

A putative lipase (CpsLip) from C. psychrerythraea strain 34H was expressed, purified and characterized. Crystallization and preliminary X-ray crystallographic analysis of CpsLip were performed.
Keywords: cold adaptation, lipases, α/β-hydrolase fold, CpsLip, Colwellia psychrerythraea
Abstract
The putative lipase CpsLip from the psychrophilic bacterium Colwellia psychrerythraea 34H encodes a 34 538 Da, 308-amino-acid protein. In this study, CpsLip (UniProtKB code Q486T5) was expressed as an N-terminal hexahistidine fusion protein in Escherichia coli and purified by affinity and size-exclusion chromatography. The expression and purification of CpsLip enabled characterization of the lipase enzymatic properties of the protein. The optimal activity temperature and pH of the recombinant protein were 298 K and pH 7, respectively. CpsLip maintained over 80% activity in the low-temperature range (278–288 K), thereby suggesting that CpsLip is a cold-active lipase. Substrate-specificity analysis demonstrated that CpsLip exhibits maximum activity towards the C12 acyl group. In addition, sequence-alignment results revealed that CpsLip has a highly conserved catalytic triad in the active site consisting of residues Ser111, Asp135 and His283. Moreover, purified CpsLip was successfully crystallized using the hanging-drop vapour-diffusion method and a complete diffraction data set was collected to 4.0 Å resolution using synchrotron radiation on the BL-5A beamline of the Photon Factory.
1. Introduction
Temperature is a critical factor for living organisms as it dictates the catalytic activities of their enzymes and thereby their growth limits. Cold-active enzymes exhibit higher catalytic activity at lower temperatures than homologues from mesophilic and thermophilic organisms (Feller & Gerday, 2003 ▶). The psychrophilic microorganism Colwellia psychrerythraea strain 34H can live in subzero-temperature environments owing to its cold-active enzymes (Methé et al., 2005 ▶). A few enzyme structures from C. psychrerythraea, including an extracellular cold-active aminopeptidase (Bauvois et al., 2008 ▶) and a phenylalanine hydroxylase (Leiros et al., 2007 ▶), have been solved and analysed in detail in order to reveal the molecular mechanisms of cold-adapted enzymes.
Cold-active enzymes have become the subject of intense research because of their potential industrial applications. More specifically, we are interested in the mechanisms of cold-active lipases. Lipases (EC 3.1.1.3) are ubiquitous enzymes that are expressed in diverse organisms and hydrolyse the ester bonds of long-chain acyl glycerides. They hydrolyse phospholipids and triglycerides to generate fatty acids for energy production. These proteins have gained considerable importance as biocatalysts that are able to catalyse hydrolysis and synthetic reactions (Jaeger et al., 1994 ▶, 1999 ▶; Joseph et al., 2008 ▶). The crystal structures of various lipases which have been elucidated from bacteria, fungi, yeast and mammals all share a common α/β-hydrolase fold in their core structure and have a characteristic catalytic triad consisting of a serine residue, an aspartic/glutamic acid residue and a histidine residue. The catalytic serine residue lies in a cleft on the surface of the enzyme and acts as the nucleophile. In the closed conformation, the active site is covered by an α-helical flexible ‘lid’ that maintains the catalytic triad in a hydrophobic environment (Jaeger et al., 1999 ▶). Although several lipases have been structurally characterized, very little work has examined the structure or the structure–function relationship of cold-adapted lipases. Molecular modelling of the Pseudomonas immobilis lipase (Arpigny et al., 1997 ▶) and structural studies of the M37 lipase from Photobacterium lipolyticum (Jung et al., 2008 ▶) revealed several features of cold-adapted lipases. Their structures are believed to be flexible, loose and maintained by a relatively small number of weak intramolecular bonds. These structural assets would explain the enhanced lability of cold-active enzymes. The current understanding is that their increased flexibility allows psychrophilic enzymes to be active at low temperatures (D’Amico et al., 2002 ▶; Spiwok et al., 2007 ▶).
