

Selenophosphate synthetases from L. major and T. brucei were crystallized for the first time using limited proteolysis and microbatch techniques and suitable crystals for X-ray diffraction experiments were obtained.
Keywords: selenocysteine, selenophosphate synthetase, Trypanosoma brucei, Leishmania major
Abstract
Selenophosphate synthetase (SPS) plays an indispensable role in selenium metabolism, being responsible for catalyzing the activation of selenide with adenosine 5′-triphosphate (ATP) to generate selenophosphate, the essential selenium donor for selenocysteine synthesis. Recombinant full-length Leishmania major SPS (LmSPS2) was recalcitrant to crystallization. Therefore, a limited proteolysis technique was used and a stable N-terminal truncated construct (ΔN-LmSPS2) yielded suitable crystals. The Trypanosoma brucei SPS orthologue (TbSPS2) was crystallized by the microbatch method using paraffin oil. X-ray diffraction data were collected to resolutions of 1.9 Å for ΔN-LmSPS2 and 3.4 Å for TbSPS2.
1. Introduction
Selenophosphate synthetase (SPS; EC 2.7.9.3) is a central enzyme in selenocysteine (Sec) biosynthesis (Leinfelder et al., 1990 ▶; Donovan & Copeland, 2010 ▶), catalyzing the production of monoselenophosphate (SeP). The reaction occurs by the activation of selenide with ATP and water, in a 1:1:1 stoichiometry, resulting in the formation of adenosine 5′-monophosphate (AMP), orthophosphate (Pi) and the biologically active form of selenium, monoselenophosphate (SeP) (Glass et al., 1993 ▶; Veres et al., 1994 ▶; Noinaj et al., 2012 ▶).
Recent reports have demonstrated that SeP is derived from the transfer of the γ-phosphate group of ATP to selenide, while the β-phosphate group is released as orthophosphate (Itoh et al., 2009 ▶; Wang et al., 2009 ▶; Noinaj et al., 2012 ▶).
SPSs are found in all domains of life (Su et al., 2009 ▶) and have a conserved cysteine or selenocysteine residue in the N-terminal glycine-rich region corresponding to the catalytic site (Persson et al., 1997 ▶; Sculaccio et al., 2008 ▶). The kinetoplastid SPSs from Trypanosoma brucei (TbSPS2) and Leishmania major (LmSPS2), which share 26 and 28% amino-acid sequence identity with the Escherichia coli orthologue, respectively, are not selenoproteins (Sculaccio et al., 2008 ▶), in contrast to the mammalian SPS2 selenoprotein. Initially considered to be irrelevant for T. brucei metabolism (Aeby et al., 2009 ▶), TbSPS2 is indeed involved in oxidative stress protection. In this organism, Sps2 silencing reduced cell viability under hydrogen peroxide treatment or excessive growth, establishing its importance in cellular metabolism (Costa et al., 2011 ▶).
Crystal structures have been reported of Aquifex aeolicus SPS (AaSPS; 27 and 29% amino-acid sequence identity to TbSPS2 and LmSPS2, respectively) in the apo form and in complex with the nonhydrolyzable AMPCPP (α,β-methyleneadenosine 5′-triphosphate) (Matsumoto et al., 2008 ▶; Itoh et al., 2009 ▶). For human SPS1 (HsSPS1; 42% amino-acid sequence identity to both TbSPS2 and LmSPS2), structures of complexes with AMPCPP, ADP and Pi products have been reported (Wang et al., 2009 ▶). Recently, the structure of the E. coli SPS (EcSPS) C17S mutant (the mutation site corresponds to positions Cys42 and Cys46 in TbSPS2 and LmSPS2, respectively) was solved in the apo form (Noinaj et al., 2012 ▶). In these structures, metal ions such as Na+ and K+ were demonstrated to mediate the binding of the highly electronegative substrate phosphate group. In AaSPS structures, both open and closed forms of the N-terminal Gly-rich loop have been observed and there is evidence that ATP binding, metal binding and the formation of their binding sites are interdependent (Itoh et al., 2009 ▶). AaSPS and HsSPS1 are selective for monovalent cations, with a preference for K+ over Na+ or Li+ (Wang et al., 2009 ▶).
Here, we report the first crystallization and preliminary X-ray diffraction analysis of SPS2 from the kinetoplastid parasites L. major and T. brucei, which are not selenoproteins. Our structures may provide new information on the reaction mechanism of these enzymes. Moreover, comparison of the TbSPS2 and LmSPS2 structures with those of AaSPS, HsSPS1 and EcSPS may contribute to an understanding of the evolution of the selenocysteine-biosynthesis pathway.
