgenic mouse was created by Serrano and colleagues who deleted this locus in mouse, resulting in the loss of both P16INK4a and P19ARF and the development of tumors at an early age [56]. This particular study demonstrates that INK4a/ARF locus plays a key role in suppressing tumor growth. Interestingly, Kamijo and colleagues [57] used a conventional targeting vector to replace ARF exon 1β with a neomycin resistance gene in mouse embryonic stem cells so that the mice expressed only the transcript encoding p16INK4a but not P19ARF. Surprisingly, these ARF−/− mice developed tumors that are virtually identical to that of INK4a/ARF null mice early in life, suggesting loss of ARF plays a dominant role in glioma pathogenesis. The genetic effect of INK4a/ARF is often studied in conjunction with other mutations such as constitutively active EGFR overexpression [34, 36, 37]. Uhrbom and colleagues also combined INK4a/ARF loss in addition to one of their models in which K-Ras and AKT signaling are activated [49]. They used the RCAS/tv-a system to combine gene transfer of activated forms of Akt and KRas into nestin-expressing neural progenitor cells and observed glioma formation with histopathological features resembling human GBM from neural progenitor cells [48]. However, these induced GBMs were only derived from neural progenitor cells but not from differentiated astrocytes.
TP53
The tumor suppressor gene TP53 regulates the cell cycle and is commonly inactivated in various types of cancer. TP53 mutation creates genomic instability and inhibits apoptosis. Unlike other genetic alterations, TP53 mutations occur more frequently in secondary GBMs (>60%) than in primary GBMs (~10%) [58]. In order to explore the role of TP53 in glioma, Reilly and colleagues constructed the Nf1+/−p53+/− model [59]. Since Nf1 mutation is associated with increased risk of optic gliomas, astrocytomas and gliobasltoams as well as its close proximity with TP53 on chromosome 11 [60, 61], they hypothesized that Nf1 and TP53 are rarely separated by recombination and can be treated as a single genetic event in gliomagenesis. As they predicted, the combination of Nf1 and TP53 mutations gave rise to malignant tumors. In a similar study, Zhu et al [62] manipulated Nf1 and TP53 under the control of human GFAP promoter in three different mouse strains P53−/−Nf1−/−GFAP-CreP53+/−; Nf1−/−GFAP-Cre, and P53+/−Nf1+/−GFAP-Cre. Not only did they find TP53 inactivation along with Nf1 loss can lead to the development of human malignant astrocytomas, they came to the conclusion that TP53 loss prior to or concomitant to Nf1 loss is essential for malignant astrocytoma formation. This finding is consistent with previously studies regarding TP53 mutation as an early genetic event in glioma formation [1].
The Canine model
Employing dogs as model in cancer research has been documented for over 40 years [63]. The canine genome exhibits closer evolutionary relationship to human compared to the mouse counterpart. The infiltrative nature of spontaneous GBM in dogs as well as their larger brain size make them an appealing model for testing novel therapeutic treatment for GBM [64]. Viral-induced non-spontaneous brain tumor has been used in canine models [65, 66]; however, spontaneous brain tumor represents the true nature of tumor initiation and progression in humans. Intracranial neoplasm in dogs have an incidence rate of approximately 20 per 100,000 according to a study in United Kingdom [67]. Among brain tumor types, meningiomas and gliomas are the most frequently reported in canine models with 43% and 32% of incidence, respectively. Dogs that are seven years old or older have the highest incidence of brain tumors among domestic animals [68]. The average age of dogs at the time of onset of astrocytomas is 8.6 years [69]. Two common mutations associated with human astrocytomas, TP53 and EGFR, are expressed in an aberrant level in 35% and 23% of canine astrocytomas models respectively. Interestingly, intracranial neoplasia occurs far more frequently in dogs than human counterpart with almost a 4-fold increase in incidence rate with 14.5 per 100,000 canines every year [70, 71].