A database search of the C. psychrerythraea strain 34H genome identified a number of putative lipases (UniProtKB codes Q486T5, Q487S5, Q486T0, Q482I9, Q481Z4, Q484K3 and Q482D4). We have recently cloned the Q486T5 gene that encodes a putative cold-adapted lipase (CpsLip) and purified the recombinant protein. To obtain information about the molecular mechanisms of CpsLip, we have performed enzyme-activity assays and initial X-ray crystallographic experiments.
2. Materials and methods
2.1. Cloning, expression and purification of CpsLip
The full-length putative lipase gene (308 amino-acid residues; 35 kDa) was amplified by PCR from the genomic DNA of C. psycherythraea 34H using forward (5′-CGATAAGAGCTCATGGCAGATACCAACGAA-3′) and reverse (5′-CGATAACTCGAGTCAAACGCTTGTTTTAATTTC-3′) primer sequences. The PCR product was digested with SacI and XhoI, and then subcloned into the pCold-I vector (Takara Bio Inc.). The resulting construct encoded a translation-enhancing element (TEE), an N-terminal hexahistidine tag and a factor Xa cleavage site (MNHKVHHHHHHIEGRHMEL) followed by the complete coding region of CpsLip. Protein expression was induced in the cells using 1 mM IPTG, and the Escherichia coli cells were incubated at 288 K overnight and collected by centrifugation at 3540g for 15 min. The cell pellet was suspended in lysis buffer (50 mM sodium phosphate pH 8.0, 300 mM NaCl, 5 mM imidazole) and lysed by sonication. The lysate was pelleted by centrifugation at 13 600g for 30 min. The supernatant was then loaded onto a gravity-flow column (Bio-Rad) packed with Ni–NTA affinity resin (Peptron) that had been pre-equilibrated with lysis buffer. After washing the column and matrix with ten column volumes of lysis buffer, the N-terminally His-tagged fusion protein was eluted using a solution consisting of 50 mM sodium phosphate pH 8.0, 300 mM NaCl, 300 mM imidazole. The eluted fractions were checked for purity by SDS–PAGE and concentrated using Centriprep ultrafiltration devices (10 kDa molecular-mass cutoff; Amicon) prior to factor Xa cleavage. During concentration, imidazole was removed via buffer exchange using chilled enzyme-cleavage buffer (20 mM Tris–HCl pH 8.0, 100 mM NaCl). To remove the N-terminal hexahistidine tag, the fusion protein (10 mg) was digested with 10 µl factor Xa (1 mg ml−1; New England Biolabs) at 277 K overnight on a rotating rack. After cleavage of the hexahistidine tag, the protein solution was concentrated using a Centricon 10K (Amicon) and applied onto a Superdex 200 column (Amersham Biosciences) equilibrated with 20 mM Tris–HCl pH 8.0, 150 mM NaCl. The fractions containing the putative lipase were collected and concentrated by ultrafiltration to a concentration of 25.0 mg ml−1 using a Centricon Plus-70 10 kDa centrifugal filter unit (Amicon). Following factor Xa cleavage, the final purified construct contained an additional HMEL sequence at its N-terminus.
2.2. Characterization of the lipase activity of CpsLip
The specific lipase activity was determined by a spectrophotometric method using various p-nitrophenyl fatty-acid esters as the substrate (Vorderwülbecke et al., 1992 ▶). In brief, 100 µl enzyme solution (20 mg ml−1) was mixed with 100 µl substrate solution (10 mM p-nitrophenyl fatty-acid esters dissolved in 100% ethanol) and 800 µl 50 mM Tris–HCl buffer pH 8.0. The absorbance increase caused by the release of p-nitrophenol from p-nitrophenyl fatty-acid esters by enzymatic hydrolysis at 298 K was spectrophotometrically measured for at least 2 min at 410 nm against an enzyme-free control.
The substrate specificity of the lipase was studied using 58 µM lipase protein and p-nitrophenyl fatty-acid esters with various acyl-chain lengths under the activity-assay conditions described earlier. The final concentration of substrates used in Tris–HCl buffer pH 8.0 were 1 mM p-nitrophenyl butyrate (p-NPB, C4), 1 mM p-nitrophenyl caprylate (p-NPC, C8), 1 mM p-nitrophenyl laurate (p-NPL, C12) and 1 mM p-nitrophenyl palmitate (p-NPP, C16).