2. Materials and methods
2.1. Overexpression and purification of LmSPS2 and TbSPS2
LmSPS2 (UniProt code Q4Q0M0) and TbSPS2 (UniProt code Q38A34) cloned into pET28a were transformed in E. coli BL21 (DE3) cells, expressed and purified by adapting a previously described protocol (Sculaccio et al., 2008 ▶). Briefly, cells were grown in LB medium containing 50 µg ml−1 kanamycin with agitation at 310 K until the OD at 600 nm of the cell culture reached 0.5–0.6. Protein expression was induced by the addition of isopropyl β-d-1-thiogalactopyranoside (IPTG) to a final concentration of 1.0 mM for 16 h under the same conditions. The cells were harvested by centrifugation at 4000g for 40 min and the cell pellet was suspended in 50 ml lysis buffer [50 mM Tris–HCl pH 8.0, 300 mM NaCl, 5%(v/v) glycerol, 1 mM β-mercaptoethanol and one tablet of Complete Protease Inhibitor Cocktail (Roche)]. The cells were disrupted by six cycles of 30 s sonication at 35 W (550 Sonic Dismembrator) and 30 s resting on ice.
The cell lysate was clarified by centrifugation at 20 000g for 30 min (277 K) and the cleared supernatant was applied onto a 5 ml Ni Sepharose High Performance HisTrap HP column (GE Healthcare) pre-equilibrated with buffer A (50 mM Tris–HCl pH 8.0, 300 mM NaCl, 1 mM β-mercaptoethanol, 10 mM imidazole). The column was washed with ten column volumes of buffer A and the bound protein was eluted using a linear 0–500 mM imidazole gradient in buffer A. The eluted fractions (5 ml) were dialyzed against 1 l buffer B (50 mM Tris–HCl pH 8.0, 300 mM NaCl, 1 mM β-mercaptoethanol) for 16 h at 277 K. The protein was purified and the buffer exchanged for buffer C (50 mM Tris–HCl pH 8.0, 50 mM NaCl, 1 mM β-mercaptoethanol) by size-exclusion chromatography using a HR 10/30 Superdex 200 column (GE Healthcare). The purity of each of the protein samples was verified by 15% SDS–PAGE and the proteins were concentrated to 5–10 mg ml−1 for crystallization and in situ proteolysis experiments. The protein concentration was determined using a NanoDrop 2000c UV–Vis spectrophotometer (Thermo Scientific) at 280 nm using predicted extinction coefficients (calculated using ProtParam; http://web.expasy.org/cgi-bin/protparam/protparam; Gasteiger et al., 2005 ▶) of 28 920 and 34 755 M −1 cm−1 for LmSPS2 and TbSPS2, respectively. The purified recombinant proteins have additional 6×His tag and thrombin-cleavage site residues, resulting in molecular weights of 44.7 and 45.0 kDa and isoelectric points of 5.83 and 5.98 for LmSPS2 and TbSPS2, respectively.
2.2. In situ proteolysis and N-terminal sequencing of LmSPS2
After unsuccessful efforts to crystallize full-length LmSPS2, we used an in situ proteolysis technique, a strategy employed to rescue proteins that are recalcitrant to crystallization (Wernimont & Edwards, 2009 ▶). The JBS Floppy-Choppy kit (Jena Bioscience) was used according to the manufacturer’s instructions with the following proteases: α-chymotrypsin, trypsin, subtilisin and papain. The best result was achieved using α-chymotrypsin incubated with 5 mg ml−1 LmSPS2 at an α-chymotrypsin:LmSPS2 ratio of 1:50(w:w) for 24 h at 291 K in buffer C and the generated fragments were analyzed by SDS–PAGE. The α-chymotrypsin-treated LmSPS2 was purified by size-exclusion chromatography (Superdex 200 10/30 HR, GE Healthcare) and transferred to a polyvinylidene fluoride membrane for N-terminal sequencing (Protein Sequence Analysis at the Department of Biochemistry, University of Cambridge). A new LmSPS2 construct consisting of residues 70–398, ΔN-LmSPS2, was cloned into the pET28a expression vector (Novagen) using the 5′-AGCATATGTCGGCAACGCCTGGGCAGAAG-3′ forward and 5′-AGCTCGAGTCACACCTCCACAATTTCATACCCGTC-3′ reverse primers.