Dog model has been used to investigate the cell-mediated immunity against spontaneous gliomas [72]. Ingram and colleagues [72] studied the immunotherapy for recurrent malignant glioma in dogs by implanting stimulated autologous lymphocytes into the tumor bed following surgical debulking, and they observed clinical improvement and decrease in tumor size in all dogs. The therapeutic value of brachytherapy was investigated by Stubbs et al [73]. The authors implanted the brachytherapy applicator, an inflatable balloon catheter, in the resection cavity in dog brain and inflated the balloon catheter with Iotrex containing iodine-125 as a reliable radiation delivery method to target canine brain tumors. Chauvet et al [74] injected the recombinant adenovirus vector bearing the Escherichia coli beta-galactosidase reporter gene into meningioma of canine models as a gene therapy for spontaneous CNS neoplasia. Xiong et al [46] developed a conditional cytotoxic/immunotherapeutic approach employing adenoviral vector (ads) that encodes human soluble fms-like tyrosine kinase 3 ligand (hsFlt3L) in dogs bearing spontaneous GBM and found that dogs received subcutaneous injections of human recombinant Flt3L displayed circulating dendritic cells, demonstrating hsFlt3L’s ability to induce the proliferation of canine dendritic cells. The resulting induced dendritic cells are similar to that induced by canine cytokine cocktail consisting of IL-4 and GM-CSF. Canine models were also adopted to study the convection-enhanced delivery (CED) that utilized pressure gradient on the tip to allow an increase volume of drugs past the blood-brain barrier with minimal toxicity [75]. Stoica and colleague [76] also identified cancer stem cell (CSC) in dog GBMs and examined the self-renewal ability of these glioblastoma cells with single colony formation and found both clone formation rate and subclone formation rate to be 100%. These dog GBM cells expressed CD133, the CSC surface marker reported in human gliomas. To elucidate the ability of CSC’s ability to initiate tumorigenesis upon xenograft transplantation, they injected the tumor cells intracranially into nude mice, which all develop tumors characterized with high cellular heterogeneity, neovascularization and necrosis. Higgins and colleague [77] identified overexpression of EGFR, PDGFRα and IGFBP2 in spontaneous canine glioma, which is consistent with their human counterpart.
The dogs have become a valuable model to study brain tumor because of their closer revolutionary relationship to human as well as their larger brain size compared to the murine models [78]. Due to the complexity of human tumor environment and host immune interactions, the majority of successful cancer therapies administered in mice fails to show the same efficacy in their human counterpart while some drug toxicities observed later in human were not first identified in mice. Since there’s no treatment regimen deemed the “gold standard”, trials of various veterinary treatment in canine cancer are advantageous of early testing of novel therapies [79]. Therefore, the canine model could be a more suitable candidate for preclinical drug screening as well as clinical trials for other therapeutic strategies. Yet, the consistency of experimental studies relies heavily on pet owners’ compliance and consent, which can limit the accessibility of these canine models by clinicians. The number of canine cases available for study is still much lower compared to that of murine models despite the higher rate of spontaneous brain tumor formation in dogs. Nowadays the canine model for brain cancer research employs only the spontaneous model. Unlike the murine models in which the tumor can be induced by genetic alteration, the same progress has not been made in canine model due to technical challenges, higher cost and ethical dilemmas [79]. Nevertheless, the canine model holds great potential for human cancer research due to its phenotypic diversity as well as its closer cancer pathogenesis resemblance to humans.
Other Animal Models
Other animal models employed in brain tumor include zebrafish and Drosophila Melanogaster (Fruit Fly). Shorter gestation time of 3–4 months, large brood size, transparent embryos for better visual observation and many other advantages make zebrafish an excellent model for medical research. Feitsma et al [80] investigated the role of defective mismatch repair (MMR) in cancer and found brain neoplasms the MMR-deficient zebrafish models. Both TP53 mutant and PTEN mutant zebrafish have also been created and found to develop cancer, however, the cancer grown from these zebrafish models are not glioma-specific.
Using the drosophila model, Read and colleagues [81] created the Gal4-UAS system with the repo-Gal driver to co-activate EGFR-Ras and PI3K in a glial-specific manner. The glial precursors in this drosophila model developed tumor resembling to human glioma. Witte and colleagues [82] also created similar glioma models using the Gal4-UAS system to overexpress homolog of human tyrosine kinase receptors under control of the glia-specific promoter reversed polarity (repo) in Drosophila. The authors found the overexpression of activated EGFR and PI3K both resulted in enhanced proliferation and migration of larval glial cells, promoting their tumor-like overgrowth. Efforts in using zebrafish as well as Drosophilas have facilitated many significant discoveries and will continue to shed new lights in cancer research.
Conclusion
Glioma has plagued approximately 30,000 patients in the United States every year. Despite vigorous research efforts, the overall survival rate has struggled to improve, lingering at approximately a year after initial diagnosis. Animal models have provided a glimpse into various aspects of brain tumor biology, becoming an indispensible tool for cancer research. Mouse models, both xenograft and genetically engineered, are used most commonly for cancer research due to their similar mammalian genetic makeup and relatively fast generation time. Many genes that contribute to gliomagenesis have been manipulated in genetically engineered mouse models to elucidate the genetic contribution as well as the intricate mechanisms of glioma development. Interestingly, the canine model is of emerging significance due to its even closer evolutionary relationship to humans. The infiltrative nature and high spontaneous rate of canine GBM make them an appealing model to assess novel therapeutic GBM interventions. Zebrafish and the fruit fly are among other models used in brain cancer research, offering many advantages such as lower cost and even shorter generation time although they exhibit less human cancer histopathological features than those of the mouse and canine models. Despite the dismal prognosis of GBM, these animal models could potentially lead to groundbreaking discoveries in cancer research.
