

Abstract
Androgen receptor (AR) is a ligand-activated transcription factor and a validated drug target for all stages of prostate cancer. Antiandrogens compete with physiological ligands for AR ligand-binding domain (LBD). High-throughput screening of a marine natural product library for small molecules that inhibit AR transcriptional activity yielded the furanoditerpenoid spongia-13(16),-14-dien-19-oic acid, designated terpene 1 (T1). Characterization of T1 and the structurally related semi-synthetic analogues (T2 and T3) revealed that these diterpenoids have antiandrogen properties that include inhibition of both androgen-dependent proliferation and AR transcriptional activity by a mechanism that involved competing with androgen for AR LBD and blocking essential N/C interactions required for androgen-induced AR transcriptional activity. Structure activity relationship analyses revealed some chemical features of T1 that are associated with activity and yielded T3 as the most potent analogue. In vivo, T3 significantly reduced the weight of seminal vesicles, which are an androgen-dependent tissue, thereby confirming T3’s on-target activity. The ability to create analogues of diterpenoids that have varying antiandrogen activity represents a novel class of chemical compounds for the analysis of AR ligand-binding properties and therapeutic development.
Keywords: androgen receptor, prostate cancer, antiandrogens, small molecules, diterpenoids
INTRODUCTION
Localized prostate cancer is effectively managed by surgery, radiation, or active surveillance. Unfortunately as many as 47% of these patients will experience recurrence and require systemic therapy for effective management of advanced disease (1). Generally androgen ablation therapy by either chemical or surgical castration is provided to these patients to reduce levels of testicular androgen. After an initial and effective response to castration, resistance will inevitably occur with the development of lethal castration-resistant prostate cancer (CRPC). In spite of reduced levels of testicular androgen, there is compelling evidence to support a role of androgen receptor (AR) in CRPC (2).
AR functions as a ligand-activated transcription factor that regulates gene expression in response to androgen. Binding of androgen to the ligand-binding domain (LBD) of the AR initiates a series of events that involve conformational changes that promote the dissociation of AR from heat shock proteins and chaperones, resulting in phosphorylation, nuclear translocation, and AR dimerization. In the nucleus, AR directly binds to specific DNA sequences known as androgen response elements (ARE) that are present in the regulatory regions of androgen-regulated genes, and DNA-bound AR recruits co-activators and assembly of transcriptional machinery to initiate transcription. Genes regulated by AR contribute to proliferation and survival of prostate cancer (3). Thus, expression and function of AR are essential for proliferation and growth of prostate cancer cells (4). Importantly, AR signaling remains functionally active in CRPC (5, 6). Molecular mechanisms suspected to be involved in the continued AR activity in CRPC include: 1) AR gene amplification and/or increased expression of AR (7–9); 2) gain-of-function mutations in AR that result in activation by non-androgenic steroidal ligands and antiandrogens (10, 11); 3) expression of constitutively active AR splice variants that lack ligand-binding domain(12); 4) ligand-independent activation of AR by alternative pathways such as the cAMP-dependent protein kinase, interleukin-6, and other factors (13–17); 5) increased expression of AR coactivators (18–21); and 6) intratumoral de novo synthesis of androgens (22). Together these findings suggest that targeting AR is a viable approach for clinical management of all stages of prostate cancer including CRPC.
AR is targeted indirectly by androgen ablation therapy that reduces androgen that binds to the AR LBD. LHRH analogues, orchiectomy, and inhibitors of androgen synthesis are standard approaches used clinically to reduce levels of androgen. Abiraterone is an irreversible inhibitor of CYP17 that is involved in androgen synthesis. Abiraterone increases survival by 3.9 months in CRPC patients who have previously failed androgen ablation and docetaxel therapies (23). Antiandrogens competitively bind to AR LBD to antagonize the action of androgens and thereby attenuate AR transcriptional activity. Non-steroidal antiandrogens used clinically for prostate cancer include bicalutamide (BIC), flutamide, nilutamide, and enzalutamide (MDV3100). The Phase 3 AFFIRM trial showed that enzalutamide has a median overall survival advantage of 4.8-months compared to placebo in patients with CRPC post docetaxel treatment (24). In spite of the survival benefits of a potent antiandrogen such as enzalutamide, all antiandrogens ultimately fail. However, once an antiandrogen fails, changing to an alternative second line antiandrogen can be clinically effective with improved survival (25, 26) thereby supporting the quest to discover additional antiandrogens for the clinical management of CRPC. Here we report that the furanoditerpenoid spongia-13(16),-14-dien-19-oic acid (T1) and the two semisynthetic derivatives T2 and T3 are antiandrogens.
MATERIALS AND METHODS
Cell lines, proliferation assay, and transfection for luciferase assay
LNCaP human prostate cancer cells were maintained in RPMI 1640 supplemented with 10% (v/v) fetal bovine serum (FBS) (Invitrogen™ by Life Technologies, Carlsbad, CA). PC3 cells were maintained in DMEM with 5% (v/v) FBS. CV-1 monkey kidney cells were maintained in MEM medium with 10% (v/v) FBS and 1% L-glutamine. VCaP cells were maintained in DMEM containing 10% (v/v) FBS. All four cell lines were obtained from American Type Culture Collection (Rockville, MD). After acquiring these cell lines, the cells were frozen at −80C° and were resuscitated immediately before experiments. LNCaP95, an androgen independent cell line derived from the parental LNCaP cells, were maintained in RPMI 1640 containing 10% (v/v) dextran-coated charcoal-stripped serum. We obtained the LNCaP95 cells from Dr. Stephen R. Plymate (University of Washington), who has recently published studies performed on these cells (27). All cells are maintained in culture no more than 10–15 passages, and regularly tested to ensure they are mycoplasma-free. No cell line authentication was conducted in our lab. Cellular proliferation assay, and plasmids and transfection for luciferase assay have been described previously (28).
