

The cloning, expression, purification, crystallization and preliminary X-ray diffraction analysis of a novel AB5 toxin EcxAB are described.
Keywords: AB5 toxins, Escherichia coli, co-expression, proteases
Abstract
AB5 toxins are key virulence factors found in a range of pathogenic bacteria. AB5 toxins consist of two components: a pentameric B subunit that targets eukaryotic cells by binding to glycans located on the cell surface and a catalytic A subunit that disrupts host cellular function following internalization. To date, the A subunits of AB5 toxins either have RNA-N-glycosidase, ADP-ribosyltransferase or serine protease activity. However, it has been suggested that a novel AB5 toxin produced by clinical isolates of Escherichia coli and Citrobacter freundii has an A subunit with metalloproteinase activity. Here, the expression, purification and crystallization of this novel AB5 toxin from E. coli (EcxAB) and the collection of X-ray data to 1.9 Å resolution are reported.
1. Introduction
AB5 toxins are of immense importance because they are key virulence determinants of pathogenic bacteria, notably Escherichia coli, Shigella dysenteriae and Vibrio cholerae, which collectively cause massive global morbidity and mortality. These toxins comprise a catalytic A subunit linked to a pentameric B subunit. They exert their effects in a two-step process: the B subunit pentamer first binds to specific glycan receptors on the cell surface, triggering uptake and intracellular trafficking of the holotoxin; this is followed by inhibition or corruption of essential host functions mediated by the enzymatic activity of the A subunit (Beddoe et al., 2010 ▶).
Until a decade ago, the enzymatic activities ascribed to these A subunits were either ADP-ribosyltransferase or RNA-N-glycosidase. However, a novel AB5 toxin termed subtilase cytotoxin (SubAB) with a proteolytic A subunit has since been described (Paton et al., 2004 ▶). The sole molecular target of SubAB was identified as the endoplasmic reticulum (ER) molecular chaperone BiP/GRP78, a master regulator of ER function, while its B subunit had unique specificity for a sugar acquired from dietary intake (Paton et al., 2006 ▶; Byres et al., 2008 ▶). Since the discovery of SubAB, the enzymatic activity of SubA has been exploited as a powerful cell-biological tool to probe the role of BiP in diverse cellular processes (Hu et al., 2009 ▶; Eletto et al., 2012 ▶; Schäuble et al., 2012 ▶). Previous studies have also identified additional putative AB5 toxin operons in E. coli and Citrobacter freundii diarrhoea isolates (designated ecxAB and cfxAB, respectively) by cross-reaction with antibodies to the B subunits of cholera toxin (CtxB) and heat-labile enterotoxin (LTB) (Karasawa et al., 2002 ▶). The expressed B subunits of these novel toxins (EcxB and CfxB) are 87% identical to each other and 72–74% identical to both CtxB and LTB. The crystal structure of CfxB superimposes closely on CtxB, and CfxB bound glycans known to interact with CtxB and LTB (Jansson et al., 2010 ▶). Interestingly, EcxA and CfxA both contain a sequence motif (HEXGHXXGXXH) that is characteristic of the active site of bacterial metalloproteinases (Bode et al., 1999 ▶). However, it has not been shown that EcxA or CfxA can associate with their cognate B subunits.
To investigate whether EcxAB represents a novel AB5 toxin, we have prepared and crystallized the EcxAB complex. Structural determination of EcxAB will provide valuable insight into the mechanistic function of this novel putative AB5 toxin.
2. Materials and methods
2.1. Cloning
The cloning of the ecxA and ecxB genes into modified pBAD18 vector (Guzman et al., 1995 ▶) was performed by chemical synthesis of both genes (GenScript, New Jersey, USA), replacing the native signal sequence with the pelB signal sequence and with the addition of restriction sites NdeI and XmaI and NcoI and XhoI flanking the ecxA and the ecxB open reading frames, respectively. The ecxA sequence encoding residues 22ERTPN…SGGDL285 of the mature protein was directly cloned into pBAD-18 using NdeI and XmaI restriction sites, while the ecxB sequence encoding residues 22ATPQN…IELSN125 of the mature protein was cloned into pET-23 (Novagen) via NcoI and XhoI sites, resulting in a protein with a C-terminal poly-His tag. To produce the co-expression vector for EcxAB, the ribosome binding site and ecxB were excised by restriction enzymes XbaI and HindIII and cloned downstream of ecxA (Fig. 1 ▶ a). The clone obtained was confirmed by DNA sequencing.
