

Abstract
Monoclonal antibodies (mAbs) have been used for decades as tools to probe the biology and pharmacology of receptors in cells and tissues. They are also increasingly being developed for clinical purposes against a broad range of targets, albeit to a lesser extent for G protein-coupled receptors (GPCRs) relative to other therapeutic targets. Recent pharmacological, structural and biophysical data have provided a great deal of new insight into the molecular details, complexity and regulation of GPCR function. Whereas GPCRs used to be viewed as having either “on” or “off” conformational states, it is now recognized that their structures may be finely tuned by ligands and other interacting proteins, leading to the selective activation of specific signaling pathways. This information coupled with new technologies for the selection of mAbs targeting GPCRs will be increasingly deployed for the development of highly selective mAbs that recognize conformational determinants leading to novel therapeutics.
Keywords: GPCR, ligand induced conformation, biased signaling, monoclonal antibody, therapeutic antibody
Introduction
G protein-coupled receptors (GPCRs) comprise the largest family of membrane receptors in the mammalian genome with approximately 850 members [1]. Moreover, it has been estimated that individual cell types, including cancer cells express, > 100 of the approximately 370 non-chemosensory GPCRs [2]. It is not surprising then, that GPCRs have long been targeted for drug discovery and development such that in 2012, it was estimated that 40–50% of marketed drugs act on GPCRs [3].
Another important family that constitutes a large fraction of the mammalian proteome is the immunoglobulin/antibody superfamily [4]. A unique feature of antibodies is their ability to adapt their ligand-binding domain in response to antigenic stimulation, making them a versatile framework for binding to virtually any macromolecule. In 1975, Kohler and Milstein published one of the most remarkable papers in the history of modern immunology that described an immunization and cell culture technique for the generation of antigen specific monoclonal antibodies (mAbs) [5]. As a result, mAbs have become powerful research tools and the most common form of biologics for therapeutic purposes [6]. Although the cost of making mAbs is higher than that for small molecule drugs, mAbs are more selective and therefore tend to have fewer non-specific or off target toxicity issues. Due to their long half-life in plasma, mAbs also have a longer duration of action than small molecule drugs. Hence, they have become a very attractive class of therapeutics.
With protein therapeutics on the rise coupled with the importance of GPCRs as pharmaceutical targets, it seems obvious that there should be some therapeutic mAbs that act on GPCRs (Figure 1). However, according to the IMGT Repertoire (www.imgt.org/mAb-DB/index#Human), there are currently thirty-seven mAbs on the market, but the first mAb targeting a GPCR was only recently approved in Japan, and currently there are no GPCRs targeting mAbs in late stage clinical development. These statistics reflect a 2008 statement by Gupta et al. who described the use of GPCR mAbs in clinical applications as “underdeveloped” but with “great potential” [7]. Fortunately, the situation is changing, and there are now several GPCR mAbs in early clinical trials, mostly targeting receptors with large peptide/protein ligands such as the chemokine receptor family, or the glucagon-like peptide receptor (GLP1R) (Table 1). Since two years have elapsed since the last major review of this topic [8], it is the intent of this brief commentary to provide an update on where the field stands. In particular, we highlight new information about GPCR structure and pharmacology that has emerged regarding conformational specificity and biased signaling, and focus on methods for mAb selection that may allow more efficient generation of mAbs that recognize or stabilize specific conformational/pharmacological states of GPCRs.
Figure 1. Model representation of an Fab (blue, PDB ID 3KJ6) bound to the extracellular domain of a GPCR.

(red, PDB ID 4DJH).
Table 1A.
Development status of therapeutic mAbs targeting GPCRs
| Target | Name | Company | Therapeutic Indication | Clinical Status |
|---|---|---|---|---|
| Humanized | ||||
| CCR4 | Mogamuli zumab | Amgen/Kiowa Hakko Kirin | Leukemia, Lymphoma/Asthma | Phase I/II Approved in Japan for relapsed or refractory T cell leukemia and lymphoma |
| Human | ||||
| C3aR | Human Genome Sciences | Asthma | Preclinical | |
| C5aR | NN-8209 | G2 Therapies/Novo Nordisk | Autoimmune Disease | Phase I |
| CCR2 | MLN1202 | Takeda | Inflammation | Phase II- Discontinued |
| CCR4 | AT0009 | Affitech | Cancer | Pre-clinical |
| CCR5 | PRO140 | Progenics | HIV | Phase 2- Discontinued |
| CCR5 | HGS004 | Human Genome Sciences | HIV | Phase I- Discontinued |
| CCR5 | HGS101 | Human Genome Sciences | HIV | Discontinued |
| CCR8 | X | Lilly | Inflammation | Undisclosed |
| CCR9 | MLN3126 | Takeda | Inflammatory Bowel Disease | Discontinued |
| CRTH2 | Sosei/Abgenix | Inflammation | No Information | |
| CXCR3 | AT0010 | Affitech | Inflammation | Discovery |
| CXCR4 | MDX 1338 | Medarex/BMS | Leukemia | Phase I |
| CXCR4 | ALX-0651 | Ablynx | Cancer | Phase I- Discontinued |
| CXCR4 | LY2624587 | Lilly | Cancer | Phase I- Discontinued |
| GCG-R | AMG477 | Amgen | Diabetes | Phase I- Discontinued |
| GLP-1R | X | Abbott/Human Genome Sciences | Diabetes/Neurologic/Metabolic Disease | No information |
| LGR5 | KM4056 | Kiowa Hakko Kirin | Cancer | Pre-clinical |
| VPAC-1 | Thrombogenics | Thrombocytopenia | Pre-clinical | |
The list in the table above should not be considered exhaustive; we have used the databases listed below to update and add to information published in [8]. They are the following: www.imgt.org/mAb-DB/index#Human; www.Alxn.com; www.kyowa-kirin.co.jp/english; www.gsk.com; www.janssenrnd.com; www.lilly.com; www.amgen.com; www.hgsi.com; www.abbott.com; www.tpna.com; www.sosei.com; www.affitech.com; clinicaltrials.gov; https://eudract.ema.europa.eu
Antibody selection
Traditional immunization-based approaches
One of the fundamental reasons that mAbs against GPCRs have been slow to populate therapeutic portfolios is that they are frequently difficult to make by traditional immunization-based methods [7]. GPCRs are membrane proteins that are generally expressed at low levels in cells, have relatively small mass exposed outside of the membrane, have low stability and require detergents or phospholipid membranes for solubility. Furthermore GPCRs purified and solubilized in detergents, the most common membrane mimetic, are generally unstable. For these reasons, they are difficult to purify and obtain in sufficient amounts for immunization compared to soluble proteins [7, 8].