Endogenous expression of androgen-regulated genes
LNCaP cells (180,000 cells/well) in 6-well plates were incubated for 48 hours in serum-free RPMI prior to pre-treatment for 1 hour with DMSO vehicle or small molecules at 10 µM before addition of 1 nM R1881. VCaP cells (300,000 cells/well) were plated in 6-well plates in DMEM with 5% dextran-coated charcoal-stripped serum. Two days later, small molecules and R1881 were added to VCaP cells in the same manner as LNCaP. Total RNA was isolated after 48 hours (for LNCaP) and 16 hours (for VCaP) by using RNeasy® Micro Kit (QIAGEN, Valencia, CA), and subsequently reverse transcribed to cDNA by SuperScript®III First-Strand Synthesis System for RT-PCR (Invitrogen™). Diluted cDNA and gene-specific primers were combined with Platinum ® SYBR® Green qPCRSuperMix-UDG with ROX (Invitrogen™), and the transcripts were measured by quantitative real-time (qRT)-PCR (ABI PRISM®, Applied Biosystems by Life Technologies, Carlsbad, CA). qRT-PCR was performed separately in triplicates for each biological sample. Expression levels were normalized to housekeeping gene glyceraldehyde-3-phosphate dehydrogenase (GAPDH). Primers used were previously described (28–31). Sequence of primers used is listed: GAPDH: F’: 5’-CTGACTTCAACAGCGACACC-3’ and R’: 5’-TGCTGTAGCCAAATTCGTTG-3’; PSA: F’: 5’-CCAAGTTCATGCTGTGTGCT-3’ and R’: 5’-CCCATGACGTGATACCTTGA-3’; KLK2: F’: 5’-TGT GTG CTA GAG CTT ACT CTG A-3’ and R’: 5’-CCA CTT CCG GTA ATG CAC CA-3’; FKBP5: F’: 5’-CGCAGGATATACGCCAACAT-3’ and R’: 5’-GAAGTCTTCTTGCCCATTGC-3’; TMPRSS2: F’: 5’-GGACAGTGTGCACCTCAAAGA-3’ and R’: 5’-TCCCACGAGGAAGGTCCC-3’. AR fl: F’: 5’-CCATCTTGTCGTCTTCGGAAATGTTAT-3’ and R’: 5’-AGCTTCTGGGTTGTCTCCTCAGTGG-3’.
In vitro ligand binding assay
Androgen Receptor, Progesterone Receptor, and Estrogen Receptor -alpha PolarScreen Competitor Assay kits (Invitrogen™) were employed according to the manufacturer’s protocol. Serial dilution was done for each small molecule and solvent was compensated to ensure equal volume of DMSO and ethanol in each sample. Fluorescence polarization at excitation wavelength 470 nm and emission at 535 nm were measured in Greiner 384 black clear bottom plates by using Infinite ® M1000 (TECAN, Grödig, Austria).
Animal studies
Twelve-week-old male NOD-SCID intact mice were maintained in the Animal Care Facility at the British Columbia Cancer Agency Research Center. All animal experiments were approved by the University of British Columbia Animal Care Committee and strictly followed the ethical regulatory standards. Mice were divided into three groups: DMSO control (n = 10), bicalutamide (BIC, n = 8), and T3 (n = 8). BIC and T3 were administered by oral gavage at 10 mg/kg daily for a total of 13 doses. Initial and final body weights were recorded for each animal. Mice were sacrificed 14 days after the first treatment, and tissues were collected and weighed. Prism (GraphPad Software) was used to generate Whisker plots for the weight of seminal vesicles and testes. Change in body weight was calculated from the difference between initial and final weight as a percentage of initial weight. Student’s t test was performed.
RESULTS
Diterpenoids reduce androgen-dependent proliferation of prostate cancer cells
Diterpenoids T1, T2, and T3 share an unsubstituted tricylic perhydrophenanthrene ABC ring system fused to a five membered D ring with the physiological AR ligand dihydrotestosterone (DHT) as well as the synthetic androgen R1881 (Figure 1A). Bicalutamide (BIC) is a non-steroidal antiandrogen used clinically to block AR activity and subsequent androgen-dependent proliferation of prostate cancer cells (32). To evaluate T1, T2, and T3 on androgen-dependent proliferation, LNCaP human prostate cancer cells that express functional full-length AR, although mutated in the LBD, were treated with vehicle, or 10 µM of either BIC (positive control), T1, T2, or T3. As expected, 0.1 nM of R1881 led to increased proliferation in LNCaP cells (Figure 1B). Both T1 and T3 inhibited androgen-dependent proliferation with T1 being significantly more effective than T3 and comparable to BIC, while T2 had no effect (Figure 1B). To determine the specificity of T1 and T3 for attenuating AR-dependent proliferation, PC3 human prostate cancer cells were employed because these cells do not express a functional AR. Consistent with PC3 cells not being dependent on AR activity for growth, 0.1 nM R1881 did not alter their proliferation. Importantly, none of the diterpenoids inhibited proliferation of these cells (Figure 1C), suggesting that these diterpenoids are not generally toxic. PC3 cells treated with each diterpenoid showed increasing proliferation over 24, 48, and 72-hour time points with no difference as compared to the vehicle control (Supplemental Figure 1). Microscopic analysis of LNCaP cells exposed to 10 µM T1, or T2, or T3 for 4 days also revealed no signs of cytotoxicity (Figure 1D). Viability assays showed no indication of cytotoxicity in LNCaP, VCaP that have a wild-type AR, or PC3 cells treated for 4, 5, and 3 days, respectively, with diterpenoids or bicalutamide (Supplemental Fig 2). These viability assays mirrored proliferation data shown here and were consistent with previous studies that androgen-dependent proliferation of VCaP cells that express wild-type AR are poorly inhibited with bicalutamide (33). Together these data suggest that T1 and T3 may have specificity for cells that are dependent on AR for growth and that these diterpenoids are not generally cytotoxic.
Figure 1. Inhibition of AR-dependent proliferation by diterpenoids.

(A) Chemical structures of diterpenoids (T1, T2, and T3), DHT, synthetic androgen R1881, and BIC. (B) Proliferation of LNCaP cells after 4 days of treatment, and (C) PC3 cells after 3 days of treatment. Proliferation assays employed BrdU incorporation. Both cell lines were pre-treated for 1 hour with 10 µM of BIC, or T1, or T2, or T3, or vehicle (VEH), prior to the addition of 0.1 nM R1881 (black bars) or ethanol control (white bars). Error bars represent the mean ± SEM, n = 4 separate experiments. Student’s t test: *p < 0.05; **p < 0.01; ***p < 0.001. (D) Morphology of LNCaP cells four days after treament of 10 uM of indicated compounds with ethanol (ETOH) vehicle or 0.1 nM R1881.
