

The crystal structure of 1,5-anhydro-d-fructose reductase from S. meliloti has been determined to 1.93 Å resolution. It is compared with that of 1,5-anhydro-d-fructose reductase from S. morelense and the significance of the observed open conformation of the active site is discussed.
Keywords: carbohydrate metabolism, redox enzymes, biotechnology
Abstract
1,5-Anhydro-d-fructose (1,5-AF) is an interesting building block for enantioselective and stereoselective organic synthesis. Enzymes acting on this compound are potential targets for structure-based protein/enzyme design to extend the repertoire of catalytic modifications of this and related building blocks. Recombinant 1,5-anhydro-d-fructose reductase (AFR) from Sinorhizobium meliloti 1021 was produced in Escherichia coli, purified using a fused 6×His affinity tag and crystallized in complex with the cofactor NADP(H) using the hanging-drop technique. Its structure was determined to 1.93 Å resolution using molecular replacement. The structure displays an empty substrate-binding site and can be interpreted as an open conformation reflecting the enzyme state shortly after the release of product, presumably with bound oxidized cofactor NADP+. Docking simulations indicated that amino-acid residues Lys94, His151, Trp162, Arg163, Asp176 and His180 are involved in substrate binding, catalysis or product release. The side chain of Lys94 seems to have the ability to function as a molecular switch. The crystal structure helps to characterize the interface relevant for dimer formation as observed in solution. The crystal structure is compared with the structure of the homologue from S. morelense, which was solved in a closed conformation and for which dimer formation in solution could not be verified but seems to be likely based on the presented studies of S. meliloti AFR.
1. Introduction
The sugar 1,5-anhydro-d-fructose (1,5-anhydro-d-arabino-hex-2-ulose; 1,5-AF) is the central intermediate of the so-called anhydrofructose pathway, an alternative starch- and glycogen-degrading pathway in bacteria, fungi, plants and mammals (Yu, 2008 ▶; Yu & Fiskesund, 2006 ▶). 1,5-AF is produced by α-(1,4)-glucan lyase (EC 4.2.2.13), which catalyses the release of 1,5-AF from the nonreducing end of α-(1,4)-glucans (Lee et al., 2003 ▶; Yu et al., 1993 ▶) or via the microsomal glycosidase II as a side product during the catabolic degradation of maltose (Andersen et al., 2002b ▶; Hirano et al., 2000 ▶). In Escherichia coli, higher plants and mammalian tissues, the half-life of 1,5-AF is short as it is instantly reduced to 1,5-anhydro-d-glucitol (1,5-AG) by a specific NADPH-dependent anhydrofructose reductase (Sakuma et al., 1998 ▶). Since only a small fraction of α-(1,4)-glucans are degraded via the anhydrofructose pathway (Yu & Pedersén, 1993 ▶; Yu et al., 1993 ▶, 2004 ▶), it was assumed that 1,5-AF or 1,5-AG could play regulatory roles in glycogen metabolism (Kametani et al., 1996 ▶; Konishi et al., 2000 ▶). In E. coli 1,5-AG promotes glycogenolysis (Shiga et al., 1999 ▶) and in mammals 1,5-AF or 1,5-AG stimulates insulin secretion (Yamanouchi et al., 2003 ▶). Although little is known about the physiological importance of 1,5-AF and 1,5-AG in human glucose homeostasis, differences in 1,5-AG serum concentrations between healthy and diabetic individuals have been observed, rendering 1,5-AG an established marker in diabetic control (Dworacka & Winiarska, 2005 ▶; Kim et al., 2011 ▶). Clinical trials have been proposed to use 1,5-AF and its derivatives against sugar metabolism disorder-related diseases (Ahrén et al., 2000 ▶; Ahrén & Yu, 2002 ▶). For a recent review of the relevance of 1,5-AF to medical applications, see Fiskesund et al. (2010 ▶). 1,5-AF has considerable relevance to the field of white biotechnology since it can serve as a chiral building block for organic synthesis (Andersen et al., 2002b ▶; Lundt & Yu, 2010 ▶). In this respect, the identification and characterization of enzymes utilizing 1,5-AF as a substrate is of specific interest.