Because of the challenges in preparing intact GPCRs for immunization [8, 9], the most common approach has been to take synthetic peptides derived from the N-terminal domains and to a lesser extent the extracellular loops (ECLs), and attach them to larger, highly immunogenic carrier proteins such as ovalbumin [9]. The resulting antibodies have unpredictable specificity, avidity and pharmacological activity, necessitating considerable screening efforts to identify those with desired properties. Therefore, they have been used primarily as probes for detection of expression and/or to understand aspects of GPCR structure/pharmacology rather than as therapeutics [7, 8]. It is particularly difficult to generate mAbs using N-terminal peptides from the class A GPCRs as immunogens, as these receptors generally have small extracellular N-termini, compared to class B and C receptors, limiting their immunogenic epitopes [10]. Furthermore, even if mAbs can be generated against N-termini, they might not show the therapeutically desired functional properties or they might recognize linear epitopes increasing the risk of cross-reactivity with unrelated proteins. It is therefore noteworthy that anti-GPCR mAbs that have entered clinical development are largely directed against the ECLs, or a combination of the N-terminus and ECLs. Other issues with the use of peptide antigens are that correct folding and the presence of post-translational modifications (PTMs, e.g. tyrosine sulfation and glycosylation) that may be present in the intact GPCRs, are generally absent in peptides [11].
Because GPCRs often have high sequence and structural homology between species, they also tend to be poorly immunogenic; for example, the chemokine receptor, CXCR4, has 90% sequence identity between humans and mice. This high degree of conservation impacts the ability of the immune system of the immunized host to produce antibodies against such GPCRs, as they are seen as “self”. B-cells that target self proteins are either deleted during embryonic development or are under very tight regulatory control, so that such ‘forbidden clones’ are never allowed to expand into antibody producing plasma cells [12].
The final issue is that one can never be certain of obtaining the types of mAbs that are desired with this approach [7, 8]. For example, following a typical immunization protocol, mAbs may or may not be generated that recognize inactive, active with agonist, constitutively active or other specific structural/conformational determinants. However, this latter issue highlights one of the reasons that GPCRs may be such good mAbs targets--because of the possibility for “state-specific” recognition [13].
New Immunization Methods
Recently, new and improved immunization technologies have emerged that should increase the success rate over classical peptide immunization [8] and enhance the potential for the resulting mAbs to be used as therapeutics, rather than simply as pharmacological reagents. These approaches include GPCRs over-expressed on whole cells and cell membranes that allow recognition of the GPCR extracellular domains in their native conformation including non-contiguous epitopes and PTMs, as well as DNA immunizations. The downside is that the abundance of other potentially more immunogenic cell surface components can result in a high background of antibodies that recognize unwanted targets, making selection akin to finding a needle in a haystack unless the background can be eliminated by depletion steps [14]. One very recent approach that addresses the issue of background head-on involves the use of purified receptors. As a pre-requisite to the flurry of GPCR structures that have been solved since 2007 [15], there have been major advances in the ability to express GPCRs in heterologous systems, and to purify and reconstitute them in vitro. As described later, this technology will allow purified receptors to be used in selections, enabling one to manipulate conditions to enhance the selection of mAbs with desired properties.