Diterpenoids inhibit AR transcriptional activity
The chemical structures, together with the observed reduction of androgen-dependent proliferation of AR-positive LNCaP cells, suggest that T1 and T3 may be inhibitors of AR. To test this hypothesis, the effects of diterpenoids on the transcriptional activity of endogenous AR in LNCaP cells was tested using the AR-driven ARR3-LUC reporter gene construct. Androgen-induced ARR3-LUC reporter activity was potently blocked to approximately baseline levels by T1 and T3 at 10 µM concentration and this inhibition was comparable to that achieved with an equal concentration of BIC (Figure 2A). Interestingly, 10 µM T2 also significantly inhibited AR transcriptional activity although it was less potent than BIC, T1 or T3 with only 50% inhibition. Dose-response curves (Figure 2B) estimated the IC50 values for inhibition of endogenous AR transcriptional activity in LNCaP cells to be 0.21 µM for BIC, 4.2 µM for T10.41 µM for T3, and greater than or equal to 10 µM for T2. LNCaP95 cells, an androgen-independent subline of LNCaP cells, has a functional AR that can be activated by androgen but its proliferation is not altered by androgen (27). R1881 weakly induced ARR3-LUC reporter activity in LNCaP95 cells by approximately 4-fold over control levels, which was significantly attenuated by BIC, T1 and T3 (Supplemental Fig 3A). T2 had no significant effect on androgen-induced ARR3-LUC activity in this cell line. LNCaP95 cell growth/viability was not altered by R1881, BIC or any of the diterpenoids (Supplemental Fig 3A). Together these data reveal that these diterpenoids were effective in blocking transcriptional activity of endogenous AR in LNCaP cells with T2 being less potent than T1 and T3.
Figure 2. Inhibition of AR transcriptional activity.

(A) LNCaP cells were transfected with ARR3-LUC and treated with 10 µM of BIC, or T1, or T2, or T3, or vehicle (VEH) in the absence or presence of 1 nM R1881 (-- or +) for 48 hours. (B) LNCaP cells transfected with ARR3-LUC were treated with diterpenoids at 10 µM, 1 µM, 0.1 µM, and 0 µM in the presence of 1 nM R1881 for 48 hours. (C) CV1 cells were cotransfected with pSV-AR0, the expression vector for wild-type AR, and ARR3-LUC, prior to treatment with 10 µM of BIC, or T1, or T2, or T3, or VEH in the absence or presence of 1 nM R1881 (-- or +) for 24 hours. (D) CV1 cells transiently cotransfected with pSV-AR0 (wild-type AR) and ARR3-LUC were treated with diterpenoids at 10 µM, 5 µM, 2.5 µM, 1 µM, and 0 µM in the presence of 1 nM R1881 for 24 hours. Error bars represent the mean ± SEM, n = 3 independent experiments. Student’s t test: *p < 0.05; **p < 0.01; ***p < 0.001.
Endogenous AR in LNCaP cells has the T877A mutation in the AR LBD (AR-T877A) that alters both ligand specificity and affinity (34). Therefore, to investigate the effect of diterpenoids on wild-type AR transcriptional activity, CV1 cells that do not express endogenous AR were transiently co-transfected with ARR3-LUC reporter and an optimized amount of an expression vector for wild-type human AR that resulted in expression of AR protein within endogenous levels measured in prostate cancer cells (Supplemental Fig 4). Surprisingly, at 10 µM, T1 and T2 had no significant effect on the transcriptional activity of wild-type AR activated by androgen, while T3 caused potent inhibition that was comparable to BIC (Figure 2C). The IC50 values for wild-type AR in CV1 cells were 0.39 µM for BIC and 0.96 µM for T3, whereas T1 had an IC50 greater than 30 µM (Figure 2D) and the IC50 for T2 could not be calculated due to only minor inhibition in this concentration range. These IC50 values were consistent with the trends observed for the potency of compounds on wild-type AR observed at the single concentration of 10 µM (Figure 2C). Importantly, the presence of a furan or the reduced tetrahyrofuran in the chemical structures of the diterpenoids appear to alter activities obtained with wild-type AR and mutant AR-T877A as measured for T1 and T2.
Diterpenoids inhibit endogenous expression of androgen-regulated genes
AR regulates the transcription of hundreds of genes in prostate cells with several well- characterized genes such as PSA, KLK2, FKBP5 and TMPRSS2 shown to have functional AREs (35–38). To test the effects of diterpenoids on endogenous expression of androgen-regulated genes, RT-QPCR was employed to measure the levels of these transcripts in cells exposed to 10 µM of each diterpenoid. First LNCaP cells with mutated AR were tested. Among the three compounds, T1 was consistently the most effective inhibitor of androgen-induced gene expression with a potency comparable to, or better than BIC (Figure 3A). T3 also inhibited androgen-induced gene expression, whereas T2 showed no significant effects in blocking androgen-induced expression of this set of genes. Importantly, in the absence of androgen, T1 and T2 decreased basal levels of transcripts while T3 significantly increased basal expression suggesting that T3 may be a partial agonist of mutated AR-T877A similar to BIC (39).
Figure 3. Reduction of endogenous expression of androgen-regulated genes.

(A) Levels of mRNA of four androgen-regulated genes (PSA, KLK2, FKBP5, and TMPRSS2) in LNCaP cells were measured by qRT-PCR. LNCaP cells were pre-treated for 1 hour with 10 µM of BIC, or T1, or T2, or T3, or vehicle (VEH), prior to the addition of 1 nM R1881 (black bars) or ethanol control (white bars) for 48 hr. Endogenous expression of AR in LNCaP cells under the same treatment conditions as mentioned above were detected by (B) qRT-PCR for AR mRNA levels and (C) Western blot analysis for AR protein levels. (D) Levels of PSA, FKBP5, and TMPRSS2 mRNAs were also measured in VCaP cells. Levels of expression of each gene were normalized to levels of GAPDH mRNA. Error bars represent the mean ± SEM, n = 3 separate experiments. Student’s t test: *p < 0.05; **p < 0.01; ***p < 0.001.