The optimum temperature for lipase activity was determined with 1 mM p-nitrophenyl butyrate as the substrate using a spectrophotometer (Ultrospec 3300 pro, Amersham Bioscience) pre-adjusted to the correct temperature. The assay buffer was pre-incubated for at least 2 min at the given temperature prior to the assay. The assay included 58 µM lipase protein and 1 mM p-nitrophenyl butyrate as the substrate. To determine the optimal pH for lipase activity, various buffers at 50 mM (sodium acetate buffer for pH 4–5, sodium phosphate buffer for pH 6–7, Tris–HCl buffer for pH 8 and Gly–NaOH buffer for pH 9), 1 mM p-nitrophenyl butyrate and 58 µM lipase protein were used.
2.3. Crystallization and data collection
An initial crystallization screening for CpsLip was performed using the crystallization solution kits Wizard Classic (Emerald Bio), Crystal Screen, Crystal Screen 2, PEG/Ion, SaltRx and Index (Hampton Research) with the sitting-drop vapour-diffusion method at 293 K. Initial microcrystals were obtained from Wizard Classic I condition No. 38 (0.1 M CHES–NaOH pH 9.5, 1 M potassium sodium tartrate, 0.2 M lithium sulfate), Index condition No. 12 (0.1 M Tris–HCl pH 8.5, 3 M NaCl), Crystal Screen 2 condition Nos. 23 [0.1 M MES monohydrate pH 6.5, 1.6 M ammonium sulfate, 10%(v/v) 1,4-dioxane] and 37 [0.1 M HEPES–NaOH pH 7.5, 10%(w/v) PEG 8000], and SaltRx 2 condition No. 21 (0.1 M Tris–HCl pH 8.0, 0.8 M lithium sulfate monohydrate). These conditions were optimized by varying the pH and the precipitant and salt concentrations. In addition, the drop volume was increased from 0.5 to 1 µl to generate larger crystals. From these results, optimal crystals were obtained after 3 d and grew to a largest dimension of 0.6 mm in a drop consisting of 1 µl protein solution (25.0 mg ml−1 protein in 20 mM Tris–HCl pH 8.0, 150 mM NaCl) and 1 µl reservoir solution (0.1 M CHES–NaOH pH 8.5, 1.3 M lithium sulfate).
For the cryogenic experiments, a suitable cryoprotectant was determined to be the reservoir solution plus 30%(v/v) glycerol. The crystals were directly transferred from the drop to the cryosolvent and were allowed to equilibrate for 1 min. The crystals were then flash-cooled by plunging the loop into liquid nitrogen and equilibrating for 1 min. A native data set to 4.0 Å resolution was collected on the BL-5A beamline of the Photon Factory, Tsukuba, Japan using the X-ray beam at a single wavelength (1.0000 Å). The data set was indexed and processed using HKL-2000 (Otwinowski & Minor, 1997 ▶).
2.4. Sequence analysis of CpsLip
A multiple sequence alignment of CpsLip and other putative lipases, including the lipases from Pseudomonas syringae (UniProtKB code F3EHA6) and Legionella pneumophila (UniProtKB code Q5ZYA4), the putative lipase from L. drancourtii (UniProtKB code C6MYG3), the α/β-hydrolase fold protein from Shewanella paeleana (UniProtKB code A8H5D2) and the α/β-superfamily hydrolase from Idiomarina baltica (UniProtKB code A3WNW3), was performed using ClustalX (Thompson et al., 1997 ▶).
3. Results and discussion
3.1. Expression and purification of CpsLip
The recombinant CpsLip was expressed in E. coli BL21 (DE3) in the soluble fraction. Oligomeric state analysis by means of size-exclusion chromatography showed that recombinant CpsLip exists in a dimeric state in solution with an apparent molecular weight of 75 kDa. The protein was purified using affinity and size-exclusion chromatography with a yield of approximately 20 mg per litre of culture (Fig. 1 ▶).