State-specific recognition of GPCRs
Despite the fact that immunization approaches as generally practiced have no inherent bias, mAbs with desirable properties, such as specificity for GPCRs in agonist, antagonist and inverse agonist conformational states, have been identified using these methods [7, 13, 14, 16] (Table 2). The earliest examples came from studies of rhodopsin where a mAb to a peptide from the N-terminus recognized the activated receptor more avidly than the inactive form [17]. Gupta reported anti-N-terminal peptide mAbs or anti-sera, first to the μ opioid receptor and later to a variety of GPCRs (α2A and β2 adrenergic receptors, the cannabinoid receptor, CB1, and the angiotensin AT1 receptor) that recognized the agonist activated state of the receptor [7, 13]. Furthermore, the authors could follow increases in antibody binding to the μ opioid receptor following ligand binding in vivo. As another example, Goetzl et al., reported the generation of rat mAbs to the human sphingolipid S1P1 receptor using transfected whole cells, one of which recognized a conformational determinant [18]. Mancia et al., were also able to isolate mAbs that recognized conformational determinants of the 5HT2c receptor only when bound to ligand [14]. From these and other studies (Table 2) it is apparent that it is possible to generate mAbs that recognize specific ligand-bound conformational states (Figure 2). As well, some mAbs recognize specific conformations independent of the presence of agonists or antagonists [7, 19]. That it is possible to have mAbs with state-specific recognition is not surprising because it has been known for a long time that natural agonists generally have a preference for the active G-protein coupled state of GPCRs over other states. Similarly, ligands can have “functional selectivity” or “ligand bias”, meaning that they can activate different signaling pathways (e.g. G protein- versus β-arrestin-dependent pathways) [20–22] or even change the trafficking behavior of receptors [23]. With the recent explosion in structural information on GPCRs, it has become clear that these functional consequences are due to the ability of the ligands to stabilize different conformational states of the receptors as illustrated in Figure 2. These findings have generated great excitement in the GPCR community because of the concept that biased ligands may prove to be superior therapeutics by minimizing signaling events that cause adverse side effects in certain diseases [24, 25]. As an example, G protein biased ligands that do not activate β-arrestin mediated desensitization are predicted to avoid β2 adrenergic receptor induced tachyphylaxis in asthma, and μ opioid receptor mediated tolerance to analgesics [24].
Table 1B.
Development status of therapeutic mAbs targeting GPCR ligands
| Target | Name | Company | Therapeutic Indication | Clinical Status |
|---|---|---|---|---|
| Humanized | ||||
| C5 (C5aR ligand) | Soliris™ | Alexion | Paroxysmal Nocturnal Hemoglobinuria | Approved |
| Human | ||||
| IL-8 | ABX-IL-8 | Abgenix | Autoimmune Disease | Phase IIb |
| CCL2 | Carlumumab | Janssen | Pulmonary Fibrosis | Phase II |
Figure 2. State-specific recognition of GPCRs.
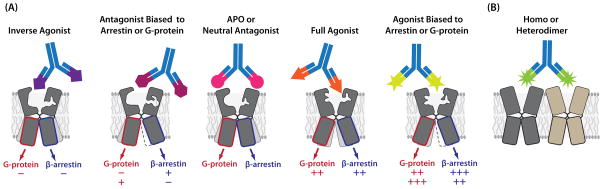
Functionally selective ligands can stabilize GPCRs in conformations that confer unique signaling properties. In principle, it should be possible to generate mAbs that recognize specific ligand-bound conformational states (A), as well as conformations induced by homo- or hetero-dimerization (B).
The realization that it is possible to generate mAbs that recognize specific ligand-induced conformational states of GPCRs, suggests that it may be possible to develop antibodies that are sensitive to other parameters that affect GPCR function; these include association with other receptors and interaction with different intracellular proteins (i.e. G protein coupling), PTMs etc. For example, in addition to agonist and antagonist bound states, GPCRs may form homo- and hetero-oligomers (Figure 2). Rios and coworkers showed agonist mediated increases in the recognition of μ opioid receptors by a mAb when the cells were exposed to a CB1 receptor agonist [26]. These results suggest the existence of heteromers between the two GPCRs and that the agonist induced structural changes in one partner of the heteromer could be transmitted to the other partner and detected by the mAb. Extrapolating state-specific recognition further, one could envision a mAb that recognizes a GPCR in a cell-context dependent manner -- e.g. only in cancer cells but not in normal cells. One could also imagine mAbs that induce specific conformational states and ligand bias as they may have unique therapeutic value.
In vitro selections and immunization-free approaches to mAb generation
The problem with immunogen-based approaches for generating antibodies with conformation-specific recognition is that extensive screening is required; and even then, antibodies with the desired properties may not be represented in the pool. This is because there is little or no control over the conformational state of the receptor in vivo, there is no selection present in the in vivo setting to enrich for the antibodies of interest, and because of the generally poor immunogenicity of GPCRs. To get around these issues and realize the full potential of mAbs with respect to recognition of defined states, new strategies are being developed. These strategies capitalize on the power of in vitro selections of recombinant antibody libraries. Clever library designs from naïve, immune and synthetic antibody repertoires combined with selection methods using phage display, yeast display, mammalian display, ribosome or cis display enable selection of high affinity antibodies with desired functional properties [27–33]. Furthermore, enhancement of screening approaches such as the use of next generation sequencing [34] empowers selection of superior mAbs.
As described above, the reliance on in vivo selection through classical immunization approaches largely constrains one to a “you take what you can get” result [35]. By contrast, phage display of antibody Fabs (antigen-binding fragments) or scFvs (single-chain variable fragments) coupled with in vitro selection allows one to manipulate the system in a variety of ways such that antibodies can be identified with specificities that would be difficult to find by traditional immunization techniques [27]. Purified, homogeneous antigens can be used to enhance the probability of identifying antibodies with pre-defined properties and can facilitate the use of anti-selection/depletion steps to further ensure specificity. Since in vitro antibody display technologies links the phenotype (protein) with genotype (cDNA), one also has the opportunity to conduct rounds of mutagenesis following initial selections for affinity or specificity maturation.
Key to the success of the antibody display approaches is the library composition and diversity. Antibody libraries randomly combine heavy and light chain cDNAs obtained through RT-PCR from mRNA of B cell pools into expression vectors that afford a compartmental or physical linkage of phenotype and genotype. Large antibody libraries can mimic natural human antibody repertoires of >1010 members in terms of molecular diversity [36, 37]. For therapeutic purposes, the strategy of using human naïve libraries alleviates the need for humanization, since the scaffold is already fully humanized, but might require affinity maturation. Libraries derived from immunized mice can also be highly useful by yielding a larger choice of high affinity immunogen-specific antibodies that can be selected by in vitro display methods. Multiple antibodies derived from such libraries have been humanized and have entered the clinic.