Acknowledgments
This work was supported in part by the National Institutes of Health (NIH) grant R21CA133604, R01CA138701, Dr. Marnie Rose Foundation, and the William and Ella Owens Medical Research Foundation (M. Li).
Footnotes
Conflict of Interest
The authors declare no conflict of interest.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Holland EC. Gliomagenesis: genetic alterations and mouse models. Nat Rev Genet. 2001;2:120–129. doi: 10.1038/35052535. [DOI] [PubMed] [Google Scholar]
- 2.Stupp R, Mason WP, van den Bent MJ, Weller M, Fisher B, Taphoorn MJ, Belanger K, Brandes AA, Marosi C, Bogdahn U, Curschmann J, Janzer RC, Ludwin SK, Gorlia T, Allgeier A, Lacombe D, Cairncross JG, Eisenhauer E, Mirimanoff RO. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. The New England journal of medicine. 2005;352:987–996. doi: 10.1056/NEJMoa043330. [DOI] [PubMed] [Google Scholar]
- 3.Brat DJ, Van Meir EG. Vaso-occlusive and prothrombotic mechanisms associated with tumor hypoxia, necrosis, and accelerated growth in glioblastoma. Lab Invest. 2004;84:397–405. doi: 10.1038/labinvest.3700070. [DOI] [PubMed] [Google Scholar]
- 4.Maher EA, Furnari FB, Bachoo RM, Rowitch DH, Louis DN, Cavenee WK, DePinho RA. Malignant glioma: genetics and biology of a grave matter. Genes Dev. 2001;15:1311–1333. doi: 10.1101/gad.891601. [DOI] [PubMed] [Google Scholar]
- 5.Ohgaki H, Dessen P, Jourde B, Horstmann S, Nishikawa T, Di Patre PL, Burkhard C, Schuler D, Probst-Hensch NM, Maiorka PC, Baeza N, Pisani P, Yonekawa Y, Yasargil MG, Lutolf UM, Kleihues P. Genetic pathways to glioblastoma: a population-based study. Cancer research. 2004;64:6892–6899. doi: 10.1158/0008-5472.CAN-04-1337. [DOI] [PubMed] [Google Scholar]
- 6.Lathrop AE, Loeb L. Further investigations on the origin of tumors in mice. III. On the part played by internal secretion in the spontaneous development of tumors. The Journal of cancer research. 1916;1:1–19. [PubMed] [Google Scholar]
- 7.Dai C, Holland EC. Glioma models. Biochim Biophys Acta. 2001;1551:M19–27. doi: 10.1016/s0304-419x(01)00027-0. [DOI] [PubMed] [Google Scholar]
- 8.Salvati M, Ramundo Orlando E, Causo R, Innocenzi G, Capone R, Cosentino F, Jr, Cosentino F. Tumors of the central nervous system induced by ionizing radiation. Update on their pathology and presentation of a case. G Ital Oncol. 1990;10:15–18. [PubMed] [Google Scholar]
- 9.Kaye AH, Morstyn G, Gardner I, Pyke K. Development of a xenograft glioma model in mouse brain. Cancer research. 1986;46:1367–1373. [PubMed] [Google Scholar]
- 10.Ponten J. Neoplastic human glia cells in culture. Human Tumor Cells in Vitro. 1975:175–185. [Google Scholar]
- 11.Camphausen K, Purow B, Sproull M, Scott T, Ozawa T, Deen DF, Tofilon PJ. Orthotopic growth of human glioma cells quantitatively and qualitatively influences radiation-induced changes in gene expression. Cancer research. 2005;65:10389–10393. doi: 10.1158/0008-5472.CAN-05-1904. [DOI] [PubMed] [Google Scholar]
- 12.Jacobs VL, Valdes PA, Hickey WF, De Leo JA. Current review of in vivo GBM rodent models: emphasis on the CNS-1 tumour model. ASN neuro. 2011;3:e00063. doi: 10.1042/AN20110014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Radaelli E, Ceruti R, Patton V, Russo M, Degrassi A, Croci V, Caprera F, Stortini G, Scanziani E, Pesenti E, Alzani R. Immunohistopathological and neuroimaging characterization of murine orthotopic xenograft models of glioblastoma multiforme recapitulating the most salient features of human disease. Histology and histopathology. 2009;24:879–891. doi: 10.14670/HH-24.879. [DOI] [PubMed] [Google Scholar]