By microbial screening using 1,5-AF as the sole carbon source, a novel 1,5-AF reductase (AFR; EC 1.1.1.292) was found in Sinorhizobium morelense S-30.7.5 (Kühn et al., 2006 ▶) which catalyses the stereoselective and quantitative reduction of 1,5-AF to 1,5-anhydro-d-mannitol (1,5-AM; Fig. 1 ▶). In contrast to the regulatory role of AFR in mammals, this bacterial AFR is a metabolic enzyme.
Figure 1.
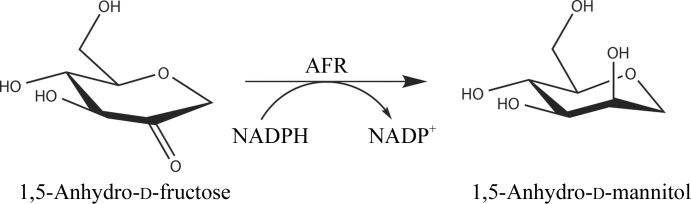
The reaction catalyzed by 1,5-anhydro-d-fructose reductase (AFR; EC 1.1.1.292) from S. meliloti. 1,5-Anhydro-d-fructose (1,5-AF) is a cyclic ether and not a hemiacetal since it lacks the hydroxyl group at the C1 position. It is stereoselectively reduced to 1,5-anhydro-d-mannitol (1,5-AM) by AFR. For the enzymatic reduction the enzyme utilizes the cofactor NADPH. This is the first step in the 1,5-AF pathway as first shown for S. morelense (Kühn et al., 2006 ▶).
The AFR from S. morelense was assigned to the GFO/IDH/MocA family and its general occurrence among the Rhizobiaceae has been demonstrated (Kühn et al., 2006 ▶). This family is composed of enzymes that utilize NADP or NAD as a redox cofactor. Glucose–fructose oxidoreductase (GFOR; Zachariou & Scopes, 1986 ▶; Kingston et al., 1996 ▶), myo-inositol dehydrogenase (IDH) and the dehydrogenase MocA of rhizopine catabolism are representative members of this family. 1,5-AF can be synthesized biocatalytically from starch by 1,4-glucanlyase and pullulanase and is a cheap resource for various chemical and biocatalytical transformations, e.g. as a building block for enantioselective and stereoselective organic synthesis (Andersen et al., 2002b ▶; Lichtenthaler et al., 1980 ▶; Lundt & Yu, 2010 ▶). 1,5-AF can also be used for a variety of applications in the food and pharmaceutical industries, e.g. as a low-calorie sweetener (Andersen et al., 2002a ▶) and antioxidant (Fujisue et al., 2003 ▶; Susumu et al., 2003 ▶) or as a humectant (Susumu et al., 2002 ▶). The crystal structure of AFR from S. morelense has recently been determined and published (Dambe et al., 2006 ▶). In contrast to S. morelense, the complete genome of S. meliloti 1021 is known (Capela et al., 2001 ▶), which provides access to putative enzymes in 1,5-AF metabolism. Therefore, this organism is currently used as a model organism for further characterization of the 1,5-anhydro-d-fructose pathway in Rhizobiaceae. The afr gene from S. meliloti has 1002 base pairs and the derived polypeptide showed 86% identity to the AFR from S. morelense. The enzyme was cloned and heterologously expressed in E. coli for further biochemical characterization and comparison with the S. morelense reductase. To provide a solid basis for rational protein design, we prepared crystals for determination of its three-dimensional structure. Here, we present the crystal structure of AFR from S. meliloti and describe the observed differences from AFR from S. morelense and GFOR.
2. Materials and methods
Recombinant AFR from S. meliloti 1021 was produced in E. coli BL21(DE3)Gold cells fused with an N-terminal 6×His affinity tag including a factor Xa protease cleavage site using pET-24a(+) as the expression vector with a stop codon prior to its C-terminal 6×His affinity tag. After purification by Ni–NTA affinity chromatography and cleavage of the N-terminal 6×His affinity tag by 24 h incubation with the protease factor Xa, the protein was purified to homogeneity by Q Sepharose anion-exchange chromatography.