Levels of transcripts of androgen-regulated genes are sensitive to changes in the levels of AR. Therefore levels of AR were measured in LNCaP cells treated with the diterpenoids. In the absence of androgen, levels of AR mRNA were significantly decreased by both T1 and T3 (Figure 3B). The decrease in AR mRNA by T1 was consistent with the observed decrease in AR protein by this compound in the absence of androgen by a currently unknown mechanism (Figure 3C). However, in the presence of androgen, the diterpenoids had no significant effect on AR levels thereby suggesting that inhibition of androgen-induced transcription of AR-regulated genes is not by a mechanism that involves decreased levels of AR.
Analysis of AR-regulated gene expression was next examined in VCaP cells that endogenously express wild-type AR. As expected, levels of PSA transcript were weakly increased in response to androgen (Figure 3D) and neither BIC or any of the diterpenoids significantly reduced these levels. However, levels of transcript for FKBP5 and TMPRSS2 were robustly increased in response to androgen and both were significantly blocked by BIC and T3 while T1 and T2 had no significant effect. Together these data are consistent with the trends observed with wild-type AR using a reporter gene assay in CV1 cells (Figure 2C).
Diterpenoids bind to AR LBD and inhibit AR N/C interaction
The chemical structures of diterpenoids resemble steroids thereby suggesting that they may physically interact with AR LBD. To determine if these diterpenoids bind the AR LBD and if so, to assess their binding affinities to the AR LBD, we performed in vitro ligand competitor assays using recombinant wild-type AR LBD. R1881 had strong affinity for AR LBD with a half maximal effective concentration (EC50) calculated at 5.3 nM (Figure 4A). T1 showed relatively weaker binding with an EC50 of 27.1 µM, whereas T2 was much weaker at 398.4 µM. However, T3 which has the carboxylic acid functionality present in T1 reduced to a primary alcohol displayed improved binding with an EC50 of 1.6 µM, which was comparable with that achieved by BIC at 1.1 µM. EC50s for diterpenoids on wild-type recombinant AR LBD were consistent with trends observed in cells with wild-type AR at 10 uM diterpenoid (Figure 2C) as well as the IC50s for wild-type AR transcriptional activity (Figure 2D).
Figure 4. Binding affinities for AR and inhibition of AR N/C interaction.

(A) Recombinant AR LBD was tested for the binding affinity of the diterpenoids by measuring fluorescence polarization (mP) with an excitation wavelength of 470 nm and emission wavelength of 535 nm. Serial dilution was performed for the test compounds using synthetic androgen R1881 as a positive control. A representative plot from at least 3 independent assays is shown, and error bars represent the mean ± SEM. (B) CV1 cells transfected with 5XGAL4Luc reporter vector, VP16-AR-NTD, and GAL4DBD-AR-LBD (wild-type) were pre-treated for 1 hour with 10 µM of BIC, or T1, or T2, or T3, or vehicle (VEH), prior to the addition of 1 nM synthetic androgen R1881 for 24 hours. Error bars represent the mean ± SEM, n=5 independent experiments. Student’s t test: *p < 0.05; **p < 0.01; ***p < 0.001.
AR transcriptional activity in response to androgen requires interaction between the N-terminal domain and the C-terminus LBD of AR (N/C interaction) (40). Antiandrogen such as BIC inhibits androgen-induced N/C interaction (41). To determine if diterpenoids also inhibit androgen-induced AR N/C interaction, the mammalian two-hybrid system was employed using wild-type LBD. T1 and T3 significantly inhibited androgen-induced N/C interaction (Figure 4B). Inhibition of N/C interaction by T3 was comparable to that achieved with BIC. T2 had no effect, which was consistent with its poor affinity for wild-type AR. Together, these data suggest that T1 and T3 inhibit N/C interaction of AR by directly binding to AR LBD.
Specificity of Diterpenoids
AR is a member of the steroid receptor superfamily and its LBD shares substantial sequence identity with the LBDs of progesterone receptor (PR) and glucocorticoid receptor (GR) at 55% and 51% respectively. Some antiandrogens such as BIC are potent inhibitors of PR transcriptional activity (42). To test receptor specificity of diterpenoids for AR, the transcriptional activities of PR and GR were examined using their respective reporter gene constructs transiently transfected into LNCaP cells, which do not endogenously express PR and GR. Induction of PR transcriptional activity by its ligand 4-pregnene-3,20 dione (Preg) was inhibited by BIC and RU486, a potent PR inhibitor, as well as by T1 and T3, whereas T2 showed no significant effect (Figure 5A). The three diterpenoids and BIC had no effect on the transcriptional activity of GR induced by dexamethasone (DEX) (Figure 5B). These results suggest that diterpenoids have some specificity for AR and PR, and that they do not have general inhibitory effects on transcription and translation. Consistent with T1 and T2 inhibiting PR transcriptional activity, these compounds competed for the PR LBD as shown using the in vitro ligand competitor assay with recombinant human PR LBD (Figure 5C). RU486 had strong binding affinity to PR LBD with an EC50 of 45 nM, comparable to Preg at 35 nM, whereas BIC and T1 had similar EC50 values at 5,100 and 5,600 nM respectively. T3 had a binding affinity to PR LBD at ~500 nM, which agreed with its potent inhibition on PR transcriptional activity (Figure 5A). T2 EC50 could not be assessed due to low activity and poor solubility at high concentrations. Consistent with estrogen receptor alpha (ERα) having little sequence homology to AR, none of the AR ligands (R1881, BIC and the diterpenoids) were efficient in competing with fluoromone for ERα (Figure 5D).
Figure 5. Effects of diterpenoids on transcriptional activity of related steroid receptors.

