

Two crystal forms of β-carbonic anhydrase psCA3 from P. aeruginosa were grown. Crystal form A belonged to space group P212121, with unit-cell parameters a = 81.9, b = 84.9, c = 124.2 Å, and diffracted X-rays to 2.9 Å resolution; crystal form B belonged to space group P21212, with unit-cell parameters a = 69.9, b = 77.7, c = 88.5 Å, and diffracted X-rays to 3.0 Å resolution.
Keywords: carbonic anhydrases, β-class carbonic anhydrases, Pseudomonas aeruginosa
Abstract
Pseudomonas aeruginosa is a Gram-negative bacterium that causes life-threatening infections in susceptible individuals and is resistant to most clinically available antimicrobials. Genomic and proteomic studies have identified three genes, pa0102, pa2053 and pa4676, in P. aeruginosa PAO1 encoding three functional β-carbonic anhydrases (β-CAs): psCA1, psCA2 and psCA3, respectively. These β-CAs could serve as novel antimicrobial drug targets for this pathogen. X-ray crystallographic structural studies have been initiated to characterize the structure and function of these proteins. This communication describes the production of two crystal forms (A and B) of β-CA psCA3. Form A diffracted to a resolution of 2.9 Å; it belonged to space group P212121, with unit-cell parameters a = 81.9, b = 84.9, c = 124.2 Å, and had a calculated Matthews coefficient of 2.23 Å3 Da−1 assuming four molecules in the crystallographic asymmetric unit. Form B diffracted to a resolution of 3.0 Å; it belonged to space group P21212, with unit-cell parameters a = 69.9, b = 77.7, c = 88.5 Å, and had a calculated Matthews coefficient of 2.48 Å3 Da−1 assuming two molecules in the crystallographic asymmetric unit. Preliminary molecular-replacement solutions have been determined with the PHENIX AutoMR wizard and refinement of both crystal forms is currently in progress.
1. Introduction
Carbonic anhydrases (CAs) are mainly zinc metalloenzymes that catalyze the reversible hydration of carbon dioxide to bicarbonate and a proton (Krishnamurthy et al., 2008 ▶). CAs are ubiquitously expressed and are found in almost all living organisms, where they play a vital role in various physiological processes. Such processes include respiration, pH regulation, CO2 fixation, photosynthesis and ion transport (Supuran, 2008 ▶). There are five evolutionary and structurally distinct classes of CA: α-CA (expressed in vertebrates and the only class found in mammals), β-CA (found in plants, fungi and prokaryotes), γ-CA (found only in archaebacteria and diatoms) and δ-CA and ζ-CA (found in diatoms) (Aggarwal et al., 2013 ▶).
Depending on the coordination of the Zn2+ in the active site, β-CAs are divided into two subclasses or clades designated type I and type II (Lotlikar et al., 2013 ▶). Type I β-CAs have an ‘open’ active site with a Zn2+ ion tetrahedrally coordinated by two Cys residues, one His residue and a water molecule for catalysis, and show activity both above and below pH 8 (Smith & Ferry, 1999 ▶; Covarrubias et al., 2005 ▶, 2006 ▶; Nishimori et al., 2010 ▶). Type II β-CAs have a ‘closed’ active site with the Zn2+ coordinated by a His, an Asp and two Cys residues (Cronk et al., 2001 ▶, 2006 ▶; Covarrubias et al., 2005 ▶, 2006 ▶) and show no catalytic activity below pH 8 owing to the absence of a coordinated water molecule. However, above pH 8 a nearby conserved Arg residue forms a salt bridge to the Zn2+-coordinated Asp residue and this liberates this position, allowing a solvent molecule to coordinate to the Zn2+, which allows catalytic activity (Covarrubias et al., 2006 ▶).