In order to obtain reasonably sized single crystals of AFR, microseeding and macroseeding steps were performed. In the end, small crystals of AFR grew in a solution composed of approximately 5.5 mg ml−1 protein in 50 mM bis-tris pH 5.9, 20 mM ammonium sulfate, 7% PEG 3350, 25 mM 1,5-AF, 1 mM NADPH, which was equilibrated at 291 K as a hanging drop against reservoir solution consisting of 100 mM bis-tris pH 5.5, 50 mM ammonium sulfate, 20%(w/v) PEG 3350. Crystals belonging to the orthorhombic space group C2221, with unit-cell parameters a = 68.9, b = 89.7, c = 94.5 Å, grew within 20 d. Prior to data collection, the crystals were soaked in a cryoprotection solution [100 mM bis-tris pH 5.5, 50 mM ammonium sulfate, 20%(w/v) PEG 3350, 28 mM 1,5-AF, 2 mM NADPH, 20%(v/v) PEG 400] and flash-cooled by rapid transfer into liquid nitrogen.
X-ray data were collected using a marμX system (MAR Research GmbH, Norderstedt, Germany) equipped with a mardtb desktop-beamline goniometer system, an IμS microfocus source (Incoatec, Geesthacht, Germany) for Cu Kα radiation, a mar345 image-plate detector and a liquid-nitrogen Cryostream 700 (Oxford Cryosystems, Oxford, England).
The collected diffraction data were indexed, integrated and scaled using XDS (Kabsch, 1993 ▶, 2010a ▶,b ▶). The phases were obtained by molecular replacement using MOLREP (Vagin & Teplyakov, 2010 ▶). Using the crystal structure of AFR from S. morelense (PDB entry 2glx; Dambe et al., 2006 ▶) as a template, a homology model with the correct amino-acid annotation was automatically generated using the SWISS-MODEL server (Schwede et al., 2003 ▶). The search model for molecular replacement did not contain water molecules or bound cofactor. Several cycles of refinement with REFMAC5 (Murshudov et al., 2011 ▶) as implemented in the CCP4 program suite (Winn et al., 2011 ▶) and manual inspection and correction of the model with Coot (Emsley et al., 2010 ▶) were performed to build the complete protein model with the NADP(H) cofactor. Structural representations were generated using PyMOL v.1.5 (DeLano, 2002 ▶).
3. Results and discussion
3.1. Structure determination
The bacterial AFR from S. meliloti 1021 was crystallized in complex with the cofactor NADP(H) in space group C2221 with one molecule per asymmetric unit (V M = 1.84 Å3 Da−1). Diffraction data were recorded to 1.93 Å resolution. For convenience, the crystal structure was determined by molecular replacement using a homology model derived from the structure of the homologous AFR from S. morelense as a search template (PDB entry 2glx; Dambe et al., 2006 ▶). Refinement of the structure resulted in final crystallographic R factors of R work = 15.4% and R free = 20.7% with good geometry according to the Ramachandran plot (Laskowski et al., 1993 ▶). The final model includes amino-acid residues 1–333, one NADP(H) cofactor and 291 water molecules. The protein structure was refined with individual temperature factors (B factors), resulting in an average crystallographic temperature factor over all atoms of 23.5 Å2 (Table 1 ▶).
Table 1. Diffraction data-collection and refinement statistics.
Values in parentheses are for the highest resolution shell.
| Wavelength (Å) | 1.5418 |
| Temperature (K) | 100 |
| Space group | C2221 |
| Unit-cell parameters (Å) | a = 68.9, b = 89.7, c = 94.5 |
| Resolution (Å) | 19.5–1.93 (1.98–1.93) |
| Unique reflections | 22202 (1487) |
| Completeness (%) | 99.2 (91.5) |
| Multiplicity | 14.3 (12.6) |
| 〈I/σ(I)〉 | 26.9 (4.7) |
| Wilson B factor (Å2) | 24.6 |
| R meas † (%) | 10.4 (63.9) |
| R work ‡ (%) | 15.4 (18.6) |
| R free § (%) | 20.7 (24.7) |
| No. of non-H atoms | |
| Protein | 2449 |
| Solvent | 291 |
| Cofactor | 48 |
| Average B factor (Å2) | |
| Protein | 22.3 |
| Waters | 28.5 |
| Cofactor | 69.5 |
| Cruickshank DPI¶ (Å) | 0.141 |
| Ramachandran plot†† | |
| Most favoured (%) | 90.0 |
| Additionally and generously allowed (%) | 9.6 |
| Outliers (%) | 0.4 |
| R.m.s.d. from ideal values‡‡ | |
| Bond lengths (Å) | 0.02 |
| Bond angles (°) | 2.05 |
| PDB code | 4koa |
R
meas = 100 × 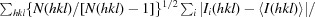
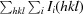 , where I
i(hkl) is the intensity of the ith individual measurement of the reflection with Miller indices hkl and 〈I(hkl)〉 is the mean intensity of all measurements of I(hkl) calculated for I ≥ 3σ(I); N(hkl) is the redundancy or multiplicity of the observed reflection (Diederichs & Karplus, 1997 ▶; Weiss, 2001 ▶).