In an even more dramatic departure from traditional approaches, synthetic or semi-synthetic libraries have been engineered to introduce additional sequence diversity; these are based on mAb frameworks that are already fully human or humanized, and that have been demonstrated to have high stability and to express well [28, 38–40]. Diversity in the range of 1010 can be achieved from single scaffolds by introducing different amino acids through degenerate codon usage at specific sites in the hypervariable regions (CDRs) of the Fab. The synthetic nature of these libraries may have some advantages over natural libraries in that they are less biased against self-antigens. Interestingly, severe restriction of CDR diversity by designing libraries limited to as few as two (Ser and Tyr) to four (Ser, Tyr, Ala, Asp) amino acid types in the CDRs can also yield single digit nanomolar affinity binders [41, 42]. However, while providing insight into antibody/target interactions and providing great research tools, it has proven difficult to further affinity mature mAbs with severely restricted amino acid usage in the CDRs, rendering this approach less suitable for development of therapeutic mAbs. Other approaches restrict amino acid usage, but also add rational design principles against specific target classes such as carbohydrates. For example, semisynthetic libraries, have been designed by introducing basic amino acid residues at specific locations in hCDR3 to better interact with negatively charged carbohydrates, thereby expanding options to select mAbs against self antigens [43].
To our knowledge, antibodies generated against GPCRs using exclusively in vitro technologies have yet to be reported in the primary literature, but they are on the horizon. However, the successful results of antibodies targeting other protein classes provide useful illustrations of the power of these new technologies. To generate antibodies against a specific conformational state, one can use compounds to stabilize the purified antigen in that particular conformation (Figure 2). For example, antibodies were generated to different “on” and “off” states of caspase-1 using covalent small molecules that stabilized the respective states [44]. After rounds of selection and anti-selection coupled with affinity maturation, Fabs were identified that stabilized the two states with nanomolar affinities and selectivities of 20–500-fold over the alternate conformer. Similarly, linkage specific antibodies that recognize polyubiquitin through either K48 or K63 linked chains were generated by phage display and selection against surface immobilized polyubiquitin containing the linkage of interest; anti-selection against monoubiquitin or the alternative linkage was achieved by adding these proteins to the solution [45]. These antibodies were based on a synthetic Tyr/Ser library; and notably, attempts to generate such antibodies using traditional immunization methods did not work, a result that the authors attributed to the inability of the naturally occurring antibody repertoire to recognize such highly conserved epitopes. Similar issues may contribute to difficulties with GPCRs and, thus, these new technologies may be highly amenable to these and other membrane proteins. Consequently, GPCRs may be well suited to in vitro selections so that they can be maintained under conditions that favor structural integrity, and in the presence of additives that stabilize specific conformations.
Additional emerging technologies
There are also additional technologies that may be applied to GPCRs. To stabilize GPCRs for use with phage display and in vitro selections of mAbs or for immunization based approaches, Heptares developed a proprietary StaR® (Stabilized Receptor) technology to engineer mutations into GPCRs that stabilize them into specific pharmacological conformations. They have applied it to the development of mAbs with agonist activity (www.heptares.com). This approach has and will be greatly facilitated by structural data on GPCRs.
Advances have also been made with constrained peptides. Pepscan’s Clip Technology (www.pepscan.com) allows expression of large number of constrained peptides mimicking epitopes in the loops of receptors. The risk of such an approach is that the conformation may not completely replicate the loop structure, but a number of mAbs have been selected using this approach.
GPCRs have been expressed in liposomes and magnetic nanoparticles [46, 47]. These technologies have the advantage of generating high levels of receptor compared to cellular expression, which is highly desirable for in vitro selections. However, this approach does not orient the receptor, resulting in internal epitopes being exposed to mAb selection. This limitation has been overcome using virus like particles.
Nanodiscs are a new class of model membranes that consist of a single phospholipid bilayer surrounded by a protein belt derived from apolipoprotein A-I [48–50]. They have been used to functionally reconstitute several membrane proteins including the β2 adrenergic receptor and μ opioid receptor [51, 52]. Superior stability and functionality with respect to G protein coupling has also observed for nanodisc solubilized the β2 adrenergic receptor compared to detergent solubilized receptor and, thus, nanodiscs may prove useful for in vitro selections [53].
In addition, advances have been made with regard to increasing expression of GPCRs at the cell surface such as using ZFP transcription factor technology (www.sangamo.com), Selexis SureTechnology (www.selexis.com) among others. Cells with high level of GPCR expression are amenable to cell panning of mAb libraries. However, this methodology is still substantially more cumbersome than selecting on purified protein, but ensures selection of mAbs against conformationally correct epitopes.
Beyond technologies involved in the generation of mAbs, more attention is being given to the incorporation of effector functions such as antibody-dependent cellular cytotoxicity (ADCC) via binding to Fc receptors. One example is a glyco-engineered mAb (mogamulizumab, POTELIGEO®) that targets the chemokine receptor CCR4 for the treatment of T cell leukemias and lymphomas [54]. This mAb was developed with Kyowa Hakko Kirin’s Potelligent® technology to eliminate fucose from the glycan PTMs and thereby enhance ADCC and anti-tumor activity. Similarly, their Complegent® Technology enhances complement mediated cytotoxicity (CDC). Xencor has also developed XmAb® and Xtend™ mAb technology for enhancing ADCC and extending half-life, respectively, although to our knowledge, they have not applied these technologies to mAbs against GPCRs (www.xencor.com).