Pseudomonas aeruginosa is commonly associated with biofilm infections of the lungs, heart, wounds and urinary tract and is of great concern in immunocompromised individuals (Richard et al., 1994 ▶; Mesaros et al., 2007 ▶). These infections are becoming increasingly difficult to treat using traditional antibiotic therapy and are often not eradicated by host defensive processes (Jesaitis et al., 2003 ▶; Walters et al., 2003 ▶). Therefore, the development of alternative drug therapies is of high importance. Earlier sequence analysis of the P. aeruginosa PAO1 genome has revealed three genes, pa0102, pa2053 and pa4676, encoding β-CAs which share 28–45% amino-acid sequence identity and belong to the two clades of β-CAs. We also showed that all three enzymes are expressed in PAO1 cells, contain Zn2+ and hydrate CO2 (Lotlikar et al., 2013 ▶). These enzymes may be involved in the formation of CaCO3 deposits and thus cause soft-tissue calcification, which is commonly associated with chronic bacterial infections (Banks et al., 2010 ▶). Therefore, β-CAs could serve as potential targets for developing alternatives to the conventional antibiotic, inhibitor-based treatments of P. aeruginosa infections. The design and development of inhibitors that exhibit a high affinity for the bacterial β-CA, but have no effect on human α-CAs, require detailed analysis of the crystal structures of the P. aeruginosa β-CAs.
2. Materials and methods
Cloning, protein expression and purification were carried out as described previously (Lotlikar et al., 2013 ▶). Briefly, a pET15b plasmid construct containing the pa4676 gene PCR-amplified using P. aeruginosa PAO1 genomic DNA as a template was transformed into Escherichia coli Tuner(DE3) cells for the production of a His-fusion protein. The cells were grown at 310 K in LB medium containing 100 µg ml−1 ampicillin and 0.05 mM ZnSO4. At an A 600 of 0.6, fusion-protein expression was induced for 3 h at 310 K by the addition of isopropyl β-d-1-thiogalactopyranoside (IPTG) to a final concentration of 1 mM and of 0.5 mM ZnSO4. Cells were harvested by centrifugation and resuspended in 20 mM Tris pH 7.9, 5 mM imidazole, 150 mM NaCl. They were then lysed by sonication. The supernatant was loaded onto Ni2+-charged IMAC columns and washed with 20 mM Tris pH 7.9, 60 mM imidazole, 150 mM NaCl. Protein was eluted with 20 mM Tris pH 7.9, 300 mM imidazole, 150 mM NaCl. To ensure purity from other proteins, the elution fractions were resolved using SDS–PAGE followed by Coomassie Blue R-250 staining. The selected fractions were then dialyzed against 20 mM Tris–HCl pH 7.9, 150 mM imidazole, 100 mM NaCl, 10% glycerol for 2 h and then against 20 mM Tris–HCl pH 7.9, 50 mM imidazole, 50 mM NaCl, 5% glycerol for 1 h, with final dialysis (three times, 1 h each) and storage in 20 mM Tris–HCl pH 8.3. The β-CA psCA3 was concentrated using an Amicon Ultra-15 Centrifugal Filter Unit (Millipore, Billerica, Massachusetts, USA) and its concentration was determined by UV–Vis spectroscopy at 280 nm using a molar extinction coefficient of 36 440 M −1 cm−1.
Initial crystallization screening was performed in 96-well Intelli-Plate sitting-drop vapor-diffusion crystallization plates (Art Robbins Instruments, Sunnyvale, California, USA) using Hampton Research Crystal Screen 2, PEG/Ion, PEG/Ion 2 and an in-house sodium citrate screen (screening conditions varied from 1.1 to 1.8 M sodium citrate, Tris–HCl pH 7.1–8.1) prepared by the Rigaku Alchemist DT. Drops consisting of protein solution at 10 mg ml−1 (in 20 mM Tris–HCl pH 8.3) and precipitant solution at two different ratios (1:1 and 2:3 protein:precipitant solution) were equilibrated at 290 K against a 60 µl reservoir containing precipitant solution.
Two crystal forms (A and B) of β-CA psCA3 were obtained. Crystals of both forms A and B were cryoprotected using the precipitant solution with 20% glycerol prior to cooling in a nitrogen stream.