, where I
i(hkl) is the intensity of the ith individual measurement of the reflection with Miller indices hkl and 〈I(hkl)〉 is the mean intensity of all measurements of I(hkl) calculated for I ≥ 3σ(I); N(hkl) is the redundancy or multiplicity of the observed reflection (Diederichs & Karplus, 1997 ▶; Weiss, 2001 ▶).
R
cryst = 100 × 
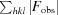 , where F
obs and F
calc are the observed and calculated structure-factor amplitudes, respectively.
, where F
obs and F
calc are the observed and calculated structure-factor amplitudes, respectively.
R free is equivalent to R cryst but is calculated from reflections (5%) that were omitted from the refinement process (Brünger, 1992 ▶; Tickle et al., 2000 ▶).
Diffraction-component precision index (Cruickshank, 1999 ▶) calculated using the program SFCHECK (Vaguine et al., 1999 ▶).
Calculated using the program PROCHECK (Laskowski et al., 1993 ▶).
From standard geometry using the Engh and Huber library (Engh & Huber, 1991 ▶).
3.2. Overall structure
AFR from S. meliloti shares a sequence identity of 86% with AFR from S. morelense and features the same three-dimensional fold (Figs. 2 ▶ and 3 ▶). The enzyme is composed of two structural domains: an N-terminal domain and a larger C-terminal domain. The NADP(H) cofactor-binding site is located between the two domains (Fig. 3 ▶ b). The domain formed by the first 120 amino-acid residues displays the typical dinucleotide-binding motif referred to as the Rossmann fold (Rossmann et al., 1974 ▶), consisting of a central β-sheet composed of two β–α–β–α–β motifs. The cofactor NADP(H) is bound in a deep cleft above this β-sheet. The larger C-terminal domain (amino-acid residues 121–333) also possesses α/β topology and exhibits structural homology to members of the family of glyceraldehyde-3-phosphate dehydrogenase-like proteins. Biochemical characterization of the AFR from S. morelense revealed a monomeric enzyme in solution (Kühn et al., 2006 ▶). In contrast, biochemical characterization of the AFR from S. meliloti using multi-angle laser light-scattering (MALLS; data not shown), size-exclusion chromatography and MALDI mass spectrometry (Stosik, 2008 ▶) revealed a dimeric molecule in solution. In the crystal structure of S. morelense AFR, the asymmetric unit contains two enzyme molecules displaying a tight dimer. In the presented crystal structure of S. meliloti AFR, the asymmetric unit contains only one enzyme molecule; however, a tight dimer is generated with a symmetry mate. Interestingly, this dimer shows the same arrangement as the two enzyme molecules within the asymmetric unit of the S. morelense AFR structure. In order to evaluate the interfaces formed between the individual monomers within the crystal structure, we analyzed the intermolecular contacts formed by crystal packing using the PISA server at the European Bioinformatics Institute (PDBePISA v.1.46; Krissinel & Henrick, 2007 ▶). The analysis revealed one extended interface formed with the (x, −y, −z) symmetry mate, covering an interface area of 1642 Å2 with a solvation-energy gain Δi G of −66.1 kJ mol−1 and a complexation significance score (CSS) of 0.622, suggesting that the protein should exist as a dimer in solution. Interestingly, the interface analysis for AFR from S. morelense (PDB entry 2glx) revealed an even larger interface area of 1822 Å2 but with a smaller solvation-energy gain Δi G of −60.3 kJ mol−1 and thus a lower complexation significance score of 0.452. For both proteins the dimer interface is formed between the large and extended β-sheet moiety of the C-terminal domain (Fig. 3 ▶ b). Therefore, based on the individual crystal structures both enzymes might exist as dimers in solution, but this could only be verified biochemically for AFR from S. meliloti (Stosik, 2008 ▶).
Figure 2.