Summary
The recognition that small molecule ligands can selectively modulate signaling properties of GPCRs by conformational stabilization has tremendous significance for the development of therapeutic mAbs that do the same. In addition, the fact that the conformations of GPCRs can be allosterically modulated by a variety of factors such as hetero-oligomerization, and that these states can be distinguished by antibodies also bodes well for the development of more finely tuned therapeutic mAbs. In the antibody field, the increased emphasis on in vitro assays that select for specific conformational/functional features of targets, the ability to bypass immunization methods and the development of methods for engineering effector functions should all contribute to the more efficient development of mAbs with precisiely defined properties. As these two fields come together, we can anticipate an acceleration in the pace at which GPCR mAbs enter clinical trials. The rate limiting step for the success of such therapeutics should remain at the level of fully understanding the underlying biology of specific diseases, and here, GPCR mAbs should accelerate the validation of specific targets in pre-clinical studies.
Table 2.
mAb that recognize specific conformations of GPCRs
| Receptor | mAb-Source/composition | Epitope Recognized | Receptor State Detected | Pharmacology | Ref |
|---|---|---|---|---|---|
| CCR5 | MC-4, MC-5, MC-7 | N-terminal domain | Agonist conformation with multiple ligands bound | Agonist Inhibition | [19] |
| CCR5 | MC-1 | ICL-2 | Agonist conformation | Multiple agonist inhibition | [19] |
| CCR5 | MC-6 | ECL- conformation determinant | Active conformation | Stabilizes active conformation | [19] |
| CXCR4 | 12G5 | ECL-1,2 conformational | Cell type and donor specific conformations | Inhibition of chemotaxis | [55] |
| CXCR4 | 44717.111 (12G5 also examine in this paper) | Proteoliposome (PMPLs) containing CXCR4 | Binds to SDF-1 bound or unbound R | Abolished binding of GP120 | [56] |
| CXCR4 | Llama immunization with CXCR4- expressing HEK293; followed by phage library selection | Antagonist and inverse agonist, binds to ECL2 and ECL3 | [57] | ||
| CCR5 | 3A9 | Phage Displayed CCR5 epitopes inferred by alanine mapping to extracellular loops and computer modeling | Not indicated | Not Indicated | [58] |
| β2AR | Camelid Nanobody Nb80 | Cytoplasmic end of receptor projecting into the lattice | By modeling and Crystallography -the agonist active state | Intracellular loop binding no pharmacological activity noted | [59] |
| Rhodopsi n | K42–41L antibody selected by reactivity with a peptide in a phage display library | Epitope found in dark-adapted rhodopsin and metarhodopsin I not in metarhodopsin II and is a sequence in C3 loop | Inhibits for formation of metarhodpsin II while stabilizing metarhodpsin I- locks C3 loop into an extended conformation | Not studied | [60] |
Acknowledgments
The authors acknowledge support from the National Institutes of Health/National Institute of General Medical Sciences PSI:Biology program as awards U54 GM094618 (to R.C.S.) and U01 GM094612 and R21 AI101687 (to T.M.H.). The authors thank Yekaterina Kadyshevskaya for assistance with figure preparation, and Angela Walker for assistance with manuscript preparation.
Footnotes
Classification: Inflammation and Immunopharmacology
References
- 1.Fredriksson R, Lagerstrom MC, Lundin LG, Schioth HB. The G-protein-coupled receptors in the human genome form five main families. Phylogenetic analysis, paralogon groups, and fingerprints. Mol Pharmacol. 2003;63:1256–1272. doi: 10.1124/mol.63.6.1256. [DOI] [PubMed] [Google Scholar]
- 2.Insel PA, Snead A, Murray F, Zhang L, Yokouchi H, Katakia T, Kwon O, Dimucci D, Wilderman A. GPCR expression in tissues and cells: are the optimal receptors being used as drug targets? Br J Pharmacol. 2012;165:1613–1616. doi: 10.1111/j.1476-5381.2011.01434.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chen L, Jin L, Zhou N. An update of novel screening methods for GPCR in drug discovery. Expert Opin Drug Discov. doi: 10.1517/17460441.2012.699036. [DOI] [PubMed] [Google Scholar]
- 4.Williams AF, Barclay AN. The immunoglobulin superfamily--domains for cell surface recognition. Annu Rev Immunol. 1988;6:381–405. doi: 10.1146/annurev.iy.06.040188.002121. [DOI] [PubMed] [Google Scholar]
- 5.Kohler G, Milstein C. Continuous cultures of fused cells secreting antibody of predefined specificity. Nature. 1975;256:495–497. doi: 10.1038/256495a0. [DOI] [PubMed] [Google Scholar]
- 6.Golay J, Introna M. Mechanism of action of therapeutic monoclonal antibodies: Promises and pitfalls of in vitro and in vivo assays. Arch Biochem Biophys. 2012 doi: 10.1016/j.abb.2012.02.011. [DOI] [PubMed] [Google Scholar]