- 14.Candolfi M, Curtin JF, Nichols WS, Muhammad AG, King GD, Pluhar GE, McNiel EA, Ohlfest JR, Freese AB, Moore PF, Lerner J, Lowenstein PR, Castro MG. Intracranial glioblastoma models in preclinical neuro-oncology: neuropathological characterization and tumor progression. Journal of neuro-oncology. 2007;85:133–148. doi: 10.1007/s11060-007-9400-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.de Vries NA, Beijnen JH, van Tellingen O. High-grade glioma mouse models and their applicability for preclinical testing. Cancer treatment reviews. 2009;35:714–723. doi: 10.1016/j.ctrv.2009.08.011. [DOI] [PubMed] [Google Scholar]
- 16.Finkelstein SD, Black P, Nowak TP, Hand CM, Christensen S, Finch PW. Histological characteristics and expression of acidic and basic fibroblast growth factor genes in intracerebral xenogeneic transplants of human glioma cells. Neurosurgery. 1994;34:136–143. [PubMed] [Google Scholar]
- 17.Zhao Y, Xiao A, diPierro CG, Carpenter JE, Abdel-Fattah R, Redpath GT, Lopes MB, Hussaini IM. An extensive invasive intracranial human glioblastoma xenograft model: role of high level matrix metalloproteinase 9. Am J Pathol. 2010;176:3032–3049. doi: 10.2353/ajpath.2010.090571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Giannini C, Sarkaria JN, Saito A, Uhm JH, Galanis E, Carlson BL, Schroeder MA, James CD. Patient tumor EGFR and PDGFRA gene amplifications retained in an invasive intracranial xenograft model of glioblastoma multiforme. Neuro Oncol. 2005;7:164–176. doi: 10.1215/S1152851704000821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lee J, Kotliarova S, Kotliarov Y, Li A, Su Q, Donin NM, Pastorino S, Purow BW, Christopher N, Zhang W, Park JK, Fine HA. Tumor stem cells derived from glioblastomas cultured in bFGF and EGF more closely mirror the phenotype and genotype of primary tumors than do serum-cultured cell lines. Cancer cell. 2006;9:391–403. doi: 10.1016/j.ccr.2006.03.030. [DOI] [PubMed] [Google Scholar]
- 20.Bello L, Giussani C, Carrabba G, Pluderi M, Lucini V, Pannacci M, Caronzolo D, Tomei G, Villani R, Scaglione F, Carroll RS, Bikfalvi A. Suppression of malignant glioma recurrence in a newly developed animal model by endogenous inhibitors. Clinical cancer research : an official journal of the American Association for Cancer Research. 2002;8:3539–3548. [PubMed] [Google Scholar]
- 21.Brinster RL, Chen HY, Messing A, van Dyke T, Levine AJ, Palmiter RD. Transgenic mice harboring SV40 T-antigen genes develop characteristic brain tumors. Cell. 1984;37:367–379. doi: 10.1016/0092-8674(84)90367-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Fomchenko EI, Holland EC. Mouse models of brain tumors and their applications in preclinical trials. Clinical cancer research : an official journal of the American Association for Cancer Research. 2006;12:5288–5297. doi: 10.1158/1078-0432.CCR-06-0438. [DOI] [PubMed] [Google Scholar]
- 23.Fisher GH, Orsulic S, Holland E, Hively WP, Li Y, Lewis BC, Williams BO, Varmus HE. Development of a flexible and specific gene delivery system for production of murine tumor models. Oncogene. 1999;18:5253–5260. doi: 10.1038/sj.onc.1203087. [DOI] [PubMed] [Google Scholar]
- 24.Danks RA, Orian JM, Gonzales MF, Tan SS, Alexander B, Mikoshiba K, Kaye AH. Transformation of astrocytes in transgenic mice expressing SV40 T antigen under the transcriptional control of the glial fibrillary acidic protein promoter. Cancer research. 1995;55:4302–4310. [PubMed] [Google Scholar]
- 25.Chen J, Tobin GJ, Pipas JM, Van Dyke T. T-antigen mutant activities in vivo: roles of p53 and pRB binding in tumorigenesis of the choroid plexus. Oncogene. 1992;7:1167–1175. [PubMed] [Google Scholar]