Diffraction data for crystal form A were collected on an in-house R-AXIS IV++ image-plate detector using an RU-H3R rotating Cu-anode generator (λ = 1.5418 Å) operating at 50 kV and 22 mA. Data were collected with a crystal-to-detector distance of 150 mm and a 0.5° oscillation angle with an exposure time of 1800 s per image over 180 frames.
Diffraction data for crystal form B were collected at the F1 station at Cornell High Energy Synchrotron Source (CHESS F1; λ = 0.9177 Å) on an ADSC Q-270 detector using the microfocused beam. Data were collected with a crystal-to-detector distance of 150 mm and a 0.5° oscillation angle with an exposure time of 20 s per image over 110 frames.
The data sets were integrated, merged and scaled using HKL-2000 (Otwinowski & Minor, 1997 ▶). Phasing was carried out with the PHENIX suite of programs (Adams et al., 2010 ▶) using the auto molecular-replacement procedure to obtain the initial phases using a previously solved β-CA structure with water molecules removed (PDB entry 1g5c; Strop et al., 2001 ▶) and with 18% sequence identity to psCA3.
3. Results and discussion
The β-CA psCA3 was overexpressed and isolated with a typical yield of 50 mg per litre of bacterial culture. Two crystal forms (A and B) of psCA3 were grown by the sitting-drop vapor-diffusion method.
Crystal form A was grown from a precipitant consisting of 1.63 M sodium citrate pH 7.4 (Fig. 1 ▶ a). Diffraction data to 2.9 Å resolution were collected in-house (Fig. 2 ▶). The crystals were shown to belong to space group P212121, with unit-cell parameters a = 81.9, b = 84.9, c = 124.2 Å and an R merge of 11.8%. Data-collection statistics and processing parameters are summarized in Table 1 ▶. Considering the P212121 space group and a molecular mass of 24 200 Da, a Matthews coefficient (V M; Matthews, 1968 ▶) of 2.23 Å3 Da−1 was calculated assuming the presence of four molecules in the crystallographic asymmetric unit.