(A) LNCaP cells were transiently cotransfected with an expression plasmid for full-length human progesterone receptor-beta (PRβ) and a PR-driven (PRE) luciferase reporter. Cells were pre-treated for 1 hour with 10 µM of BIC, or RU486, or each diterpenoid, or vehicle (VEH), prior to the addition of 10 nM 4-pregnene-3,20 dione (Preg) as indicated by black bars (+), or ethanol control (white bars and --). (B) LNCaP cells were transiently cotransfected with an expression plasmid for full-length human glucocorticoid receptor (GR) and GR-driven (GRE) reporter and pre-treated with 10 µM of BIC, or diterpenoids, or VEH, prior to the addition of 10 nM dexamethasone (Dex) as indicated by black bars (+), or ethanol control (white bars and --). 48 hours after treatment, luciferase assay was performed and relative luminescent unit (RLU) per minute was measured and normalized to protein concentration. Error bars represent the mean ± SEM, n ≥ 3 independent experiments. Student’s t test: *p < 0.05; **p < 0.01; ***p < 0.001. (C) Recombinant human PR LBD and (D) recombinant human full-length ERα were tested for the binding affinity of the diterpenoids by measuring fluorescence polarization (mP) with an excitation wavelength of 470 nm and emission wavelength of 535 nm. Serial dilution was performed for the testing compounds. A representative plot from at least 3 independent assays is shown for each receptor.
In vivo effects of diterpenoid T3 on androgen-dependent tissue
In vivo, blocking the androgen axis results in atrophy of androgen-dependent tissues such as the seminal vesicles and thereby provides an indication of on-target activity (43, 44). Of the 3 diterpenoids tested, T3 was consistently the most potent inhibitor of wild-type AR and was therefore chosen for in vivo evaluation for effects on benign tissue that would harbor wild-type AR. Mature male mice treated with 13 daily oral doses of T3 had a significant decrease in seminal vesicle weight (Figure 6A) which was consistent with the properties of an antiandrogen. No changes in the testes weight (Figure 6B) or body weight (Figure 6C) were observed thereby suggesting a relatively specific effect of T3 on androgen-dependent tissue as opposed to it merely being toxic.
Figure 6. Efficacy of diterpenoids in vivo.
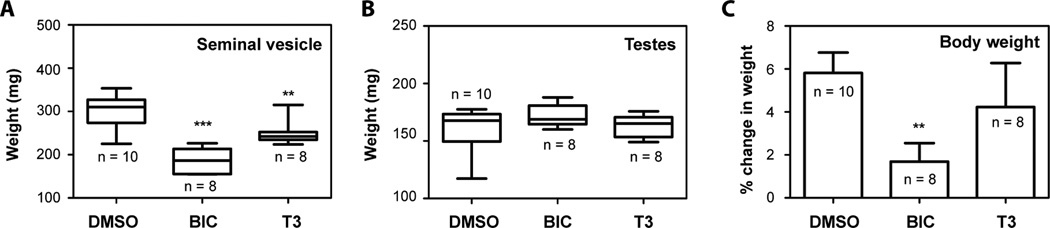
Male NOD-SCID intact mice were treated with DMSO, or BIC, or T3 at 10 mg/kg body weight for two weeks. Tissues were collected, and their weights were measured. The weight of seminal vesicle (A) and testes (B) were plotted as Whisker plots. Body weight (C) was plotted as the percentage change between initial and final weights. Error bars represent the mean ± SEM, n = 8 to 10 as indicated. Student’s t test: *p < 0.05; **p < 0.01; ***p < 0.001.
DISCUSSION
AR is a ligand-activated transcription factor that plays an important role in prostate cancer. Drug development to block AR transcriptional activity for the treatment of this disease has yielded steroidal and non-steroidal antiandrogens. Here T1 was originally isolated from a crude extract of a marine sponge based upon its activity in our screen for inhibitors of AR transcriptional activity. T1 belongs to a family of compounds called spongian diterpenoids, which are commonly found in marine sponges and shell-less mollusks that feed on the sponges (45). T2 and T3 are semisynthetic compounds produced by reducing either the furan or 17β carboxylic acid functionalities in T1, respectively. Spongian diterpenoids have been previously investigated for in vitro antiviral and antitumor activities (46). This is the first report that diterpenoids have antiandrogen activity as shown by: 1) inhibition of androgen-dependent proliferation while having no effect on cells that do not depend on AR for growth and survival; 2) blocking AR transcriptional activity measured by reporter gene constructs and endogenous gene expression; 3) specificity for AR with no effect on GR transcriptional activity and no effect on the proliferation of cells that do not express functional AR; 4) competing with androgen for the LBD; 5) blocking androgen-induced N/C interaction; and 6) reducing the weight of androgen-dependent tissue in vivo while having no effect on body weight. The mechanism of action of diterpenoids to inhibit AR transcriptional activity involved binding to the AR LBD as shown in vitro ligand competition binding assays. Consistent with other antiandrogens for blocking N/C interaction induced by androgen (47), the diterpenoids also inhibited this interaction.
AR mediates the effects of androgen, which is the major mitogen for prostate cancer thereby providing the rationale for targeting AR for the treatment of prostate cancer. Here, furano diterpenoids T1 and T3 inhibited androgen-dependent proliferation of LNCaP human prostate cancer cells that are androgen sensitive and express functional AR. The antiproliferative effect of T1 and T3 was not observed in PC3 cells that do not have a functional AR thereby suggesting potential specificity of these diterpenoids for cells that are dependent on AR for growth and survival. Consistent with this interpretation, administration of T3 to mature male mice reduced the weight of androgen-dependent seminal vesicles while having no effect on body weight. T3 had no effect on the weight of testis, similar to BIC and consistent with other inhibitors of the androgen axis (48). Other data supporting specificity of the diterpenoids for blocking AR transcriptional activity include that they did not broadly inhibit transcription and translation or affect all steroid hormone receptors as indicated by a lack of effect on the transcriptional activity of the closely structurally related GR. However, similar to BIC, T1 and T3 inhibited PR transcriptional activity, while T2 did not. PR is not known to be associated with any essential biological function in mature men and BIC, a PR inhibitor, has been used clinically for many years with an acceptable safety profile (32). Consistent with its relatively modest biological activity, T2 showed very weak binding affinity to AR and PR. However, both T1 and T3 were shown to bind to AR and PR, T3 clearly demonstrated stronger binding affinity, which was comparable to BIC for AR. Together these data suggest these spongian diterpenoids are novel small molecule inhibitors of AR with T3 having the best potency of the 3 compounds.