- 7.Gupta A, Heimann AS, Gomes I, Devi LA. Antibodies against G-protein coupled receptors: novel uses in screening and drug development. Comb Chem High Throughput Screen. 2008;11:463–467. doi: 10.2174/138620708784911465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hutchings CJ, Koglin M, Marshall FH. Therapeutic antibodies directed at G protein-coupled receptors. MAbs. 2010;2:594–606. doi: 10.4161/mabs.2.6.13420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mackrill JJ. Generation, use, and validation of receptor-selective antibodies. Methods Mol Biol. 2004;259:47–65. doi: 10.1385/1-59259-754-8:047. [DOI] [PubMed] [Google Scholar]
- 10.Bockaert J, Pin JP. Molecular tinkering of G protein-coupled receptors: an evolutionary success. EMBO J. 1999;18:1723–1729. doi: 10.1093/emboj/18.7.1723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Duvernay MT, Filipeanu CM, Wu G. The regulatory mechanisms of export trafficking of G protein-coupled receptors. Cell Signal. 2005;17:1457–1465. doi: 10.1016/j.cellsig.2005.05.020. [DOI] [PubMed] [Google Scholar]
- 12.Cancro MP, Kearney JF. B cell positive selection: road map to the primary repertoire? J Immunol. 2004;173:15–19. doi: 10.4049/jimmunol.173.1.15. [DOI] [PubMed] [Google Scholar]
- 13.Gupta A, Decaillot FM, Gomes I, Tkalych O, Heimann AS, Ferro ES, Devi LA. Conformation state-sensitive antibodies to G-protein-coupled receptors. J Biol Chem. 2007;282:5116–5124. doi: 10.1074/jbc.M609254200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mancia F, Brenner-Morton S, Siegel R, Assur Z, Sun Y, Schieren I, Mendelsohn M, Axel R, Hendrickson WA. Production and characterization of monoclonal antibodies sensitive to conformation in the 5HT2c serotonin receptor. Proc Natl Acad Sci U S A. 2007;104:4303–4308. doi: 10.1073/pnas.0700301104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jacobson KA, Costanzi S. New Insights for Drug Design from the X-ray Crystallographic Structures of GPCRs. Mol Pharmacol. 2012 doi: 10.1124/mol.112.079335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hicks D, Barnstable CJ. Different rhodopsin monoclonal antibodies reveal different binding patterns on developing and adult rat retina. J Histochem Cytochem. 1987;35:1317–1328. doi: 10.1177/35.11.3655327. [DOI] [PubMed] [Google Scholar]
- 17.Bailey BW, Mumey B, Hargrave PA, Arendt A, Ernst OP, Hofmann KP, Callis PR, Burritt JB, Jesaitis AJ, Dratz EA. Constraints on the conformation of the cytoplasmic face of dark-adapted and light-excited rhodopsin inferred from antirhodopsin antibody imprints. Protein Sci. 2003;12:2453–2475. doi: 10.1110/ps.03233703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Goetzl EJ, Dembrow D, Van Brocklyn JR, Graler M, Huang MC. An IgM-kappa rat monoclonal antibody specific for the type 1 sphingosine 1-phosphate G protein-coupled receptor with antagonist and agonist activities. Immunol Lett. 2004;93:63–69. doi: 10.1016/j.imlet.2004.02.007. [DOI] [PubMed] [Google Scholar]
- 19.Blanpain C, Vanderwinden JM, Cihak J, Wittamer V, Le Poul E, Issafras H, Stangassinger M, Vassart G, Marullo S, Schlndorff D, Parmentier M, Mack M. Multiple active states and oligomerization of CCR5 revealed by functional properties of monoclonal antibodies. Mol Biol Cell. 2002;13:723–737. doi: 10.1091/mbc.01-03-0129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Shukla AK, Violin JD, Whalen EJ, Gesty-Palmer D, Shenoy SK, Lefkowitz RJ. Distinct conformational changes in beta-arrestin report biased agonism at seven-transmembrane receptors. Proc Natl Acad Sci U S A. 2008;105:9988–9993. doi: 10.1073/pnas.0804246105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kenakin T. Functional selectivity through protean and biased agonism: who steers the ship? Mol Pharmacol. 2007;72:1393–1401. doi: 10.1124/mol.107.040352. [DOI] [PubMed] [Google Scholar]
- 22.Kenakin T. G protein coupled receptors as allosteric proteins and the role of allosteric modulators. J Recept Signal Transduct Res. 2010;30:313–321. doi: 10.3109/10799893.2010.503964. [DOI] [PubMed] [Google Scholar]
- 23.Hartley O, Gaertner H, Wilken J, Thompson D, Fish R, Ramos A, Pastore C, Dufour B, Cerini F, Melotti A, Heveker N, Picard L, Alizon M, Mosier D, Kent S, Offord R. Medicinal chemistry applied to a synthetic protein: development of highly potent HIV entry inhibitors. Proc Natl Acad Sci U S A. 2004;101:16460–16465. doi: 10.1073/pnas.0404802101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Whalen EJ, Rajagopal S, Lefkowitz RJ. Therapeutic potential of beta-arrestin- and G protein-biased agonists. Trends Mol Med. 2012;17:126–139. doi: 10.1016/j.molmed.2010.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kenakin T. Functional selectivity and biased receptor signaling. J Pharmacol Exp Ther. 336:296–302. doi: 10.1124/jpet.110.173948. [DOI] [PubMed] [Google Scholar]