Figure 1.

Expression and purification of recombinant CpsLip. (a) 12% SDS–PAGE analysis of the expression and purification of CpsLip. Lane M, molecular-weight markers (labelled in kDa); lane I, induced fraction; lane S, soluble fraction; lane P, pellet; lane R, remaining fraction after flowing through the resin; lane W, washing fraction; lane E, elution fraction. (b) The size-exclusion chromatography profile of CpsLip from a Superdex 200 column. The injection sample (I) and several fractions, indicated by a red bar (fraction numbers from 25–35) in the chromatography profile, were visualized using a 12% SDS–PAGE gel (inset). (c) The column was calibrated at 298 K with globular protein standards that included thyroglobulin (669 kDa, V e = 58.4 ml), apoferritin (443 kDa, V e = 65.1 ml), β-amylase (200 kDa, V e = 71.4 ml) and BSA (67 kDa, V e = 80.8 ml). The void volume of the column (V o) was determined to be 42.5 ml using blue dextran (2000 kDa); V e is the peak elution volume. The V e/V o values were used to generate a standard curve; the molecular mass of CpsLip was determined from the standard curve. The elution volume indicated that CpsLip was a dimer in solution with an apparent molecular weight of 75 kDa (V e = 80.5 ml).
3.2. Effects of temperature, pH and substrate specificity on lipase activity
After completion of its genome sequencing, the marine psychrophilic bacterium C. psychrerythraea 34H living in cold environments has been considered as a model for cold-adaptation studies (Methé et al., 2005 ▶). The molecular characterization of the detailed biochemical mechanisms involved in cold adaptability have been studied using the cold-adapted enzyme phenylalanine hydroxylase (Leiros et al., 2007 ▶) and the cold-active aminopeptidase from C. psychrerythraea 34H (Bauvois et al., 2008 ▶). Because CpsLip was isolated from a cold marine habitat, it has also attracted attention owing to the potential use of its psychrophilic properties in industrial applications. As expected, the purified recombinant CpsLip protein showed cold-adapted lipase activity in our standard functional assays based on the hydrolysis of p-NPB. The CpsLip protein displayed maximum activity at 298 K and maintained substantial activity (over 80%) in the low-temperature range (278–288 K; Fig. 2 ▶ b). To examine the substrate specificity of purified CpsLip, various p-nitrophenyl substrates, namely p-NPB, p-NPC, p-NPL and p-NPP, were tested. p-NPL (C12) showed the highest activity among the substrates used, while p-NPP (C16) was a poor substrate (Fig. 2 ▶ a). Next, the pH-dependence of CpsLip was investigated in the pH range 4–9. As shown in Fig. 2 ▶(c), CpsLip displayed broad pH-range activity, with maximum activity at pH 7.
Figure 2.

Lipase activity of recombinantly expressed and purified CpsLip. (a) Substrate specificity of purified CpsLip. p-Nitrophenyl butyrate (p-NPB, C4), p-nitrophenyl caprylate (p-NPC, C8), p-nitrophenyl laurate (p-NPL, C12) and p-nitrophenyl palmitate (p-NPP, C16) were used in Tris–HCl buffer pH 8.0. (b) Effect of temperature on the activity of CpsLip. The activity of the enzyme was assayed at each temperature in 50 mM Tris–HCl buffer pH 8.0 by measuring p-nitrophenyl butyrate hydrolysis. (c) The effect of pH on the activity of the enzyme was determined at 298 K in sodium acetate (pH 4 and 5), sodium phosphate (pH 6 and 7), Tris–HCl (pH 8) and Gly–NaOH (pH 9) buffer with p-nitrophenyl butyrate as the substrate.