Figure 1.
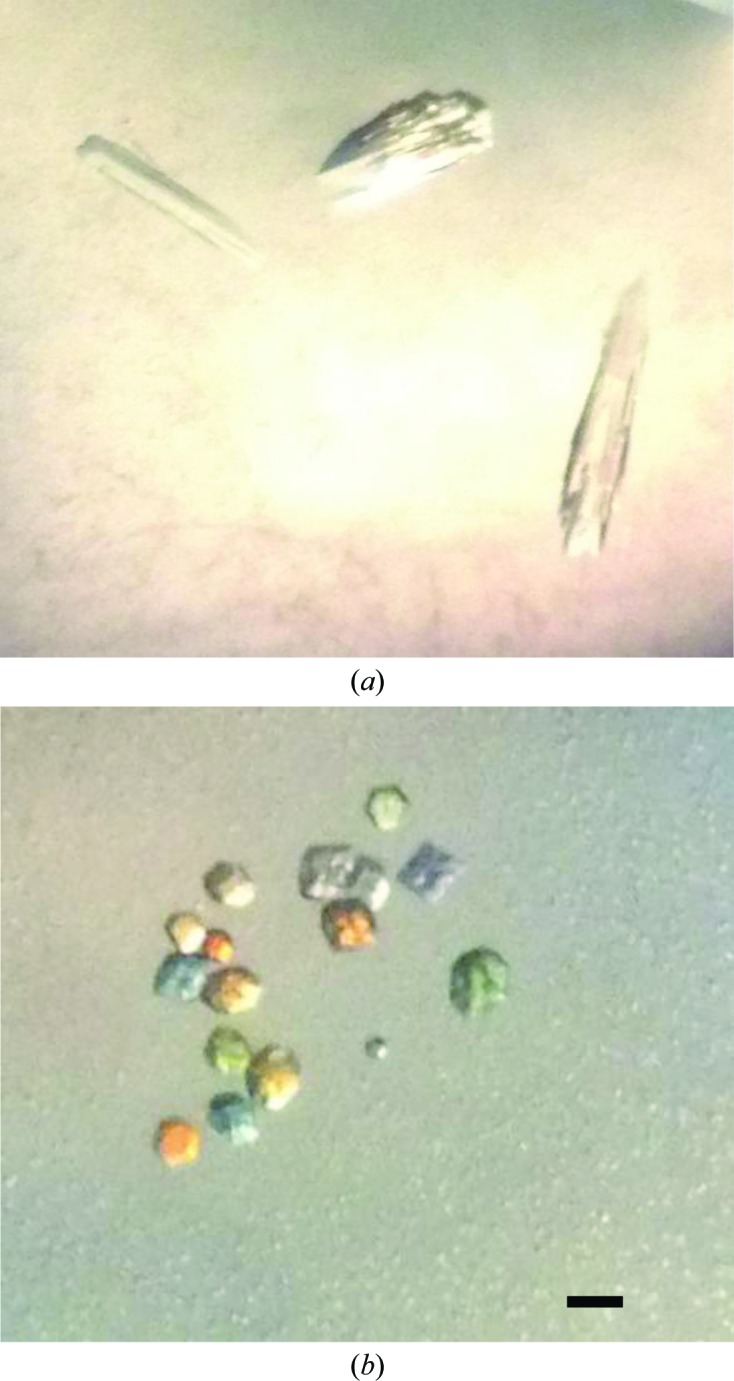
Optical photograph of β-CA psCA3 crystals from P. aeruginosa. (a) Form A, grown in 1.2 M sodium citrate, 20 mM Tris–HCl pH 7.2 at 290 K. (b) Form B, grown in 2.2 M ammonium sulfate, 0.1 M malic acid, 0.1 M imidazole pH 7.5 at 290 K. The bar indicates 0.05 mm.
Figure 2.

X-ray diffraction image of crystal form A of β-CApsCA3. The image was collected in-house on an R-AXIS IV++ image-plate detector using an RU-H3R rotating Cu-anode generator (λ = 1.5418 Å) operating at 50 kV and 22 mA. The crystal-to-detector distance was 150 mm, with 0.5° oscillation angle and an exposure time of 1800 s. The circle represents 2.9 Å resolution.
Table 1. Data-collection statistics and processing parameters for crystal forms A and B .
Values in parentheses are for the outermost resolution shell.
| Crystal form | A | B |
|---|---|---|
| Beamline | Rotating Cu anode | CHESS F1 |
| Detector | R-AXIS IV++ | ADSC Q270 CCD |
| Crystal-to-detector distance (mm) | 150 | 150 |
| Wavelength (Å) | 1.518 | 0.9177 |
| Temperature (K) | 100 | 100 |
| Resolution range (Å) | 20.00–2.90 (3.00–2.90) | 20.00–3.00 (3.11–3.00) |
| Space group | P212121 | P21212 |
| Unit-cell parameters (Å) | a = 81.9, b = 84.9, c = 124.2 | a = 69.9, b = 77.7, c = 88.5 |
| No. of measured reflections | 19275 | 20439 |
| No. of unique reflections | 18118 | 7679 |
| Multiplicity | 2.7 (2.5) | 2.7 (2.7) |
| Completeness (%) | 95.0 (90.6) | 76.1 (81.5) |
| Mean I/σ(I) | 13.8 (3.2) | 24.5 (12.3) |
| R merge † (%) | 11.8 (37.9) | 7.6 (12.6) |
| V M (Å3 Da−1) | 2.23 | 2.48 |
R
merge is defined as 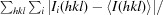
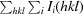 × 100, where I
i(hkl) is the intensity of an individual reflection and 〈I(hkl)〉 is the average intensity for this reflection; the summation is over all intensities.