Figure 1.

(a) Schematic of the co-expression vector for EcxAB. (b) 18% SDS–PAGE of purified non-boiled EcxAB. Molecular weights (in kDa) are indicated on the left.
2.2. Overexpression of recombinant protein
Recombinant protein expression of EcxAB was performed in E. coli strain BL21(DE3). A starter culture was grown overnight in LB medium containing ampicillin (100 µg ml −1) at 310 K. The starter culture was used at a dilution of 1:100 to inoculate 800 ml fresh LB medium containing 100 µg ml−1 ampicillin and was grown at 310 K until the OD600 reached 0.6. Expression of EcxAB was induced with 0.2%(w/v) l-arabinose and the cells were allowed to grow for a further 4 h at 310 K. The cells were collected following centrifugation at 5000g for 20 min at 277 K and stored at 193 K.
2.3. Purification of recombinant protein
Cells were thawed and resuspended in Tris-buffered saline (TBS; 20 mM Tris pH 8, 150 mM NaCl) and lysed using a cell disruptor (Avestin) in the presence of 0.2 µM phenylmethanesulfonylfluoride (PMSF). Cell debris was removed by centrifugation at 30 000g for 30 min at 277 K. All subsequent purification steps were performed at 293 K. High-speed supernatant was collected and loaded onto 5 ml nickel Sepharose beads (GE Healthcare) equilibrated in binding buffer (50 mM Tris–HCl pH 8.0, 500 mM NaCl, 60 mM imidazole). Unbound material was removed by washing with two column volumes of binding buffer. The protein was eluted using four column volumes of elution buffer (50 mM Tris–HCl pH 8.0, 500 mM NaCl, 1 M imidazole). Fractions containing EcxAB were pooled, buffer-exchanged into TBS and concentrated to 5 ml by low-speed centrifugation using a Vivaspin-2 50 000 Da molecular-weight cutoff (Sartorius Stedim Biotech). EcxAB was further purified by size-exclusion chromatography using an ÄKTA Basic Fast Protein Liquid Chromatography (FPLC) system with a Superdex S200 16/60 gel-filtration column (GE Healthcare Life Sciences) equilibrated in TBS. The molecular weight, purity and identity of the EcxAB preparation were confirmed by SDS–PAGE (Fig. 1 ▶ b), Western blotting with anti-hexahistidine antibody (R&D systems) and N-terminal sequencing. The purified protein was concentrated to 5 mg ml−1 and stored at 277 K for use in crystallization trials. The concentration of EcxAB was determined spectrophotometrically (NanoDrop 1000, Thermo Scientific) at 280 nm and was calculated using an extinction coefficient of 81 430 M −1 cm−1.
2.4. Crystallization
The initial crystallization experiments involved screening 192 conditions in commercially available kits from Hampton Research (Crystal Screen HT and PEG/Ion HT) using the CrystalMation integrated robotic workstation (Rigaku) and the sitting-drop vapour-diffusion technique. Initial crystals were obtained in condition E9 of the PEG/Ion HT screen [4%(v/v) Tacsimate pH 4, 12%(w/v) PEG 3350]. Optimization of the EcxAB crystallization conditions was performed by fine-screening the initial condition by varying the protein and precipitant concentrations using the hanging-drop method in 24-well Linbro plates (Hampton Research) with drops consisting of 1 µl protein solution and 1 µl precipitant solution and a reservoir volume of 500 µl. Crystals appeared in most conditions; however, the best crystals appeared after 1 d in 4%(v/v) Tacsimate pH 4.5, 10%(w/v) PEG 3350 (Fig. 2 ▶), which was found after several optimization plates.
Figure 2.