3.3. Crystallization and data collection
To determine the crystal structure of CpsLip, we found and optimized crystallization conditions for the full-length protein concentrated to 25.0 mg ml−1. Native crystals of approximate dimensions 0.05 × 0.05 × 0.3 mm were obtained within 3 d using a reservoir solution consisting of 0.1 M CHES–NaOH pH 8.5, 1.3 M lithium sulfate (Fig. 3 ▶ a). The CpsLip crystal belonged to a primitive hexagonal space group and diffracted to 4.0 Å resolution (Fig. 3 ▶ b). Firstly, the diffraction images were indexed and integrated in space group P3 with approximate unit-cell parameters a = b = 129.0, c = 98.5 Å. However, further analysis of the integrated intensities with POINTLESS (Evans, 2006 ▶) suggested that the space group was either P6122 or its enantiomer P6522. Thus, the data were reindexed in space group P6122/P6522 and were scaled using HKL-2000 (Otwinowski & Minor, 1997 ▶; Table 1 ▶). Assuming two molecules per asymmetric unit, the Matthews coefficient (V M) was calculated to be 3.48 Å3 Da−1, which corresponds to a solvent content of 64.7% (Matthews, 1968 ▶).
Figure 3.

Crystallization of recombinant CpsLip. (a) A crystal of CpsLip grown for 3 d using 0.1 M CHES–NaOH pH 8.5, 1.3 M lithium sulfate. The approximate dimensions are 0.05 × 0.05 × 0.3 mm. (b) The X-ray diffraction pattern (4.0 Å resolution) from a crystal of CpsLip.
Table 1. Data-collection statistics.
Values in parentheses are for the outermost resolution shell.
| X-ray source | BL-5A, PF |
| Space group | P6122 or P6522 |
| Unit-cell parameters (Å) | a = b = 129.0, c = 98.5 |
| Wavelength (Å) | 1.0000 |
| No. of frames | 180 |
| Oscillation angle (°) | 1 |
| Resolution range (Å) | 50–4.0 (4.07–4.0) |
| No. of unique reflections | 4412 (221) |
| Completeness (%) | 99.8 (100) |
| Multiplicity | 20.2 (21.2) |
| R merge † | 0.078 (0.455) |
| Average I/σ(I) | 66.0 (12.2) |
R
merge = 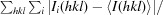
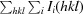 , where I
i(hkl) is the intensity of the ith measurement of reflection hkl,
, where I
i(hkl) is the intensity of the ith measurement of reflection hkl, 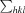 is the sum over all reflections and
is the sum over all reflections and 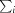 is the sum over i measurements of reflection hkl.
is the sum over i measurements of reflection hkl.
We attempted molecular-replacement methods for phase determination with the programs MOLREP (Vagin & Teplyakov, 2010 ▶) and Phaser (McCoy et al., 2007 ▶). However, molecular replacement failed to provide correct solutions for either the molecular orientation or the molecular packing. The low-resolution data and the low sequence identity to other known structures probably preclude structural determination of CpsLip by molecular replacement. Therefore, attempts to solve the structure by the multiwavelength anomalous dispersion (MAD) method using selenomethionine-labelled protein are in progress, and additional studies are planned to obtain high-resolution X-ray diffraction data of the enzyme complexed with lipase inhibitors.
3.4. Sequence analysis of CpsLip
As shown in Fig. 4 ▶, a multiple alignment of the amino-acid sequences of CpsLip and the putative lipases from several bacteria revealed that CpsLip possesses the conserved lipase catalytic triad residues Ser111, Asp135 and His283.
Figure 4.

The sequence alignment of CpsLip and selected putative lipases, including the lipase from P. syringae (UniProtKB code F3EHA6), the lipase from L. pneumophila (UniProtKB code Q5ZYA4), the putative lipase from L. drancourtii (UniProtKB code C6MYG3), the α/β-hydrolase fold protein from S. paeleana (UniProtKB code A8H5D2) and the α/β-superfamily hydrolase from I. baltica (UniProtKB code A3WNW3), using ClustalX (Thompson et al., 1997 ▶). The highly conserved residues are shaded grey and black. The secondary-structural elements of CpsLip were predicted using the Phyre server (Kelley & Sternberg, 2009 ▶) and are shown above the alignments. CpsLip has conserved catalytic residues (at positions Ser111, Asp135 and His283) which are thought to be important for lipase activity. The residues constituting the active site are indicated by filled circles.