2.3. Protein crystallization and data collection
For crystallization, the ΔN-LmSPS2 (extinction coefficient of 27 305 M −1 cm−1 calculated using ProtParam; http://web.expasy.org/cgi-bin/protparam/protparam; Gasteiger et al., 2005 ▶) and TbSPS2 recombinant proteins were concentrated to 10 mg ml−1 in buffer C. Crystallization trials of the SPS2 proteins were set up in 96-well plates with commercial screens (Qiagen) using Honeybee (Digilab) and Crystal Phoenix (Art Robbins Instruments) robots for ΔN-LmSPS2 and TbSPS2, respectively. Optimization screens were carried out in 24-well plates using the hanging-drop vapour-diffusion technique with 1 µl well solution mixed with 1 µl protein solution at 10 mg ml−1 in buffer C. The microbatch experiment for TbSPS2 was carried out in Greiner microbatch plates (Hampton Research) prepared with 2 µl TbSPS2 at 10 mg ml−1 in buffer C mixed with 2 µl crystallization solution and 20 µl paraffin oil in each well and incubated at 291 K. The final crystallization conditions for ΔN-LmSPS2 and TbSPS2 were 0.2 M ammonium sulfate, 0.1 M trisodium citrate pH 5.2, 21%(w/v) PEG 4000 and 0.1 M trisodium citrate pH 5.6, 17%(w/v) PEG 3350, respectively. Crystals were cooled and stored in liquid nitrogen after rapid transfer to a cryoprotectant solution consisting of their respective mother liquor with 25%(v/v) ethylene glycol for ΔN-LmSPS2 crystals and 25%(v/v) MPD for TbSPS2 crystals. X-ray diffraction data were collected from a single crystal at a wavelength of 0.92 Å (13 477 eV) on the I04-1 beamline at the Diamond Light Source (DLS), Harwell Science and Innovation Campus, Didcot, Oxfordshire, England (Lobley et al., 2012 ▶). Integration and scaling of data were carried out using MOSFLM (Leslie, 2006 ▶) and SCALA (Winn, 2003 ▶; Evans, 2006 ▶). Data-collection and processing statistics for the crystals of ΔN-LmSPS2 and TbSPS2 are summarized in Table 1 ▶.
Table 1. Data-collection statistics for ΔN-LmSPS2 and TbSPS2.
Values in parentheses are for the outermost resolution shell.
| ΔN-LmSPS2 | TbSPS2 | |
|---|---|---|
| Data-collection parameters | ||
| X-ray source | DLS beamline I04-1 | DLS beamline I04-1 |
| Wavelength (Å) | 0.92 | 0.92 |
| Detector | PILATUS 2M | PILATUS 2M |
| Crystal-to-detector distance (mm) | 237.0 | 325.2 |
| Oscillation range per image (°) | 0.25 | 0.35 |
| No. of images | 600 | 400 |
| Crystal parameters | ||
| Space group | I212121 | P6422 |
| No of molecules in asymmetric unit | 1 | 1 |
| Unit-cell parameters (Å) | a = 55.95, b = 81.69, c = 177.92 | a = b = 110.69, c = 142.93 |
| Solvent content (%) | 57.21 | 58.18 |
| V M (Å3 Da−1) | 2.87 | 2.94 |
| Resolution range (Å) | 40.97–1.88 (1.98–1.88) | 57.29–3.40 (3.60–3.40) |
| R p.i.m. † (%) | 3.8 (39.3) | 2.2 (15.0) |
| 〈I/σ(I)〉 | 11.4 (2.8) | 20.7 (4.8) |
| Total No. of observations | 167436 (24993) | 134256 (10398) |
| No. of unique reflections | 33416 (4849) | 9061 (1285) |
| Multiplicity | 5.0 (5.2) | 13.1 (14.3) |
| Completeness (%) | 97.7 (99.9) | 99.9 (100.0) |
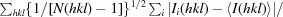
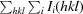
3. Results and discussion
As all attempts to crystallize LmSP2 were unsuccessful, we applied in situ proteolysis, which has been demonstrated to be a successful approach to crystallization. This technique has demonstrated efficacy in rescuing proteins that are recalcitrant to crystallization. However, the initial crystals obtained were not suitable for X-ray diffraction experiments, probably owing to heterogeneous cleavage of the protein. Therefore, to obtain reproducible crystals we cloned the stable core of the LmSPS2 protein, ΔN-LmSPS2, which lacks residues 1–69, and it was obtained with high homogeneity.