LNCaP cells have a mutated AR LBD (T877A), which reduces ligand specificity as well as alters ligand affinity, and dissociation rates (34, 49). Thus inhibitory properties of compounds that bind to AR LBD can be altered by mutations within this domain. When comparing the properties of the 3 diterpenoids on wild-type versus mutated AR-T877A, T1 activity was the most affected by this mutation. T1 had no effect on wild-type AR transcriptional activity with accompanying poor inhibition of N/C interaction and higher IC50 and EC50 values. However, with the mutated AR-T877A in LNCaP cells, T1 became a potent inhibitor of both androgen dependent proliferation and AR transcriptional activity with an IC50 of 4.2 µM. Generally, T2 had poor activity while T3 had good activity regardless of whether the AR had the T877A mutation.
T3 was synthesized with the intention of maintaining the furan ring in T1 but modifying the 17β carboxylic acid group in order to determine if a compound could be generated that had better binding affinity to AR and consequently be a more potent inhibitor. From in vitro ligand competition binding assays, T3 indeed demonstrated approximately 16-fold higher affinity to AR LBD with an EC50 of 1.6 µM as compared with T1 (27.1 µM), making T3 comparable to BIC (1.1 µM). Improved affinity of T3 compared to T1 probably involves specific interactions with a set of well-conserved amino acid residues in the LBD of AR. Hydrophobic interactions between the perhydrophenanthrene skeleton of the ligand and the amino acid residues within the ligand-binding pocket are critical for binding, as well as hydrogen-bonding which would impact affinity. Comparing with other non-steroidal small molecule inhibitors of the AR LBD such as BIC and enzalutamide, the diterpenoids represent a novel class of chemical compounds with antiandrogen activity. Thus further structure activity relationship studies are ongoing with the intention of developing more potent derivatives of T1 and T3 with optimized drug-like qualities for their potential clinical application for prostate cancer and/or other diseases involving the androgen axis.
Supplementary Material
Acknowledgments
GRANT SUPPORT: Canadian Institutes of Health Research (M.D. Sadar: MOP79308, M.D. Sadar/R.J. Andersen: MOP89902) and the U.S. National Cancer Institutes (M.D. Sadar/R.J. Andersen: 2R01 CA105304) and the Canadian Cancer Society (R.J. Andersen: 017289). We are grateful to the Beedie Foundation for partial salary support for Y.C. Yang.
Abbreviation list
- AR
androgen receptor
- ER
estrogen receptor
- GR
glucocorticoid receptor
- PR
progesterone receptor
- LBD
ligand-binding domain
- EC50
half maximal effective concentration
- IC50
half maximal inhibitory concentration
- BIC
bicalutamide
- CRPC
castration resistant prostate cancer
Footnotes
Disclosure of Potential Conflicts of Interest: No potential conflicts of interest.
REFERENCES
- 1.Rogers CG, Khan MA, Craig-Miller M, Veltri RW, Partin AW. Natural history of disease progression in patients who fail to achieve an undetectable prostate-specific antigen level after undergoing radical prostatectomy. Cancer. 2004;101:2549–2556. doi: 10.1002/cncr.20637. [DOI] [PubMed] [Google Scholar]
- 2.Attard G, Cooper CS, de Bono JS. Steroid hormone receptors in prostate cancer: a hard habit to break? Cancer Cell. 2009;16:458–462. doi: 10.1016/j.ccr.2009.11.006. [DOI] [PubMed] [Google Scholar]
- 3.Heinlein CA, Chang C. Androgen receptor in prostate cancer. Endocrine Reviews. 2004;25:276–308. doi: 10.1210/er.2002-0032. [DOI] [PubMed] [Google Scholar]
- 4.Zegarra-Moro OL, Schmidt LJ, Huang H, Tindall DJ. Disruption of androgen receptor function inhibits proliferation of androgen-refractory prostate cancer cells. Cancer Research. 2002;62:1008–1013. [PubMed] [Google Scholar]
- 5.Yuan X, Balk SP. Mechanisms mediating androgen receptor reactivation after castration. Urologic Oncology. 2009;27:36–41. doi: 10.1016/j.urolonc.2008.03.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wang Q, Li W, Zhang Y, Yuan X, Xu K, Yu J, et al. Androgen receptor regulates a distinct transcription program in androgen-independent prostate cancer. Cell. 2009;138:245–256. doi: 10.1016/j.cell.2009.04.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Visakorpi T, Hyytinen E, Koivisto P, Tanner M, Keinanen R, Palmberg C, et al. In vivo amplification of the androgen receptor gene and progression of human prostate cancer. Nature Genetics. 1995;9:401–406. doi: 10.1038/ng0495-401. [DOI] [PubMed] [Google Scholar]
- 8.Koivisto P, Kononen J, Palmberg C, Tammela T, Hyytinen E, Isola J, et al. Androgen receptor gene amplification: a possible molecular mechanism for androgen deprivation therapy failure in prostate cancer. Cancer Research. 1997;57:314–319. [PubMed] [Google Scholar]
- 9.Chen CD, Welsbie DS, Tran C, Baek SH, Chen R, Vessella R, et al. Molecular determinants of resistance to antiandrogen therapy. Nature Medicine. 2004;10:33–39. doi: 10.1038/nm972. [DOI] [PubMed] [Google Scholar]
- 10.Zhao XY, Malloy PJ, Krishnan AV, Swami S, Navone NM, Peehl DM, et al. Glucocorticoids can promote androgen-independent growth of prostate cancer cells through a mutated androgen receptor. Nature Medicine. 2000;6:703–706. doi: 10.1038/76287. [DOI] [PubMed] [Google Scholar]