- 26.Rios C, Gomes I, Devi LA. mu opioid and CB1 cannabinoid receptor interactions: reciprocal inhibition of receptor signaling and neuritogenesis. Br J Pharmacol. 2006;148:387–395. doi: 10.1038/sj.bjp.0706757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bradbury AR, Sidhu S, Dubel S, McCafferty J. Beyond natural antibodies: the power of in vitro display technologies. Nat Biotechnol. 2011;29:245–254. doi: 10.1038/nbt.1791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sidhu SS, Fellouse FA. Synthetic therapeutic antibodies. Nat Chem Biol. 2006;2:682–688. doi: 10.1038/nchembio843. [DOI] [PubMed] [Google Scholar]
- 29.Sidhu SS, Li B, Chen Y, Fellouse FA, Eigenbrot C, Fuh G. Phage-displayed antibody libraries of synthetic heavy chain complementarity determining regions. J Mol Biol. 2004;338:299–310. doi: 10.1016/j.jmb.2004.02.050. [DOI] [PubMed] [Google Scholar]
- 30.Kim J, Stroud RM, Craik CS. Rapid identification of recombinant Fabs that bind to membrane proteins. Methods. 2012;55:303–309. doi: 10.1016/j.ymeth.2011.09.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.de Haard HJ, van Neer N, Reurs A, Hufton SE, Roovers RC, Henderikx P, de Bruine AP, Arends JW, Hoogenboom HR. A large non-immunized human Fab fragment phage library that permits rapid isolation and kinetic analysis of high affinity antibodies. J Biol Chem. 1999;274:18218–18230. doi: 10.1074/jbc.274.26.18218. [DOI] [PubMed] [Google Scholar]
- 32.Hoogenboom HR. Selecting and screening recombinant antibody libraries. Nat Biotechnol. 2005;23:1105–1116. doi: 10.1038/nbt1126. [DOI] [PubMed] [Google Scholar]
- 33.Beerli RR, Rader C. Mining human antibody repertoires. MAbs. 2010;2:365–378. doi: 10.4161/mabs.2.4.12187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ravn U, Gueneau F, Baerlocher L, Osteras M, Desmurs M, Malinge P, Magistrelli G, Farinelli L, Kosco-Vilbois MH, Fischer N. Bypassing in vitro screening--next generation sequencing technologies applied to antibody display and in silico candidate selection. Nucleic Acids Res. 2010;38:e193. doi: 10.1093/nar/gkq789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Michnick SW, Sidhu SS. Submitting antibodies to binding arbitration. Nat Chem Biol. 2008;4:326–329. doi: 10.1038/nchembio0608-326. [DOI] [PubMed] [Google Scholar]
- 36.Glanville J, Zhai W, Berka J, Telman D, Huerta G, Mehta GR, Ni I, Mei L, Sundar PD, Day GM, Cox D, Rajpal A, Pons J. Precise determination of the diversity of a combinatorial antibody library gives insight into the human immunoglobulin repertoire. Proc Natl Acad Sci U S A. 2009;106:20216–20221. doi: 10.1073/pnas.0909775106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Persson MA. Twenty years of combinatorial antibody libraries, but how well do they mimic the immunoglobulin repertoire? Proc Natl Acad Sci U S A. 2009;106:20137–20138. doi: 10.1073/pnas.0912118106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Fellouse FA, Wiesmann C, Sidhu SS. Synthetic antibodies from a four-amino-acid code: a dominant role for tyrosine in antigen recognition. Proc Natl Acad Sci U S A. 2004;101:12467–12472. doi: 10.1073/pnas.0401786101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Knappik A, Ge L, Honegger A, Pack P, Fischer M, Wellnhofer G, Hoess A, Wolle J, Pluckthun A, Virnekas B. Fully synthetic human combinatorial antibody libraries (HuCAL) based on modular consensus frameworks and CDRs randomized with trinucleotides. J Mol Biol. 2000;296:57–86. doi: 10.1006/jmbi.1999.3444. [DOI] [PubMed] [Google Scholar]
- 40.Rothe C, Urlinger S, Lohning C, Prassler J, Stark Y, Jager U, Hubner B, Bardroff M, Pradel I, Boss M, Bittlingmaier R, Bataa T, Frisch C, Brocks B, Honegger A, Urban M. The human combinatorial antibody library HuCAL GOLD combines diversification of all six CDRs according to the natural immune system with a novel display method for efficient selection of high-affinity antibodies. J Mol Biol. 2008;376:1182–1200. doi: 10.1016/j.jmb.2007.12.018. [DOI] [PubMed] [Google Scholar]
- 41.Koide S, Sidhu SS. The importance of being tyrosine: lessons in molecular recognition from minimalist synthetic binding proteins. ACS Chem Biol. 2009;4:325–334. doi: 10.1021/cb800314v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Fellouse FA, Barthelemy PA, Kelley RF, Sidhu SS. Tyrosine plays a dominant functional role in the paratope of a synthetic antibody derived from a four amino acid code. J Mol Biol. 2006;357:100–114. doi: 10.1016/j.jmb.2005.11.092. [DOI] [PubMed] [Google Scholar]
- 43.Schoonbroodt S, Steukers M, Viswanathan M, Frans N, Timmermans M, Wehnert A, Nguyen M, Ladner RC, Hoet RM. Engineering antibody heavy chain CDR3 to create a phage display Fab library rich in antibodies that bind charged carbohydrates. J Immunol. 2008;181:6213–6221. doi: 10.4049/jimmunol.181.9.6213. [DOI] [PubMed] [Google Scholar]