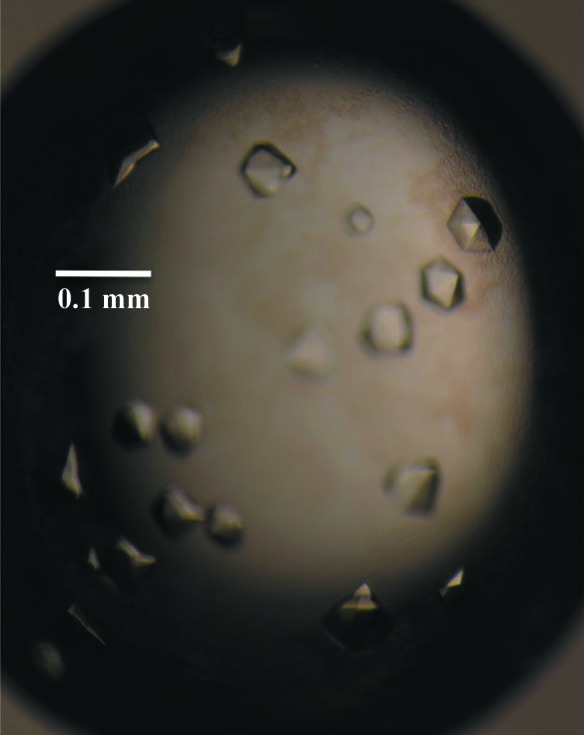
Diffraction-quality crystals of EcxAB.
2.5. X-ray data collection
For data collection, crystals were transferred to cryoprotectant solution consisting of 20%(v/v) glycerol, 4%(v/v) Tacsimate pH 4.5, 10%(w/v) PEG 3350 before cooling to 100 K in a stream of nitrogen gas. The majority of the crystals diffracted to better than 2.2 Å resolution (Fig. 3 ▶) and a complete data set was collected from a single crystal on the MX2 beamline at the Australian Synchrotron using an ADSC Quantum 315r CCD detector. The data were processed with MOSFLM (Leslie & Powell, 2007 ▶) and various programs from the CCP4 suite (Winn et al., 2011 ▶). The final statistics for data collection and processing are summarized in Table 1 ▶.
Figure 3.

A diffraction pattern of an EcxAB crystal.
Table 1. Crystal and data-collection statistics for apo EcxAB.
Values in parentheses are for the outer shell.
| Diffraction source | MX2, Australian Synchrotron |
| Detector | ADSC Quantum 315r CCD |
| Space group | P6522 |
| Temperature (K) | 100 |
| Unit-cell parameters (Å, °) | a = b = 120.7, c = 274.3, α = β = γ = 90.0 |
| Resolution range (Å) | 39.5–1.80 (1.90–1.80) |
| Wavelength (Å) | 0.9537 |
| Total No. of reflections | 109241 |
| No. of unique reflections | 15721 |
| R merge † (%) | 7.2 (55.1) |
| Mean I/σ(I)‡ | 8.2 (1.4) |
| Completeness (%) | 99.9 (100) |
R
merge = 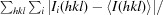
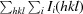 , where Ii(hkl) is the scaled intensity of the ith measurement and 〈I(hkl)〉 is the mean intensity for that reflection.
, where Ii(hkl) is the scaled intensity of the ith measurement and 〈I(hkl)〉 is the mean intensity for that reflection.
I is the intergrated intensity and σ(I) is the estimated standard deviation of that intensity.
3. Results and discussion
The ecxA and ecxB genes were successfully cloned, co-expressed, purified and crystallized using vapour-diffusion methods. The EcxAB complex was purified to homogeneity and the pentameric arrangement of EcxB was retained when the sample was not boiled before SDS–PAGE analysis (Fig. 1 ▶ b). This is a hallmark of B-subunit pentamers (Lebens et al., 1996 ▶). This expression and purification strategy routinely produced yields of 10 mg l−1. Crystals of EcxAB were obtained in 4%(v/v) Tacsimate pH 4.5, 10%(w/v) PEG 3350 (Fig. 2 ▶) and a complete diffraction data set to 1.9 Å resolution was collected at 100 K. The preliminary crystallographic analysis indicated that the crystals belonged to space group P6522, with unit-cell parameters a = b = 120.7, c = 274.3 Å, α = β = γ = 90.0°. The data-collection and processing statistics are summarized in Table 1 ▶. Based on Matthews coefficient calculations, one EcxAB complex (∼60% solvent content) could be accommodated in the asymmetric unit (Matthews, 1968 ▶). Structure determination by means of molecular replacement appears feasible as the known structure of C. freundii CfxB (PDB entry 2wv6; Jansson et al., 2010 ▶) shares 87% sequence identity with EcxB. Structure verification and model rebuilding are currently in progress.