In conclusion, we have successfully overexpressed, purified and characterized the cold-active lipase CpsLip from C. psychrerythraea 34H. This recombinant CpsLip protein has potential in biotechnological and commercial applications because of its significant activity at low temperatures and over a broad pH range. Taken together, our biochemical and X-ray crystallographic studies represent the beginning of investigations that will contribute to understanding the molecular mechanisms of this important enzyme.
Acknowledgments
We would like to thank the beamline staff at BL-5A of the Photon Factory, KEK (Tsukuba, Japan) for their kind help with the data collection. This work was supported by the Korea Polar Research Institute (KOPRI; grant No. PE13270 to HJK).
References
- Arpigny, J., Lamotte, J. & Gerday, C. (1997). J. Mol. Catal. B Enzym. 3, 29–35.
- Bauvois, C., Jacquamet, L., Huston, A. L., Borel, F., Feller, G. & Ferrer, J.-L. (2008). J. Biol. Chem. 283, 23315–23325. [DOI] [PubMed]
- D’Amico, S., Claverie, P., Collins, T., Georlette, D., Gratia, E., Hoyoux, A., Meuwis, M. A., Feller, G. & Gerday, C. (2002). Philos. Trans. R. Soc. Lond. Ser. B Biol. Sci. 357, 917–925. [DOI] [PMC free article] [PubMed]
- Evans, P. (2006). Acta Cryst. D62, 72–82. [DOI] [PubMed]
- Feller, G. & Gerday, C. (2003). Nature Rev. Microbiol. 1, 200–208. [DOI] [PubMed]
- Jaeger, K. E., Dijkstra, B. W. & Reetz, M. T. (1999). Annu. Rev. Microbiol. 53, 315–351. [DOI] [PubMed]
- Jaeger, K. E., Ransac, S., Dijkstra, B. W., Colson, C., van Heuvel, M. & Misset, O. (1994). FEMS Microbiol. Rev. 15, 29–63. [DOI] [PubMed]
- Joseph, B., Ramteke, P. W. & Thomas, G. (2008). Biotechnol. Adv. 26, 457–470. [DOI] [PubMed]
- Jung, S.-K., Jeong, D. G., Lee, M. S., Lee, J.-K., Kim, H.-K., Ryu, S. E., Park, B. C., Kim, J. H. & Kim, S. J. (2008). Proteins, 71, 476–484. [DOI] [PubMed]
- Kelley, L. A. & Sternberg, M. J. (2009). Nature Protoc. 4, 363–371. [DOI] [PubMed]
- Leiros, H. K., Pey, A. L., Innselset, M., Moe, E., Leiros, I., Steen, I. H. & Martinez, A. (2007). J. Biol. Chem. 282, 21973–21986. [DOI] [PubMed]
- McCoy, A. J., Grosse-Kunstleve, R. W., Adams, P. D., Winn, M. D., Storoni, L. C. & Read, R. J. (2007). J. Appl. Cryst. 40, 658–674. [DOI] [PMC free article] [PubMed]
- Matthews, B. W. (1968). J. Mol. Biol. 33, 491–497. [DOI] [PubMed]
- Methé, B. A. et al. (2005). Proc. Natl Acad. Sci. USA, 102, 10913–10918.
- Otwinowski, Z. & Minor, W. (1997). Methods Enzymol. 276, 307–326. [DOI] [PubMed]
- Spiwok, V., Lipovová, P., Skálová, T., Dusková, J., Dohnálek, J., Hasek, J., Russell, N. J. & Králová, B. (2007). J. Mol. Model. 13, 485–497. [DOI] [PubMed]
- Thompson, J. D., Gibson, T. J., Plewniak, F., Jeanmougin, F. & Higgins, D. G. (1997). Nucleic Acids Res. 25, 4876–4882. [DOI] [PMC free article] [PubMed]
- Vagin, A. & Teplyakov, A. (2010). Acta Cryst. D66, 22–25. [DOI] [PubMed]
- Vorderwülbecke, T., Kieslich, K. & Erdmann, H. (1992). Enzyme Microb. Technol. 14, 631–639.