Sequence alignment of S. meliloti AFR with S. morelense AFR and with GFOR (Wiegert et al., 1997 ▶). This figure was produced using ESPript (Gouet et al., 2003 ▶).
Figure 3.

Topology of S. meliloti AFR. (a) Arrangement of the secondary-structure elements within one molecule. The figure was generated using PDBsum (Laskowski, 2009 ▶). (b) Ribbon representation of AFR and the putative dimeric assembly of the active enzyme. The secondary-structure elements of the molecule within the crystallographic asymmetric unit are coloured cyan (α-helices), red (β-sheets) and magenta (loops). The bound cofactor NADP(H) is displayed in ball-and-stick representation. The molecule in green represents the (x, −y, −z) symmetry mate. Intermolecular contacts are formed between the two extended β-sheets of the C-terminal domain to build the stable dimer as observed in solution.
3.3. The active site
We compared the crystal structure of S. meliloti AFR with the recently published structure of its closest relative, S. morelense AFR (PDB entry 2glx; Dambe et al., 2006 ▶), and with the well characterized structure of glucose–fructose oxidoreductase (GFOR) from Zymomonas mobilis (PDB entry 1h6a; Nurizzo et al., 2001 ▶; Wiegert et al., 1997 ▶). The identity between S. meliloti AFR and S. morelense AFR is 86% (with an r.m.s.d. of 1.3 Å for 323 aligned residues out of 332 residues; Z-score of 50.6) and that between S. meliloti AFR and GFOR is 23% (with an r.m.s.d. of 2.4 Å for 324 aligned residues out of 381 residues; Z-score of 36.9). The comparison was performed by pairwise analysis using the DALI server (Hasegawa & Holm, 2009 ▶). The comparison with S. morelense AFR revealed only two large structural deviations within the protein fold. The two loop regions connecting β-strand S7 to α-helix H8 and connecting β-strand S8 to β-strand S9 (Fig. 4 ▶ a) have an extended conformation in S. meliloti AFR and display higher flexibility and disorder (the average B factor for amino-acid residues 154–170 and 203–210 is around 75 Å2). These two loop regions are not involved in crystal packing. They are positioned close to the putative active site and thus should influence the mode of substrate binding or product release. The conformations observed within the presented S. meliloti AFR structure can be interpreted as an open conformation, whereas the conformations observed in the structures of S. morelense AFR and GFOR display a closed conformation in respect to accessibility towards the substrate-binding site. Superimposing the crystal structures of the AFRs from S. meliloti and S. morelense reveals a concerted domain movement. Compared with S. morelense AFR, the two domains of S. meliloti AFR rotate approximately by 2° (N-terminal domain) and 2.5° (C-terminal domain), both to the outside (Fig. 4 ▶ a). As a consequence, the binding cleft for the cofactor as well as for the substrate becomes more accessible. The electron density in the active site of S. meliloti AFR is poor for some parts of the cofactor NADP (high temperature factors, suggesting high flexibility) and although the crystals were grown in the presence of 25 mM 1,5-AF no electron density for the substrate was found. In contrast, acetate is bound within the active site of S. morelense AFR and succinate and glycerol are bound in that of GFOR. Thus, the conformation of S. morelense AFR and GFOR can be regarded as closed after induced-fit binding of the substrate and cofactor has occurred. In addition to the differences within the mentioned loop regions and the overall domain arrangement, the nicotinamide moiety displays a different orientation in comparison to the NADP(H) cofactor of 2glx and GFOR (Fig. 4 ▶ b). The orientation observed in S. meliloti AFR cannot be adopted in S. morelense AFR and GFOR since the conserved residues Trp162–Arg163 (S. morelense AFR) and Trp251–Arg252 (GFOR) within the loop S7/H8 are positioned in too close a proximity to the nicotinamide when the loop is in its closed conformation. It seems to be likely that the nicotinamide moiety and the loop S7/H8 change their orientation in a concerted manner depending on the occupancy of the active site. Various strategies to obtain a structure of S. meliloti AFR with bound substrate, substrate analogue or competitive inhibitor failed. In order to obtain some insight into the molecular interactions for substrate binding, we performed docking simulations of the mode of binding of the substrate 1,5-AF and the product 1,5-AM within the active site using the program AutoDock v.4.2 (Morris