Acknowledgments
We thank the staff of the Australian Synchrotron and Monash Macromolecular Crystallization Facility for assistance with crystallization and X-ray data collection. The Australian Research Council and the National Health and Medical Research Council of Australia supported this work. TB is a Pfizer Australian Research Fellow, AWP is an Australian Research Council DORA Fellow, JCP is an NHMRC Senior Principal Research Fellow and JR is an NHMRC Australia Fellow.
References
- Beddoe, T., Paton, A. W., Le Nours, J., Rossjohn, J. & Paton, J. C. (2010). Trends Biochem. Sci. 35, 411–418. [DOI] [PMC free article] [PubMed]
- Bode, W., Fernandez-Catalan, C., Tschesche, H., Grams, F., Nagase, H. & Maskos, K. (1999). Cell. Mol. Life Sci. 55, 639–652. [DOI] [PMC free article] [PubMed]
- Byres, E., Paton, A. W., Paton, J. C., Löfling, J. C., Smith, D. F., Wilce, M. C. J., Talbot, U. M., Chong, D. C., Yu, H., Huang, S., Chen, X., Varki, N. M., Varki, A., Rossjohn, J. & Beddoe, T. (2008). Nature (London), 456, 648–652. [DOI] [PMC free article] [PubMed]
- Eletto, D., Maganty, A., Eletto, D., Dersh, D., Makarewich, C., Biswas, C., Paton, J. C., Paton, A. W., Doroudgar, S., Glembotski, C. C. & Argon, Y. (2012). J. Cell Sci. 125, 4865–4875. [DOI] [PMC free article] [PubMed]
- Guzman, L. M., Belin, D., Carson, M. J. & Beckwith, J. (1995). J. Bacteriol. 177, 4121–4130. [DOI] [PMC free article] [PubMed]
- Hu, C.-C. A., Dougan, S. K., Winter, S. V., Paton, A. W., Paton, J. C. & Ploegh, H. L. (2009). J. Exp. Med. 206, 2429–2440. [DOI] [PMC free article] [PubMed]
- Jansson, L., Angström, J., Lebens, M., Imberty, A., Varrot, A. & Teneberg, S. (2010). Biochimie, 92, 482–490. [DOI] [PubMed]
- Karasawa, T., Ito, H., Tsukamoto, T., Yamasaki, S., Kurazono, H., Faruque, S. M., Nair, G. B., Nishibuchi, M. & Takeda, Y. (2002). Infect. Immun. 70, 7153–7155. [DOI] [PMC free article] [PubMed]
- Leslie, A. G. W. & Powell, H. R. (2007). Evolving Methods for Macromolecular Crystallography, edited by R. J. Read & J. L. Sussman, pp. 41–51. Dordrecht: Springer.
- Lebens, M., Shahabi, V., Bäckström, M., Houze, T., Lindblad, N. & Holmgren, J. (1996). Infect. Immun. 64, 2144–2150. [DOI] [PMC free article] [PubMed]
- Matthews, B. W. (1968). J. Mol. Biol. 33, 491–497. [DOI] [PubMed]
- Paton, A. W., Beddoe, T., Thorpe, C. M., Whisstock, J. C., Wilce, M. C. J., Rossjohn, J., Talbot, U. M. & Paton, J. C. (2006). Nature (London), 443, 548–552. [DOI] [PubMed]
- Paton, A. W., Srimanote, P., Talbot, U. M., Wang, H. & Paton, J. C. (2004). J. Exp. Med. 200, 35–46. [DOI] [PMC free article] [PubMed]
- Schäuble, N., Lang, S., Jung, M., Cappel, S., Schorr, S., Ulucan, Ö., Linxweiler, J., Dudek, J., Blum, R., Helms, V., Paton, A. W., Paton, J. C., Cavalié, A. & Zimmermann, R. (2012). EMBO J. 31, 3282–3296. [DOI] [PMC free article] [PubMed]
- Winn, M. D. et al. (2011). Acta Cryst. D67, 235–242.