- 11.Hara T, Miyazaki J, Araki H, Yamaoka M, Kanzaki N, Kusaka M, et al. Novel mutations of androgen receptor: a possible mechanism of bicalutamide withdrawal syndrome. Cancer Research. 2003;63:149–153. [PubMed] [Google Scholar]
- 12.Haile S, Sadar MD. Androgen receptor and its splice variants in prostate cancer. Cellular and Molecular Life Sciences. 2011;68:3971–3981. doi: 10.1007/s00018-011-0766-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Culig Z, Hobisch A, Cronauer MV, Radmayr C, Trapman J, Hittmair A, et al. Androgen receptor activation in prostatic tumor cell lines by insulin-like growth factor-I, keratinocyte growth factor, and epidermal growth factor. Cancer Research. 1994;54:5474–5478. [PubMed] [Google Scholar]
- 14.Nazareth LV, Weigel NL. Activation of the human androgen receptor through a protein kinase A signaling pathway. The Journal of Biological Chemistry. 1996;271:19900–19907. doi: 10.1074/jbc.271.33.19900. [DOI] [PubMed] [Google Scholar]
- 15.Sadar MD. Androgen-independent induction of prostate-specific antigen gene expression via cross-talk between the androgen receptor and protein kinase A signal transduction pathways. The Journal of Biological Chemistry. 1999;274:7777–7783. doi: 10.1074/jbc.274.12.7777. [DOI] [PubMed] [Google Scholar]
- 16.Ueda T, Bruchovsky N, Sadar MD. Activation of the androgen receptor N-terminal domain by interleukin-6 via MAPK and STAT3 signal transduction pathways. The Journal of Biological Chemistry. 2002;277:7076–7085. doi: 10.1074/jbc.M108255200. [DOI] [PubMed] [Google Scholar]
- 17.Blaszczyk N, Masri BA, Mawji NR, Ueda T, McAlinden G, Duncan CP, et al. Osteoblast-derived factors induce androgen-independent proliferation and expression of prostate-specific antigen in human prostate cancer cells. Clinical Cancer Research. 2004;10:1860–1869. doi: 10.1158/1078-0432.ccr-0974-3. [DOI] [PubMed] [Google Scholar]
- 18.Ngan ES, Hashimoto Y, Ma ZQ, Tsai MJ, Tsai SY. Overexpression of Cdc25B, an androgen receptor coactivator, in prostate cancer. Oncogene. 2003;22:734–739. doi: 10.1038/sj.onc.1206121. [DOI] [PubMed] [Google Scholar]
- 19.Debes JD, Sebo TJ, Lohse CM, Murphy LM, Haugen DA, Tindall DJ. p300 in prostate cancer proliferation and progression. Cancer Research. 2003;63:7638–7640. [PubMed] [Google Scholar]
- 20.Comuzzi B, Nemes C, Schmidt S, Jasarevic Z, Lodde M, Pycha A, et al. The androgen receptor co-activator CBP is up-regulated following androgen withdrawal and is highly expressed in advanced prostate cancer. The Journal of Pathology. 2004;204:159–166. doi: 10.1002/path.1609. [DOI] [PubMed] [Google Scholar]
- 21.Agoulnik IU, Vaid A, Bingman WE, 3rd, Erdeme H, Frolov A, Smith CL, et al. Role of SRC-1 in the promotion of prostate cancer cell growth and tumor progression. Cancer Research. 2005;65:7959–7967. doi: 10.1158/0008-5472.CAN-04-3541. [DOI] [PubMed] [Google Scholar]
- 22.Montgomery RB, Mostaghel EA, Vessella R, Hess DL, Kalhorn TF, Higano CS, et al. Maintenance of intratumoral androgens in metastatic prostate cancer: a mechanism for castration-resistant tumor growth. Cancer Research. 2008;68:4447–4454. doi: 10.1158/0008-5472.CAN-08-0249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.de Bono JS, Logothetis CJ, Molina A, Fizazi K, North S, Chu L, et al. Abiraterone and increased survival in metastatic prostate cancer. The New England Journal of Medicine. 2011;364:1995–2005. doi: 10.1056/NEJMoa1014618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Scher HI, Fizazi K, Saad F, Taplin ME, Sternberg CN, Miller K, et al. Increased survival with enzalutamide in prostate cancer after chemotherapy. The New England Journal of Medicine. 2012;367:1187–1197. doi: 10.1056/NEJMoa1207506. [DOI] [PubMed] [Google Scholar]
- 25.Suzuki H, Okihara K, Miyake H, Fujisawa M, Miyoshi S, Matsumoto T, et al. Alternative nonsteroidal antiandrogen therapy for advanced prostate cancer that relapsed after initial maximum androgen blockade. J Urol. 2008;180:921–927. doi: 10.1016/j.juro.2008.05.045. [DOI] [PubMed] [Google Scholar]
- 26.Kassouf W, Tanguay S, Aprikian AG. Nilutamide as second line hormone therapy for prostate cancer after androgen ablation fails. J Urol. 2003;169:1742–1744. doi: 10.1097/01.ju.0000057795.97626.66. [DOI] [PubMed] [Google Scholar]
- 27.Hu R, Lu C, Mostaghel EA, Yegnasubramanian S, Gurel M, Tannahill C, et al. Distinct transcriptional programs mediated by the ligand-dependent full-length androgen receptor and its splice variants in castration-resistant prostate cancer. Cancer Research. 2012;72:3457–3462. doi: 10.1158/0008-5472.CAN-11-3892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Andersen RJ, Mawji NR, Wang J, Wang G, Haile S, Myung JK, et al. Regression of castrate-recurrent prostate cancer by a small-molecule inhibitor of the amino-terminus domain of the androgen receptor. Cancer Cell. 2010;17:535–546. doi: 10.1016/j.ccr.2010.04.027. [DOI] [PubMed] [Google Scholar]