et al., 2009 ▶). The structure of S. morelense AFR was used to simulate the enzyme with a closed conformation of loop S7/H8 and its interaction with bound substrate and product, respectively (Figs. 4 ▶ c, 4 ▶ d and 4 ▶ e). The docking simulations produced diverse conformations of bound substrate/product within the active site, with one dominant conformation. In the following we will interpret the two most likely orientations from these simulations and will put them into perspective for substrate binding and product release (Fig. 4 ▶ f). In a comparison between 1,5-AF and 1,5-AM the orientations of the 3′-OH, 4′-OH and 6′-OH hydroxyl groups are in nearly the same positions; thus, their interactions with the side chains of amino-acid residues His151, Arg163 and Asp176 are maintained. The side chain of His151 can form two alternative hydrogen bonds via its NE2 atom: one to the 6′-OH and one to the endocyclic O atom of 1,5-AF. Owing to the reduction of the carbonyl group at the 2′ C atom, the O atom at the 2′ C atom and the endocyclic O atom adopt new positions. Since His151 remains in its orientation owing to the interaction with the 6′-OH hydroxyl group, formation of the hydrogen bond between the endocyclic O atom and His151 NE2 is no longer possible. Furthermore, His180 is released since the hydrogen bond formed between the carbonyl O atom of 1,5-AF and the side-chain atom His180 NE2 is lost. In addition, the reduced O atom (2′-carbonyl→2′-hydroxy) rearranges and therefore the interacting side chain of Lys94 adopts a new conformation. This rearrangement of Lys94 might serve as a molecular switch for triggering product release. Since Lys94 also interacts with the 2′-OH group of the nicotinamide ribose moiety, the structural reorientation of Lys94 might trigger a conformational rearrangement of the ribose sugar and thus rotational reorientation of the nicotinamide ring. As a consequence of the movement of the nicotinamide moiety towards amino-acid residues Trp162 and Arg163, these side chains have to move away, releasing their interaction with the pyrophosphate moiety of the NADP cofactor and thus inducing even more flexibility to the cofactor and less strain upon reorientation of the nicotinamide moiety. Arg163 forms a hydrogen bond to the 3′-OH group of the substrate and product. Upon reorientation of Arg163 this interaction is lost and release of the product is facilitated. The amino-acid side chain of Asp176 is observed in two alternative conformations. While one conformation forms a hydrogen bond to the 3′-OH and 4′-OH groups, the other does not. Owing to the rearrangement within the region Trp162–Arg163 the second conformation is favoured, allowing the product to be pulled out from the active site, thus further reducing interaction with the product and favouring its release from the enzyme. In a final step loop S7/H8 completely rearranges and the product is released.
Figure 4.

Open and closed conformations of AFR. (a) Superposition of the Cα main-chain trace of S. meliloti AFR (light green) and S. morelense AFR (orange). The cofactor NADP(H) is displayed in ball-and-stick representation (C atoms in cyan for S. meliloti and in orange for S. morelense, O atoms in red, N atoms in blue). The angle and arrows represent the relative movement of the N-terminal and C-terminal domains of S. meliloti AFR relative to S. morelense AFR. (b) Close-up view of the cofactor binding mode. (c–f) Representation of 1,5-AF (c) and 1,5-AM (d) in the conformation identified by docking simulations. (e) Active site of AFR (S. morelense) with docked substrate 1,5-AF and product 1,5-AM. The amino-acid residues interacting with the sugar molecules are highlighted in ball-and-stick representation, with potential hydrogen bonds shown as dotted lines. The figures were produced using PyMOL v.1.5 (DeLano, 2002 ▶).
In conclusion, we propose that the observed orientations of the presented residues of S. meliloti AFR represent a conformation after the release of the product, whereas the structure of S. morelense AFR represents a structure shortly before or after reduction with bound substrate or product. The outlined stepwise rearrangement within the active site and the adjacent loop is most likely to be triggered by the reorientation of the side chain of Lys94, thus giving it the role of a molecular switch. A similar role was proposed for the corresponding Lys181 in GFOR, where the different observed conformations favour or discriminate the binding of substrate or product (Nurizzo et al., 2001 ▶).