N-terminal sequencing analysis of the approximately 36 kDa fragment allowed the identification of the cleavage point at the N-terminal flexible region. The resulting ΔN-LmSPS2 protein yielded crystals (Fig. 1 ▶ a) that diffracted to 1.9 Å resolution and belonged to space group I212121, with unit-cell parameters a = 55.95, b = 81.69, c = 177.92 Å. Matthews coefficient calculation using the cell-content analysis program from the CCP4 suite is most consistent with one molecule per asymmetric unit (V M = 2.87 Å3 Da−1, 57.21% solvent content; Winn et al., 2011 ▶; Matthews, 1968 ▶).
Figure 1.

(a) ΔN-LmSPS2 crystals observed in 0.2 M ammonium sulfate, 0.1 M trisodium citrate pH 5.2, 21%(w/v) PEG 4000 after 3 d. (b) TbSPS2 crystals obtained after two months of incubation in 0.1 M trisodium citrate pH 5.6, 17%(w/v) PEG 3350 using the microbatch method (paraffin oil) in Greiner plates. These crystals were used for X-ray data collection.
Initially, the crystals obtained of TbSPS2 were anisotropic with a high solvent content. Using the microbatch method, a crystal of TbSPS2 grew after two months of incubation and diffracted to 3.4 Å resolution (Fig. 1 ▶ b). The space group was P6422 and the unit-cell parameters were a = b = 110.69, c = 142.93 Å. The monomeric molecular weight of TbSPS2 is 45 kDa and from the Matthews coefficient the crystal asymmetric unit contains one protein molecule, with a V M of 2.94 Å3 Da−1 and a solvent content of approximately 58.18%.
The first reported SPS structure from the thermophilic A. aeolicus was also obtained as an N-terminally truncated fragment (Matsumoto et al., 2008 ▶). The selenophosphate synthetases possess a flexible N-terminal region owing to the Gly-rich loop (Itoh et al., 2009 ▶). The full-length A. aeolicus structure shows that this loop alternates between an open and a closed conformation depending on the binding-site occupation. It is probable that the flexibility of this region allows large conformational changes, suggesting that this N-terminal loop is involved in a substrate-delivery mechanism (Itoh et al., 2009 ▶). The lack of residues 1–69 of LmSPS2 in ΔN-LmSPS2 resulted in crystals that diffracted to a higher resolution compared with TbSPS2. These results indicate that the flexibility of the N-terminal region of SPS2 interferes with protein–protein contacts during crystallization. A detailed study of the SPS N-terminal sequence is necessary to understand the importance of this region and its participation in the SPS catalytic mechanism.
The catalytic residue Sec/Cys of SPS (corresponding to Cys46 and Cys42 in LmSPS2 and TbSPS2, respectively) is located in the N-terminal region of Leishmania and Trypanosoma SPS2. In the ΔN-LmSPS2 construct this residue is missing; however, it has been reported (Itoh et al., 2009 ▶) that this conserved residue is not essential for ATP binding. This shows that although the truncated enzyme is not functional for the formation of selenophosphate, it may retain ATP hydrolysis activity.
We are now working to determine the ΔN-LmSPS2 phases by the molecular-replacement methodology as implemented in Phaser (McCoy et al., 2007 ▶) using the three-dimensional structure of human SPS1 (PDB entry 3fd5; Wang et al., 2009 ▶), which shares 43% sequence identity with ΔN-LmSPS2, as a search model. The structure of ΔN-LmSPS2, with 66% amino-acid sequence identity, will be a good search model to solve the structure of TbSPS2.
Acknowledgments
This research was supported in part by Research Grant 98/14138-2 from the Fundação de Amparo à Pesquisa no Estado de São Paulo (FAPESP). We would like to express our gratitude to Dr Victor Bolanos-Garcia for help with the N-terminal sequencing analysis and to Dr Susana A. Sculaccio for technical help during this study. We also kindly acknowledge the support received from the Department of Biochemistry of the University of Cambridge and Professor Dr Tom Blundell and the technical assistance provided to us at the N-terminal sequencing facility.