- 26.Weissenberger J, Steinbach JP, Malin G, Spada S, Rulicke T, Aguzzi A. Development and malignant progression of astrocytomas in GFAP-v-src transgenic mice. Oncogene. 1997;14:2005–2013. doi: 10.1038/sj.onc.1201168. [DOI] [PubMed] [Google Scholar]
- 27.Ding H, Roncari L, Shannon P, Wu X, Lau N, Karaskova J, Gutmann DH, Squire JA, Nagy A, Guha A. Astrocyte-specific expression of activated p21-ras results in malignant astrocytoma formation in a transgenic mouse model of human gliomas. Cancer research. 2001;61:3826–3836. [PubMed] [Google Scholar]
- 28.Xiao A, Wu H, Pandolfi PP, Louis DN, Van Dyke T. Astrocyte inactivation of the pRb pathway predisposes mice to malignant astrocytoma development that is accelerated by PTEN mutation. Cancer cell. 2002;1:157–168. doi: 10.1016/s1535-6108(02)00029-6. [DOI] [PubMed] [Google Scholar]
- 29.Huang PH, Xu AM, White FM. Oncogenic EGFR signaling networks in glioma. Sci Signal. 2009;2:re6. doi: 10.1126/scisignal.287re6. [DOI] [PubMed] [Google Scholar]
- 30.Hatanpaa KJ, Burma S, Zhao D, Habib AA. Epidermal growth factor receptor in glioma: signal transduction, neuropathology, imaging, and radioresistance. Neoplasia. 2010;12:675–684. doi: 10.1593/neo.10688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sugawa N, Ekstrand AJ, James CD, Collins VP. Identical splicing of aberrant epidermal growth factor receptor transcripts from amplified rearranged genes in human glioblastomas. Proc Natl Acad Sci U S A. 1990;87:8602–8606. doi: 10.1073/pnas.87.21.8602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Schlegel J, Merdes A, Stumm G, Albert FK, Forsting M, Hynes N, Kiessling M. Amplification of the epidermal-growth-factor-receptor gene correlates with different growth behaviour in human glioblastoma. Int J Cancer. 1994;56:72–77. doi: 10.1002/ijc.2910560114. [DOI] [PubMed] [Google Scholar]
- 33.Zhu Y, Parada LF. The molecular and genetic basis of neurological tumours. Nat Rev Cancer. 2002;2:616–626. doi: 10.1038/nrc866. [DOI] [PubMed] [Google Scholar]
- 34.Holland EC. A mouse model for glioma: biology, pathology, and therapeutic opportunities. Toxicol Pathol. 2000;28:171–177. doi: 10.1177/019262330002800122. [DOI] [PubMed] [Google Scholar]
- 35.Ding H, Shannon P, Lau N, Wu X, Roncari L, Baldwin RL, Takebayashi H, Nagy A, Gutmann DH, Guha A. Oligodendrogliomas result from the expression of an activated mutant epidermal growth factor receptor in a RAS transgenic mouse astrocytoma model. Cancer research. 2003;63:1106–1113. [PubMed] [Google Scholar]
- 36.Weiss WA, Burns MJ, Hackett C, Aldape K, Hill JR, Kuriyama H, Kuriyama N, Milshteyn N, Roberts T, Wendland MF, DePinho R, Israel MA. Genetic determinants of malignancy in a mouse model for oligodendroglioma. Cancer research. 2003;63:1589–1595. [PubMed] [Google Scholar]
- 37.Zhu H, Acquaviva J, Ramachandran P, Boskovitz A, Woolfenden S, Pfannl R, Bronson RT, Chen JW, Weissleder R, Housman DE, Charest A. Oncogenic EGFR signaling cooperates with loss of tumor suppressor gene functions in gliomagenesis. Proc Natl Acad Sci U S A. 2009;106:2712–2716. doi: 10.1073/pnas.0813314106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Chu EC, Tarnawski AS. PTEN regulatory functions in tumor suppression and cell biology. Med Sci Monit. 2004;10:RA235–241. [PubMed] [Google Scholar]
- 39.Cully M, You H, Levine AJ, Mak TW. Beyond PTEN mutations: the PI3K pathway as an integrator of multiple inputs during tumorigenesis. Nat Rev Cancer. 2006;6:184–192. doi: 10.1038/nrc1819. [DOI] [PubMed] [Google Scholar]