Supplementary Material
PDB reference: 1,5-anhydro-d-fructose reductase, 4koa
Acknowledgments
We gratefully acknowledge access to the core facilities of the ZBM/LMB of the CAU. Additionally, we are grateful for access to the HTX crystallization facility at the EMBL Outstation (Hamburg, Germany). We would like to thank Drs Yvonne Carius, Tresfore Dambe and Ulrich Zander for helpful discussions. This work was supported by the Deutsche Bundesstiftung Umwelt (Az 13195-32) within the research network ChemBioTec.
References
- Ahrén, B., Holst, J. J. & Yu, S. (2000). Eur. J. Pharmacol. 397, 219–225. [DOI] [PubMed]
- Ahrén, B. & Yu, S. (2002). US Patent WO 2001051058 A1.
- Andersen, S. M., Lundt, I., Marcussen, J. & Yu, S. (2002a). Carbohydr. Res. 337, 873–890. [DOI] [PubMed]
- Andersen, S. M., Lundt, I., Marcussen, J. & Yu, S. (2002b). US Patent WO 2000056745 A1.
- Brünger, A. T. (1992). Nature (London), 355, 472–475. [DOI] [PubMed]
- Capela, D. et al. (2001). Proc. Natl Acad. Sci. USA, 98, 9877–9882.
- Cruickshank, D. W. J. (1999). Acta Cryst. D55, 583–601. [DOI] [PubMed]
- Dambe, T. R., Kühn, A. M., Brossette, T., Giffhorn, F. & Scheidig, A. J. (2006). Biochemistry, 45, 10030–10042. [DOI] [PubMed]
- Delano, W. L. (2002). PyMOL http://www.pymol.org.
- Diederichs, K. & Karplus, P. A. (1997). Nature Struct. Biol. 4, 269–275. [DOI] [PubMed]
- Dworacka, M. & Winiarska, H. (2005). Dis. Markers, 21, 127–132. [DOI] [PMC free article] [PubMed]
- Emsley, P., Lohkamp, B., Scott, W. G. & Cowtan, K. (2010). Acta Cryst. D66, 486–501. [DOI] [PMC free article] [PubMed]
- Engh, R. A. & Huber, R. (1991). Acta Cryst. A47, 392–400.
- Fiskesund, R., Abeyama, K., Yoshinaga, K., Abe, J., Yuan, Y. & Yu, S. (2010). Planta Med. 76, 1635–1641. [DOI] [PubMed]
- Fujisue, M., Muroya, K., Nozaki, K., Yajima, M. & Yoshinaga, K. (2003). US Patent WO 2001056408 A1.
- Gouet, P., Robert, X. & Courcelle, E. (2003). Nucleic Acids Res. 31, 3320–3323. [DOI] [PMC free article] [PubMed]
- Hasegawa, H. & Holm, L. (2009). Curr. Opin. Struct. Biol. 19, 341–348. [DOI] [PubMed]
- Hirano, K., Ziak, M., Kamoshita, K., Sukenaga, Y., Kametani, S., Shiga, Y., Roth, J. & Akanuma, H. (2000). Glycobiology, 10, 1283–1289. [DOI] [PubMed]
- Kabsch, W. (1993). J. Appl. Cryst. 26, 795–800.
- Kabsch, W. (2010a). Acta Cryst. D66, 133–144. [DOI] [PMC free article] [PubMed]
- Kabsch, W. (2010b). Acta Cryst. D66, 125–132. [DOI] [PMC free article] [PubMed]
- Kametani, S., Shiga, Y. & Akanuma, H. (1996). Eur. J. Biochem. 242, 832–838. [DOI] [PubMed]
- Kim, M. J., Jung, H. S., Hwang-Bo, Y., Cho, S. W., Jang, H. C., Kim, S. Y. & Park, K. S. (2011). Acta Diabetol., 10.1007/s00592-011-0302-0.
- Kingston, R. L., Scopes, R. K. & Baker, E. N. (1996). Structure, 4, 1413–1428. [DOI] [PubMed]
- Konishi, Y., Hashima, K. & Kishida, K. (2000). Biosci. Biotechnol. Biochem. 64, 2462–2465. [DOI] [PubMed]
- Krissinel, E. & Henrick, K. (2007). J. Mol. Biol. 372, 774–797. [DOI] [PubMed]
- Kühn, A., Yu, S. & Giffhorn, F. (2006). Appl. Environ. Microbiol. 72, 1248–1257. [DOI] [PMC free article] [PubMed]
- Laskowski, R. A. (2009). Nucleic Acids Res. 37, D355–D359. [DOI] [PMC free article] [PubMed]
- Laskowski, R. A., MacArthur, M. W., Moss, D. S. & Thornton, J. M. (1993). J. Appl. Cryst. 26, 283–291.