- 44.Gao J, Sidhu SS, Wells JA. Two-state selection of conformation-specific antibodies. Proc Natl Acad Sci U S A. 2009;106:3071–3076. doi: 10.1073/pnas.0812952106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Newton K, Matsumoto ML, Wertz IE, Kirkpatrick DS, Lill JR, Tan J, Dugger D, Gordon N, Sidhu SS, Fellouse FA, Komuves L, French DM, Ferrando RE, Lam C, Compaan D, Yu C, Bosanac I, Hymowitz SG, Kelley RF, Dixit VM. Ubiquitin chain editing revealed by polyubiquitin linkage-specific antibodies. Cell. 2008;134:668–678. doi: 10.1016/j.cell.2008.07.039. [DOI] [PubMed] [Google Scholar]
- 46.Jones JW, Greene TA, Grygon CA, Doranz BJ, Brown MP. Cell-free assay of G-protein-coupled receptors using fluorescence polarization. J Biomol Screen. 2008;13:424–429. doi: 10.1177/1087057108318332. [DOI] [PubMed] [Google Scholar]
- 47.Kimura T, Yeliseev AA, Vukoti K, Rhodes SD, Cheng K, Rice KC, Gawrisch K. Recombinant cannabinoid type 2 receptor in liposome model activates g protein in response to anionic lipid constituents. J Biol Chem. 2012;287:4076–4087. doi: 10.1074/jbc.M111.268425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Borch J, Hamann T. The nanodisc: a novel tool for membrane protein studies. Biol Chem. 2009;390:805–814. doi: 10.1515/BC.2009.091. [DOI] [PubMed] [Google Scholar]
- 49.Leitz AJ, Bayburt TH, Barnakov AN, Springer BA, Sligar SG. Functional reconstitution of Beta2-adrenergic receptors utilizing self-assembling Nanodisc technology. Biotechniques. 2006;40:601–602. 604, 606. doi: 10.2144/000112169. passim. [DOI] [PubMed] [Google Scholar]
- 50.Bayburt TH, Sligar SG. Membrane protein assembly into Nanodiscs. FEBS Lett. 2010;584:1721–1727. doi: 10.1016/j.febslet.2009.10.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Whorton MR, Bokoch MP, Rasmussen SG, Huang B, Zare RN, Kobilka B, Sunahara RK. A monomeric G protein-coupled receptor isolated in a high-density lipoprotein particle efficiently activates its G protein. Proc Natl Acad Sci U S A. 2007;104:7682–7687. doi: 10.1073/pnas.0611448104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kuszak AJ, Pitchiaya S, Anand JP, Mosberg HI, Walter NG, Sunahara RK. Purification and functional reconstitution of monomeric mu-opioid receptors: allosteric modulation of agonist binding by Gi2. J Biol Chem. 2009;284:26732–26741. doi: 10.1074/jbc.M109.026922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Yao XJ, Velez Ruiz G, Whorton MR, Rasmussen SG, DeVree BT, Deupi X, Sunahara RK, Kobilka B. The effect of ligand efficacy on the formation and stability of a GPCR-G protein complex. Proc Natl Acad Sci U S A. 2009;106:9501–9506. doi: 10.1073/pnas.0811437106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Beck A, Reichert JM. Marketing approval of mogamulizumab: A triumph for glyco-engineering. MAbs. 2012;4:419–425. doi: 10.4161/mabs.20996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Baribaud F, Edwards TG, Sharron M, Brelot A, Heveker N, Price K, Mortari F, Alizon M, Tsang M, Doms RW. Antigenically distinct conformations of CXCR4. J Virol. 2001;75:8957–8967. doi: 10.1128/JVI.75.19.8957-8967.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Babcock GJ, Mirzabekov T, Wojtowicz W, Sodroski J. Ligand binding characteristics of CXCR4 incorporated into paramagnetic proteoliposomes. J Biol Chem. 2001;276:38433–38440. doi: 10.1074/jbc.M106229200. [DOI] [PubMed] [Google Scholar]
- 57.Jahnichen S, Blanchetot C, Maussang D, Gonzalez-Pajuelo M, Chow KY, Bosch L, De Vrieze S, Serruys B, Ulrichts H, Vandevelde W, Saunders M, De Haard HJ, Schols D, Leurs R, Vanlandschoot P, Verrips T, Smit MJ. CXCR4 nanobodies (VHH-based single variable domains) potently inhibit chemotaxis and HIV-1 replication and mobilize stem cells. Proc Natl Acad Sci U S A. 2010;107:20565–20570. doi: 10.1073/pnas.1012865107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.O’Connor KH, Konigs C, Rowley MJ, Irving JA, Wijeyewickrema LC, Pustowka A, Dietrich U, Mackay IR. Requirement of multiple phage displayed peptide libraries for optimal mapping of a conformational antibody epitope on CCR5. J Immunol Methods. 2005;299:21–35. doi: 10.1016/j.jim.2004.11.025. [DOI] [PubMed] [Google Scholar]
- 59.Rasmussen SG, Choi HJ, Fung JJ, Pardon E, Casarosa P, Chae PS, Devree BT, Rosenbaum DM, Thian FS, Kobilka TS, Schnapp A, Konetzki I, Sunahara RK, Gellman SH, Pautsch A, Steyaert J, Weis WI, Kobilka BK. Structure of a nanobody-stabilized active state of the beta(2) adrenoceptor. Nature. 2011;469:175–180. doi: 10.1038/nature09648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Piscitelli CL, Angel TE, Bailey BW, Hargrave P, Dratz EA, Lawrence CM. Equilibrium between metarhodopsin-I and metarhodopsin-II is dependent on the conformation of the third cytoplasmic loop. J Biol Chem. 2006;281:6813–6825. doi: 10.1074/jbc.M510175200. [DOI] [PubMed] [Google Scholar]