- 40.Simpson L, Parsons R. PTEN: life as a tumor suppressor. Exp Cell Res. 2001;264:29–41. doi: 10.1006/excr.2000.5130. [DOI] [PubMed] [Google Scholar]
- 41.Zheng H, Ying H, Yan H, Kimmelman AC, Hiller DJ, Chen AJ, Perry SR, Tonon G, Chu GC, Ding Z, Stommel JM, Dunn KL, Wiedemeyer R, You MJ, Brennan C, Wang YA, Ligon KL, Wong WH, Chin L, DePinho RA. p53 and Pten control neural and glioma stem/progenitor cell renewal and differentiation. Nature. 2008;455:1129–1133. doi: 10.1038/nature07443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.T.C.G.A.R. Network. Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature. 2008;455:1061–1068. doi: 10.1038/nature07385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Tohma Y, Gratas C, Biernat W, Peraud A, Fukuda M, Yonekawa Y, Kleihues P, Ohgaki H. PTEN (MMAC1) mutations are frequent in primary glioblastomas (de novo) but not in secondary glioblastomas. J Neuropathol Exp Neurol. 1998;57:684–689. doi: 10.1097/00005072-199807000-00005. [DOI] [PubMed] [Google Scholar]
- 44.Ichimura K, Ohgaki H, Kleihues P, Collins VP. Molecular pathogenesis of astrocytic tumours. Journal of neuro-oncology. 2004;70:137–160. doi: 10.1007/s11060-004-2747-2. [DOI] [PubMed] [Google Scholar]
- 45.Koul D, Parthasarathy R, Shen R, Davies MA, Jasser SA, Chintala SK, Rao JS, Sun Y, Benvenisite EN, Liu TJ, Yung WK. Suppression of matrix metalloproteinase-2 gene expression and invasion in human glioma cells by MMAC/PTEN. Oncogene. 2001;20:6669–6678. doi: 10.1038/sj.onc.1204799. [DOI] [PubMed] [Google Scholar]
- 46.Xiao A, Yin C, Yang C, Di Cristofano A, Pandolfi PP, Van Dyke T. Somatic induction of Pten loss in a preclinical astrocytoma model reveals major roles in disease progression and avenues for target discovery and validation. Cancer research. 2005;65:5172–5180. doi: 10.1158/0008-5472.CAN-04-3902. [DOI] [PubMed] [Google Scholar]
- 47.Hu X, Pandolfi PP, Li Y, Koutcher JA, Rosenblum M, Holland EC. mTOR promotes survival and astrocytic characteristics induced by Pten/AKT signaling in glioblastoma. Neoplasia. 2005;7:356–368. doi: 10.1593/neo.04595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Holland EC, Celestino J, Dai C, Schaefer L, Sawaya RE, Fuller GN. Combined activation of Ras and Akt in neural progenitors induces glioblastoma formation in mice. Nat Genet. 2000;25:55–57. doi: 10.1038/75596. [DOI] [PubMed] [Google Scholar]
- 49.Uhrbom L, Dai C, Celestino JC, Rosenblum MK, Fuller GN, Holland EC. Ink4a-Arf loss cooperates with KRas activation in astrocytes and neural progenitors to generate glioblastomas of various morphologies depending on activated Akt. Cancer research. 2002;62:5551–5558. [PubMed] [Google Scholar]
- 50.Wei Q, Clarke L, Scheidenhelm DK, Qian B, Tong A, Sabha N, Karim Z, Bock NA, Reti R, Swoboda R, Purev E, Lavoie JF, Bajenaru ML, Shannon P, Herlyn D, Kaplan D, Henkelman RM, Gutmann DH, Guha A. High-grade glioma formation results from postnatal pten loss or mutant epidermal growth factor receptor expression in a transgenic mouse glioma model. Cancer research. 2006;66:7429–7437. doi: 10.1158/0008-5472.CAN-06-0712. [DOI] [PubMed] [Google Scholar]
- 51.Kwon CH, Zhao D, Chen J, Alcantara S, Li Y, Burns DK, Mason RP, Lee EY, Wu H, Parada LF. Pten haploinsufficiency accelerates formation of high-grade astrocytomas. Cancer research. 2008;68:3286–3294. doi: 10.1158/0008-5472.CAN-07-6867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ivanchuk SM, Mondal S, Dirks PB, Rutka JT. The INK4A/ARF locus: role in cell cycle control and apoptosis and implications for glioma growth. Journal of neuro-oncology. 2001;51:219–229. doi: 10.1023/a:1010632309113. [DOI] [PubMed] [Google Scholar]