References
- Aeby, E., Palioura, S., Pusnik, M., Marazzi, J., Lieberman, A., Ullu, E., Söll, D. & Schneider, A. (2009). Proc. Natl Acad. Sci. USA, 106, 5088–5092. [DOI] [PMC free article] [PubMed]
- Costa, F., Oliva, M., de Jesus, T., Schenkman, S. & Thiemann, O. (2011). Mol. Biochem. Parasitol. 180, 47–50. [DOI] [PubMed]
- Donovan, J. & Copeland, P. R. (2010). Antioxid. Redox Signal. 12, 881–892. [DOI] [PMC free article] [PubMed]
- Evans, P. (2006). Acta Cryst. D62, 72–82. [DOI] [PubMed]
- Gasteiger, E., Hoogland, C., Gattiker, A., Duvaud, S., Wilkins, M. R., Appel, R. D. & Bairoch, A. (2005). The Proteomics Protocols Handbook, edited by J. M. Walker, pp. 571–607. Totowa: Humana Press.
- Glass, R. S., Singh, W. P., Jung, W., Veres, Z., Scholz, T. D. & Stadtman, T. (1993). Biochemistry, 32, 12555–12559. [DOI] [PubMed]
- Itoh, Y., Sekine, S., Matsumoto, E., Akasaka, R., Takemoto, C., Shirouzu, M. & Yokoyama, S. (2009). J. Mol. Biol. 385, 1456–1469. [DOI] [PubMed]
- Leinfelder, W., Forchhammer, K., Veprek, B., Zehelein, E. & Bock, A. (1990). Proc. Natl Acad. Sci. USA, 87, 543–547. [DOI] [PMC free article] [PubMed]
- Leslie, A. G. W. (2006). Acta Cryst. D62, 48–57. [DOI] [PubMed]
- Lobley, C. M. C., Aller, P., Douangamath, A., Reddivari, Y., Bumann, M., Bird, L. E., Nettleship, J. E., Brandao-Neto, J., Owens, R. J., O’Toole, P. W. & Walsh, M. A. (2012). Acta Cryst. F68, 1427–1433. [DOI] [PMC free article] [PubMed]
- McCoy, A. J., Grosse-Kunstleve, R. W., Adams, P. D., Winn, M. D., Storoni, L. C. & Read, R. J. (2007). J. Appl. Cryst. 40, 658–674. [DOI] [PMC free article] [PubMed]
- Matsumoto, E., Sekine, S., Akasaka, R., Otta, Y., Katsura, K., Inoue, M., Kaminishi, T., Terada, T., Shirouzu, M. & Yokoyama, S. (2008). Acta Cryst. F64, 453–458. [DOI] [PMC free article] [PubMed]
- Matthews, B. (1968). J. Mol. Biol. 33, 491–497. [DOI] [PubMed]
- Noinaj, N., Wattanasak, R., Lee, D. Y., Wally, J. L., Piszczek, G., Chock, P. B., Stadtman, T. C. & Buchanan, S. K. (2012). J. Bacteriol. 194, 499–508. [DOI] [PMC free article] [PubMed]
- Persson, B. C., Böck, A., Jäckle, H. & Vorbrüggen, G. (1997). J. Mol. Biol. 274, 174–180. [DOI] [PubMed]
- Sculaccio, S., Rodrigues, E., Cordeiro, A., Magalhaes, A., Braga, A., Alberto, E. & Thiemann, O. (2008). Mol. Biochem. Parasitol. 162, 165–171. [DOI] [PubMed]
- Su, D., Hohn, M. J., Palioura, S., Sherrer, R. L., Yuan, J., Söll, D. & O’Donoghue, P. (2009). IUBMB Life, 61, 35–39. [DOI] [PubMed]
- Veres, Z., Kim, I. Y., Scholz, T. D. & Stadtman, T. C. (1994). J. Biol. Chem. 269, 10597–10603. [PubMed]
- Wang, K., Wang, J., Li, L. & Su, X. (2009). J. Mol. Biol. 390, 747–759. [DOI] [PubMed]
- Weiss, M. S. (2001). J. Appl. Cryst. 34, 130–135.
- Weiss, M. S. & Hilgenfeld, R. (1997). J. Appl. Cryst. 30, 203–205.
- Weiss, M. S., Metzner, H. J. & Hilgenfeld, R. (1998). FEBS Lett. 423, 291–296. [DOI] [PubMed]
- Wernimont, A. & Edwards, A. (2009). PLoS One, 4, e5094. [DOI] [PMC free article] [PubMed]
- Winn, M. D. (2003). J. Synchrotron Rad. 10, 23–25. [DOI] [PubMed]
- Winn, M. D. et al. (2011). Acta Cryst. D67, 235–242.