- Lee, S. S., Yu, S. & Withers, S. G. (2003). Biochemistry, 42, 13081–13090. [DOI] [PubMed]
- Lichtenthaler, F., El Ashry, E. & Göckel, V. (1980). Tetrahedron Lett. 21, 1429–1432.
- Lundt, I. & Yu, S. (2010). Carbohydr. Res. 345, 181–190. [DOI] [PubMed]
- Morris, G. M., Huey, R., Lindstrom, W., Sanner, M. F., Belew, R. K., Goodsell, D. S. & Olson, A. J. (2009). J. Comput. Chem. 30, 2785–2791. [DOI] [PMC free article] [PubMed]
- Murshudov, G. N., Skubák, P., Lebedev, A. A., Pannu, N. S., Steiner, R. A., Nicholls, R. A., Winn, M. D., Long, F. & Vagin, A. A. (2011). Acta Cryst. D67, 355–367. [DOI] [PMC free article] [PubMed]
- Nurizzo, D., Halbig, D., Sprenger, G. A. & Baker, E. N. (2001). Biochemistry, 40, 13857–13867. [DOI] [PubMed]
- Rossmann, M. G., Moras, D. & Olsen, K. W. (1974). Nature (London), 250, 194–199. [DOI] [PubMed]
- Sakuma, M., Kametani, S. & Akanuma, H. (1998). J. Biochem. 123, 189–193. [DOI] [PubMed]
- Schwede, T., Kopp, J., Guex, N. & Peitsch, M. C. (2003). Nucleic Acids Res. 31, 3381–3385. [DOI] [PMC free article] [PubMed]
- Shiga, Y., Kametani, S., Kadokura, T. & Akanuma, H. (1999). J. Biochem. 125, 166–172. [DOI] [PubMed]
- Stosik, B. (2008). PhD thesis. Saarland University, Germany.
- Susumu, H., Junichi, A., Toshiyasu, M., Kazuhiro, Y., Hideto, I. & Masamitsu, F. (2002). Japan Patent 2002-027945.
- Susumu, H., Yasuhito, T., Junichi, A., Kenkou, M., Kazuhiro, Y., Mami, F. & Hideto, I. (2003). US Patent WO 2001072124 A1.
- Tickle, I. J., Laskowski, R. A. & Moss, D. S. (2000). Acta Cryst. D56, 442–450. [DOI] [PubMed]
- Vagin, A. & Teplyakov, A. (2010). Acta Cryst. D66, 22–25. [DOI] [PubMed]
- Vaguine, A. A., Richelle, J. & Wodak, S. J. (1999). Acta Cryst. D55, 191–205. [DOI] [PubMed]
- Weiss, M. S. (2001). J. Appl. Cryst. 34, 130–135.
- Wiegert, T., Sahm, H. & Sprenger, G. A. (1997). J. Biol. Chem. 272, 13126–13133. [DOI] [PubMed]
- Winn, M. D. et al. (2011). Acta Cryst. D67, 235–242.
- Yamanouchi, T., Inoue, T., Ichiyanagi, K., Sakai, T. & Ogata, N. (2003). Biochim. Biophys. Acta, 1623, 82–87. [DOI] [PubMed]
- Yu, S. (2008). IUBMB Life, 60, 798–809. [DOI] [PubMed]
- Yu, S. & Fiskesund, R. (2006). Biochim. Biophys. Acta, 1760, 1314–1322. [DOI] [PubMed]
- Yu, S., Kenne, L. & Pedersén, M. (1993). Biochim. Biophys. Acta, 1156, 313–320. [DOI] [PubMed]
- Yu, S. & Pedersén, M. (1993). Planta, 191, 137–142. [DOI] [PubMed]
- Yu, S., Refdahl, C. & Lundt, I. (2004). Biochim. Biophys. Acta, 1672, 120–129. [DOI] [PubMed]
- Zachariou, M. & Scopes, R. K. (1986). J. Bacteriol. 167, 863–869. [DOI] [PMC free article] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
PDB reference: 1,5-anhydro-d-fructose reductase, 4koa