- 53.Ranuncolo SM, Varela M, Morandi A, Lastiri J, Christiansen S, Bal de Kier Joffe E, Pallotta MG, Puricelli L. Prognostic value of Mdm2, p53 and p16 in patients with astrocytomas. Journal of neuro-oncology. 2004;68:113–121. doi: 10.1023/b:neon.0000027741.19213.99. [DOI] [PubMed] [Google Scholar]
- 54.Nishikawa R, Furnari FB, Lin H, Arap W, Berger MS, Cavenee WK, Su Huang HJ. Loss of P16INK4 expression is frequent in high grade gliomas. Cancer research. 1995;55:1941–1945. [PubMed] [Google Scholar]
- 55.Ichimura K, Schmidt EE, Goike HM, Collins VP. Human glioblastomas with no alterations of the CDKN2A (p16INK4A, MTS1) and CDK4 genes have frequent mutations of the retinoblastoma gene. Oncogene. 1996;13:1065–1072. [PubMed] [Google Scholar]
- 56.Serrano M, Lee H, Chin L, Cordon-Cardo C, Beach D, DePinho RA. Role of the INK4a locus in tumor suppression and cell mortality. Cell. 1996;85:27–37. doi: 10.1016/s0092-8674(00)81079-x. [DOI] [PubMed] [Google Scholar]
- 57.Kamijo T, Zindy F, Roussel MF, Quelle DE, Downing JR, Ashmun RA, Grosveld G, Sherr CJ. Tumor suppression at the mouse INK4a locus mediated by the alternative reading frame product p19ARF. Cell. 1997;91:649–659. doi: 10.1016/s0092-8674(00)80452-3. [DOI] [PubMed] [Google Scholar]
- 58.Lang FF, Miller DC, Koslow M, Newcomb EW. Pathways leading to glioblastoma multiforme: a molecular analysis of genetic alterations in 65 astrocytic tumors. J Neurosurg. 1994;81:427–436. doi: 10.3171/jns.1994.81.3.0427. [DOI] [PubMed] [Google Scholar]
- 59.Reilly KM, Loisel DA, Bronson RT, McLaughlin ME, Jacks T. Nf1;Trp53 mutant mice develop glioblastoma with evidence of strain-specific effects. Nat Genet. 2000;26:109–113. doi: 10.1038/79075. [DOI] [PubMed] [Google Scholar]
- 60.Sorensen SA, Mulvihill JJ, Nielsen A. Long-term follow-up of von Recklinghausen neurofibromatosis. Survival and malignant neoplasms. The New England journal of medicine. 1986;314:1010–1015. doi: 10.1056/NEJM198604173141603. [DOI] [PubMed] [Google Scholar]
- 61.Gutmann DH. New insights into the neurofibromatoses. Curr Opin Neurol. 1994;7:166–171. doi: 10.1097/00019052-199404000-00014. [DOI] [PubMed] [Google Scholar]
- 62.Zhu Y, Guignard F, Zhao D, Liu L, Burns DK, Mason RP, Messing A, Parada LF. Early inactivation of p53 tumor suppressor gene cooperating with NF1 loss induces malignant astrocytoma. Cancer cell. 2005;8:119–130. doi: 10.1016/j.ccr.2005.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Paoloni M, Khanna C. Translation of new cancer treatments from pet dogs to humans. Nat Rev Cancer. 2008;8:147–156. doi: 10.1038/nrc2273. [DOI] [PubMed] [Google Scholar]
- 64.Louis DN. Molecular pathology of malignant gliomas. Annu Rev Pathol. 2006;1:97–117. doi: 10.1146/annurev.pathol.1.110304.100043. [DOI] [PubMed] [Google Scholar]
- 65.Warnke PC, Molnar P, Lapin GD, Kuruvilla A, Groothuis DR. The effects of dexamethasone on transcapillary transport in experimental brain tumors: II. Canine brain tumors. Journal of neuro-oncology. 1995;25:29–38. doi: 10.1007/BF01054720. [DOI] [PubMed] [Google Scholar]
- 66.Whelan HT, Clanton JA, Wilson RE, Tulipan NB. Comparison of CT and MRI brain tumor imaging using a canine glioma model. Pediatr Neurol. 1988;4:279–283. doi: 10.1016/0887-8994(88)90066-5. [DOI] [PubMed] [Google Scholar]
- 67.Dobson JM, Samuel S, Milstein H, Rogers K, Wood JL. Canine neoplasia in the UK: estimates of incidence rates from a population of insured dogs. The Journal of small animal practice. 2002;43:240–246. doi: 10.1111/j.1748-5827.2002.tb00066.x. [DOI] [PubMed] [Google Scholar]