- 29.Romanuik TL, Ueda T, Le N, Haile S, Yong TM, Thomson T, et al. Novel biomarkers for prostate cancer including noncoding transcripts. The American Journal of Pathology. 2009;175:2264–2276. doi: 10.2353/ajpath.2009.080868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Romanuik TL, Wang G, Holt RA, Jones SJ, Marra MA, Sadar MD. Identification of novel androgen-responsive genes by sequencing of LongSAGE libraries. BMC Genomics. 2009;10:476. doi: 10.1186/1471-2164-10-476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wang G, Haile S, Comuzzi B, Tien AH, Wang J, Yong TM, et al. Osteoblast-derived factors induce an expression signature that identifies prostate cancer metastasis and hormonal progression. Cancer Research. 2009;69:3433–3442. doi: 10.1158/0008-5472.CAN-08-3506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kolvenbag GJ, Blackledge GR, Gotting-Smith K. Bicalutamide (Casodex) in the treatment of prostate cancer: history of clinical development. Prostate. 1998;34:61–72. doi: 10.1002/(sici)1097-0045(19980101)34:1<61::aid-pros8>3.0.co;2-n. [DOI] [PubMed] [Google Scholar]
- 33.Tran C, Ouk S, Clegg NJ, Chen Y, Watson PA, Arora V, et al. Development of a second-generation antiandrogen for treatment of advanced prostate cancer. Science. 2009;324:787–790. doi: 10.1126/science.1168175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Veldscholte J, Ris-Stalpers C, Kuiper GG, Jenster G, Berrevoets C, Claassen E, et al. A mutation in the ligand binding domain of the androgen receptor of human LNCaP cells affects steroid binding characteristics and response to anti-androgens. Biochemical and Biophysical Research Communications. 1990;173:534–540. doi: 10.1016/s0006-291x(05)80067-1. [DOI] [PubMed] [Google Scholar]
- 35.Balk SP, Ko YJ, Bubley GJ. Biology of prostate-specific antigen. J Clin Oncol. 2003;21:383–391. doi: 10.1200/JCO.2003.02.083. [DOI] [PubMed] [Google Scholar]
- 36.Sun Z, Pan J, Balk SP. Androgen receptor-associated protein complex binds upstream of the androgen-responsive elements in the promoters of human prostate-specific antigen and kallikrein 2 genes. Nucleic Acids Research. 1997;25:3318–3325. doi: 10.1093/nar/25.16.3318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Magee JA, Chang LW, Stormo GD, Milbrandt J. Direct, androgen receptor-mediated regulation of the FKBP5 gene via a distal enhancer element. Endocrinology. 2006;147:590–598. doi: 10.1210/en.2005-1001. [DOI] [PubMed] [Google Scholar]
- 38.Lin B, Ferguson C, White JT, Wang S, Vessella R, True LD, et al. Prostate-localized and androgen-regulated expression of the membrane-bound serine protease TMPRSS2. Cancer Research. 1999;59:4180–4184. [PubMed] [Google Scholar]
- 39.Sun C, Shi Y, Xu LL, Nageswararao C, Davis LD, Segawa T, et al. Androgen receptor mutation (T877A) promotes prostate cancer cell growth and cell survival. Oncogene. 2006;25:3905–3913. doi: 10.1038/sj.onc.1209424. [DOI] [PubMed] [Google Scholar]
- 40.He B, Lee LW, Minges JT, Wilson EM. Dependence of selective gene activation on the androgen receptor NH2- and COOH-terminal interaction. The Journal of Biological Chemistry. 2002;277:25631–25639. doi: 10.1074/jbc.M202809200. [DOI] [PubMed] [Google Scholar]
- 41.Masiello D, Cheng S, Bubley GJ, Lu ML, Balk SP. Bicalutamide functions as an androgen receptor antagonist by assembly of a transcriptionally inactive receptor. The Journal of Biological Chemistry. 2002;277:26321–26326. doi: 10.1074/jbc.M203310200. [DOI] [PubMed] [Google Scholar]
- 42.Poujol N, Wurtz JM, Tahiri B, Lumbroso S, Nicolas JC, Moras D, et al. Specific recognition of androgens by their nuclear receptor. A structure-function study. The Journal of Biological Chemistry. 2000;275:24022–24031. doi: 10.1074/jbc.M001999200. [DOI] [PubMed] [Google Scholar]
- 43.Welsh M, Moffat L, Jack L, McNeilly A, Brownstein D, Saunders PT, et al. Deletion of androgen receptor in the smooth muscle of the seminal vesicles impairs secretory function and alters its responsiveness to exogenous testosterone and estradiol. Endocrinology. 2010;151:3374–3385. doi: 10.1210/en.2009-1339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Yeh S, Tsai MY, Xu Q, Mu XM, Lardy H, Huang KE, et al. Generation and characterization of androgen receptor knockout (ARKO) mice: an in vivo model for the study of androgen functions in selective tissues. Proc Natl Acad Sci USA. 2002;99:13498–13503. doi: 10.1073/pnas.212474399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Arno M, Betancur-Galvis L, Gonzalez MA, Sierra J, Zaragoza RJ. Synthesis and cytotoxic activity of novel C7-functionalized spongiane diterpenes. Bioorg Med Chem. 2003;11:3171–3177. doi: 10.1016/s0968-0896(03)00230-x. [DOI] [PubMed] [Google Scholar]
- 46.Betancur-Galvis L, Zuluaga C, Arno M, Gonzalez MA, Zaragoza RJ. Cytotoxic effect (on tumor cells) and in vitro antiviral activity against herpes simplex virus of synthetic spongiane diterpenes. J Nat Prod. 2002;65:189–192. doi: 10.1021/np010206t. [DOI] [PubMed] [Google Scholar]
- 47.Kemppainen JA, Langley E, Wong CI, Bobseine K, Kelce WR, Wilson EM. Distinguishing androgen receptor agonists and antagonists: distinct mechanisms of activation by medroxyprogesterone acetate and dihydrotestosterone. Mol Endocrinol. 1999;13:440–454. doi: 10.1210/mend.13.3.0255. [DOI] [PubMed] [Google Scholar]
- 48.Bramson HN, Hermann D, Batchelor KW, Lee FW, James MK, Frye SV. Unique preclinical characteristics of GG745, a potent dual inhibitor of 5AR. The Journal of Pharmacology and Experimental Therapeutics. 1997;282:1496–1502. [PubMed] [Google Scholar]
- 49.Berrevoets CA, Veldscholte J, Mulder E. Effects of antiandrogens on transformation and transcription activation of wild-type and mutated (LNCaP) androgen receptors. J Steroid Biochem Mol Biol. 1993;46:731–736. doi: 10.1016/0960-0760(93)90313-l. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.