× 100, where I
i(hkl) is the intensity of an individual reflection and 〈I(hkl)〉 is the average intensity for this reflection; the summation is over all intensities.
Crystal form B was grown from a precipitant consisting of 2.2 M ammonium sulfate, 0.1 M malic acid, 0.1 M imidazole pH 7.5 (Fig. 1 ▶ b). Diffraction data to 3.0 Å resolution were collected at CHESS F1. The crystals were shown to belong to space group P21212, with unit-cell parameters a = 69.9, b = 77.7, c = 88.5 Å and an R merge of 7.6%. Data-collection statistics and processing parameters are summarized in Table 1 ▶. Considering the P21212 space group and a molecular mass of 24 200 Da, a Matthews coefficient (V M; Matthews, 1968 ▶) of 2.48 Å3 Da−1 was calculated assuming the presence of two molecules in the crystallographic asymmetric unit.
Currently, we are optimizing these psCA3 crystallization conditions in an effort to obtain better diffracting crystals. One approach is to cocrystallize psCA3 with a sulfonamide inhibitor such as acetazolamide that would serve to stabilize the protein, thus promoting crystallization. Sulfonamides are potent inhibitors of CAs (Alterio et al., 2012 ▶) and acetazolamide is a known inhibitor of another β-CA: that from the unicellular green alga Coccomyxa (Huang et al., 2011 ▶). Although this approach has been shown to produce better ordered crystals, the drawback is that the active site is inhibited and the structural study to understand the enzyme mechanism is somewhat limited.
A potential reason for obtaining lower-resolution diffraction data from seemingly visually ‘good’ crystals could be that the cryoprotectant (20% glycerol with precipitant solution) used in both cases failed to successfully protect the crystals during the cooling process, as indicated by the higher than expected mosaicity spread of ∼0.8°. Using an alternative cryoprotectant such as 50%(v/v) Paratone-N and 50%(v/v) paraffin oil, a combination which works well for protein crystals (Hope, 1988 ▶), may help in minimizing damage during flash-cooling.
In conjunction with the use of different screening conditions, inhibitors and alternate cryoprotectants, microseeding is another approach that will be considered in an attempt to obtain better diffracting crystals. That is, transfer of the available submicroscopic crystals of psCA3 into the protein–precipitant drop, which may aid in the nucleation process.
Once the optimum conditions have been resolved, the hanging-drop variant of the vapor-diffusion method in 24-well plates can be adopted to grow larger crystals of psCA3.
Initial electron-density maps were viewed for both crystal forms using the graphics program Coot (Emsley & Cowtan, 2004 ▶); they show that psCA3 packs as a dimer of dimers in both crystal forms.
We are now refining the two crystal form structures and this will give us an insight into the psCA3 active site in order to understand its catalytic mechanism and also enable us to perform a comparative study to determine whether crystal packing has any effect on the structures. Furthermore, we can now start a structure-based drug-screening study to develop new inhibitors for the bacterial β-CA.
Acknowledgments
This work was supported by a grant from the NIH (GM25154). RM would also like to thank the Center of Structural Biology for support of the X-ray facility at UF. We would also like to thank the MacCHESS staff for their help during X-ray diffraction data collection at the Cornell High Energy Synchrotron (CHESS) Facility, Ithaca.
References
- Adams, P. D. et al. (2010). Acta Cryst. D66, 213–221.
- Aggarwal, M., Boone, C. D., Kondeti, B. & McKenna, R. (2013). J. Enzyme Inhib. Med. Chem. 28, 267–277. [DOI] [PubMed]
- Alterio, V., Di Fiore, A., D’Ambrosio, K., Supuran, C. T. & De Simone, G. (2012). Chem. Rev. 112, 4421–4468. [DOI] [PubMed]
- Banks, E. D., Taylor, N. M., Gulley, J., Lubbers, B. R., Giarrizzo, J. G., Bullen, H. A., Hoehler, T. M. & Barton, H. A. (2010). Geomicrobiol. J. 27, 444–454.