- 68.Snyder SA, Dewhirst MW, Hauck ML. The role of hypoxia in canine cancer. Vet Comp Oncol. 2008;6:213–223. doi: 10.1111/j.1476-5829.2008.00163.x. [DOI] [PubMed] [Google Scholar]
- 69.Keller ET, Madewell BR. Locations and types of neoplasms in immature dogs: 69 cases (1964–1989) J Am Vet Med Assoc. 1992;200:1530–1532. [PubMed] [Google Scholar]
- 70.Heidner GL, Kornegay JN, Page RL, Dodge RK, Thrall DE. Analysis of survival in a retrospective study of 86 dogs with brain tumors. J Vet Intern Med. 1991;5:219–226. doi: 10.1111/j.1939-1676.1991.tb00952.x. [DOI] [PubMed] [Google Scholar]
- 71.Gavin PR, Fike JR, Hoopes PJ. Central nervous system tumors. Semin Vet Med Surg (Small Anim) 1995;10:180–189. [PubMed] [Google Scholar]
- 72.Ingram M, Buckwalter JG, Jacques DB, Freshwater DB, Abts RM, Techy GB, Miyagi K, Shelden CH, Rand RW, English LW. Immunotherapy for recurrent malignant glioma: an interim report on survival. Neurological research. 1990;12:265–273. doi: 10.1080/01616412.1990.11739955. [DOI] [PubMed] [Google Scholar]
- 73.Stubbs JB, Frankel RH, Schultz K, Crocker I, Dillehay D, Olson JJ. Preclinical evaluation of a novel device for delivering brachytherapy to the margins of resected brain tumor cavities. J Neurosurg. 2002;96:335–343. doi: 10.3171/jns.2002.96.2.0335. [DOI] [PubMed] [Google Scholar]
- 74.Chauvet AE, Kesava PP, Goh CS, Badie B. Selective intraarterial gene delivery into a canine meningioma. J Neurosurg. 1998;88:870–873. doi: 10.3171/jns.1998.88.5.0870. [DOI] [PubMed] [Google Scholar]
- 75.Dickinson PJ, LeCouteur RA, Higgins RJ, Bringas JR, Roberts B, Larson RF, Yamashita Y, Krauze M, Noble CO, Drummond D, Kirpotin DB, Park JW, Berger MS, Bankiewicz KS. Canine model of convection-enhanced delivery of liposomes containing CPT-11 monitored with real-time magnetic resonance imaging: laboratory investigation. J Neurosurg. 2008;108:989–998. doi: 10.3171/JNS/2008/108/5/0989. [DOI] [PubMed] [Google Scholar]
- 76.Stoica G, Lungu G, Martini-Stoica H, Waghela S, Levine J, Smith R., 3rd Identification of cancer stem cells in dog glioblastoma. Vet Pathol. 2009;46:391–406. doi: 10.1354/vp.08-VP-0218-S-FL. [DOI] [PubMed] [Google Scholar]
- 77.Higgins RJ, Dickinson PJ, LeCouteur RA, Bollen AW, Wang H, Corely LJ, Moore LM, Zang W, Fuller GN. Spontaneous canine gliomas: overexpression of EGFR, PDGFRalpha and IGFBP2 demonstrated by tissue microarray immunophenotyping. Journal of neuro-oncology. 2010;98:49–55. doi: 10.1007/s11060-009-0072-5. [DOI] [PubMed] [Google Scholar]
- 78.Kirkness EF, Bafna V, Halpern AL, Levy S, Remington K, Rusch DB, Delcher AL, Pop M, Wang W, Fraser CM, Venter JC. The dog genome: survey sequencing and comparative analysis. Science. 2003;301:1898–1903. doi: 10.1126/science.1086432. [DOI] [PubMed] [Google Scholar]
- 79.Hansen K, Khanna C. Spontaneous and genetically engineered animal models; use in preclinical cancer drug development. Eur J Cancer. 2004;40:858–880. doi: 10.1016/j.ejca.2003.11.031. [DOI] [PubMed] [Google Scholar]
- 80.Feitsma H, Kuiper RV, Korving J, Nijman IJ, Cuppen E. Zebrafish with mutations in mismatch repair genes develop neurofibromas and other tumors. Cancer research. 2008;68:5059–5066. doi: 10.1158/0008-5472.CAN-08-0019. [DOI] [PubMed] [Google Scholar]
- 81.Read RD, Cavenee WK, Furnari FB, Thomas JB. A drosophila model for EGFR-Ras and PI3K-dependent human glioma. PLoS Genet. 2009;5:e1000374. doi: 10.1371/journal.pgen.1000374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Witte HT, Jeibmann A, Klambt C, Paulus W. Modeling glioma growth and invasion in Drosophila melanogaster. Neoplasia. 2009;11:882–888. doi: 10.1593/neo.09576. [DOI] [PMC free article] [PubMed] [Google Scholar]