- Covarrubias, A. S., Bergfors, T., Jones, T. A. & Högbom, M. (2006). J. Biol. Chem. 281, 4993–4999. [DOI] [PubMed]
- Covarrubias, A. S., Larsson, A. M., Högbom, M., Lindberg, J., Bergfors, T., Björkelid, C., Mowbray, S. L., Unge, T. & Jones, T. A. (2005). J. Biol. Chem. 280, 18782–18789. [DOI] [PubMed]
- Cronk, J. D., Endrizzi, J. A., Cronk, M. R., O’neill, J. W. & Zhang, K. Y. J. (2001). Protein Sci. 10, 911–922. [DOI] [PMC free article] [PubMed]
- Cronk, J. D., Rowlett, R. S., Zhang, K. Y. J., Tu, C., Endrizzi, J. A., Lee, J., Gareiss, P. C. & Preiss, J. R. (2006). Biochemistry, 45, 4351–4361. [DOI] [PubMed]
- Emsley, P. & Cowtan, K. (2004). Acta Cryst. D60, 2126–2132. [DOI] [PubMed]
- Hope, H. (1988). Acta Cryst. B44, 22–26. [DOI] [PubMed]
- Huang, S., Hainzl, T., Grundström, C., Forsman, C., Samuelsson, G. & Sauer-Eriksson, A. E. (2011). PLoS One, 6, e28458. [DOI] [PMC free article] [PubMed]
- Jesaitis, A. J., Franklin, M. J., Berglund, D., Sasaki, M., Lord, C. I., Bleazard, J. B., Duffy, J. E., Beyenal, H. & Lewandowski, Z. (2003). J. Immunol. 171, 4329–4339. [DOI] [PubMed]
- Krishnamurthy, V. M., Kaufman, G. K., Urbach, A. R., Gitlin, I., Gudiksen, K. L., Weibel, D. B. & Whitesides, G. M. (2008). Chem. Rev. 108, 946–1051. [DOI] [PMC free article] [PubMed]
- Lotlikar, S. R., Hnatusko, S., Dickenson, N. E., Choudhari, S. P., Picking, W. L. & Patrauchan, M. A. (2013). Microbiology, 10.1099/mic.0.066357-0. [DOI] [PMC free article] [PubMed]
- Matthews, B. W. (1968). J. Mol. Biol. 33, 491–497. [DOI] [PubMed]
- Mesaros, N., Nordmann, P., Plésiat, P., Roussel-Delvallez, M., Van Eldere, J., Glupczynski, Y., Van Laethem, Y., Jacobs, F., Lebecque, P., Malfroot, A., Tulkens, P. M. & Van Bambeke, F. (2007). Clin. Microbiol. Infect. 13, 560–578. [DOI] [PubMed]
- Nishimori, I., Minakuchi, T., Maresca, A., Carta, F., Scozzafava, A. & Supuran, C. T. (2010). Curr. Pharm. Des. 16, 3300–3309. [DOI] [PubMed]
- Otwinowski, Z. & Minor, W. (1997). Methods Enzymol. 276, 307–326. [DOI] [PubMed]
- Richard, P., Le Floch, R., Chamoux, C., Pannier, M., Espaze, E. & Richet, H. (1994). J. Infect. Dis. 170, 377–383. [DOI] [PubMed]
- Smith, K. S. & Ferry, J. G. (1999). J. Bacteriol. 181, 6247–6253. [DOI] [PMC free article] [PubMed]
- Strop, P., Smith, K. S., Iverson, T. M., Ferry, J. G. & Rees, D. C. (2001). J. Biol. Chem. 276, 10299–10305. [DOI] [PubMed]
- Supuran, C. T. (2008). Nature Rev. Drug Discov. 7, 168–181. [DOI] [PubMed]
- Walters, M. C. III, Roe, F., Bugnicourt, A., Franklin, M. J. & Stewart, P. S. (2003). Antimicrob. Agents Chemother. 47, 317–323. [DOI] [PMC free article] [PubMed]