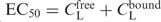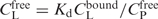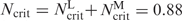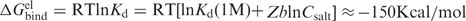An official website of the United States government
Here's how you know
Official websites use .gov
A
.gov website belongs to an official
government organization in the United States.
Secure .gov websites use HTTPS
A lock (
) or https:// means you've safely
connected to the .gov website. Share sensitive
information only on official, secure websites.
As a library, NLM provides access to scientific literature. Inclusion in an NLM database does not imply endorsement of, or agreement with,
the contents by NLM or the National Institutes of Health.
Learn more:
PMC Disclaimer
|
PMC Copyright Notice
This is an Open Access article distributed under the terms of the Creative Commons Attribution Non-Commercial License (http://creativecommons.org/licenses/by-nc/3.0), which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
The authors wish to apologize for an error in the above article. Because of the particular variant of the solid phase peptide chemistry used, the molecules displayed in Figure 1 do not have a carboxylate group at the C-terminus, but an amide group (the C-terminus should be CONH2, not CO−2). Hence, the molecular weights changed slightly and each molecule should have one unit larger positive charge. For example, the un-branched εK3 molecule (upper panel Figure 1 with n = 3) will have a charge Z = +4, not +3 as indicated in the article. We have re-calculated, re-plotted and re-analyzed all the data in the article with correct molecular weights and charges of the molecules. This has resulted in several numerical changes and Figures 1, 2, 8 and 9 have been redrawn. For some peptides, the fitting to Equation 3 also results in different numerical values for the binding parameters (see legends to Figures 8 and 9 and the related discussion of the results in the corrected text). The discussion of the interpretation of the results comparing three- and four-valent cations in the first paragraph on page 7145 of the original article is incorrect and has now been excluded. However, the majority of the text and all general conclusions are unchanged. The article (also containing corrected Supplementary Data) is reproduced in full here with all the relevant corrections.
This is an Open Access article distributed under the terms of the Creative Commons Attribution Non-Commercial License (http://creativecommons.org/licenses/by-nc/2.5/uk/) which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
We report a systematic study of the condensation of plasmid DNA by oligocations with variation of the charge, Z, from +3 to +31. The oligocations include a series of synthetic linear ε-oligo(l-lysines), (denoted εKn, n = 3–10, 31; n is the number of lysines with the ligand charge Z = n+1) and branched α-substituted homologues of εK10: εYK10, εLK10 (Z = +11); εRK10, εYRK10 and εLYRK10 (Z = +21). Data were obtained by light scattering, UV absorption monitored precipitation assay and isothermal titration calorimetry in a wide range concentrations of DNA and monovalent salt (KCl, CKCl). The dependence of EC50 (ligand concentration at the midpoint of DNA condensation) on CKCl shows the existence of a salt-independent regime at low CKCl and a salt-dependent regime with a steep rise of EC50 with increase of CKCl. Increase of the ligand charge shifts the transition from the salt-independent to salt-dependent regime to higher CKCl. A novel and simple relationship describing the EC50 dependence on DNA concentration, charge of the ligand and the salt-dependent dissociation constant of the ligand–DNA complex is derived. For the ε-oligolysines εK6–εK10, the experimental dependencies of EC50 on CKCl and Z are well-described by an equation with a common set of parameters. Implications from our findings for understanding DNA condensation in chromatin are discussed.
INTRODUCTION
DNA is arguably the most important and well-studied molecule in biology. Yet, the understanding of its physico-chemical properties and the implications of these for the biochemical function of DNA in the cell nucleus is still incomplete. One highly important property of DNA, which is still not fully understood, is its compaction and aggregation ability. For example, in the cell nucleus, DNA is present in the form of chromatin and the compaction maintained with the aid of positively charged histones. DNA compaction is also of considerable biotechnological importance in non-viral gene delivery, where DNA compaction by polycationic delivery vehicles enables penetration through the plasma cell membrane (1,2).
Given the high negative surface charge, with an expected strong repulsive free energy barrier to compaction, it may seem surprising that DNA in solution, spontaneously undergoes a transition from an extended random coil conformation to a condensed state upon addition of polycationic ligands LZ+ (Z > 2) (3–7). DNA condensation may proceed as a collapse of single DNA molecules or as intermolecular aggregation. The outcome depends on DNA molecular weight and DNA concentration (6,8,9). Several factors contribute to the condensation of DNA with electrostatic repulsion between the highly negatively charged DNA polyions being the major force opposing formation of compact DNA structures (3,4). Therefore, the major prerequisite for DNA compaction by oligocations is a sufficient neutralization of the DNA negative charge. Additionally, in the presence of multivalent cations, ion-ion correlations create net DNA–DNA attraction (10,11). Hydration forces have also been suggested to contribute to DNA–DNA repulsion as well as attraction (12). Both DNA neutralization by oligocations and cation–cation correlation are expected to decrease with increase of monovalent salt, due to increasing competition with K+ (Na+) for binding to DNA (7,13). Another favorable contribution to the DNA condensation is the formation of DNA–DNA bridges by oligocations when the ligand is a flexible molecule of sufficient length [e.g. polyamines, N-terminal domains of the histones, protamines (6,14–17)].
Already 30 years ago, Bloomfield and co-workers established a simple rule for polycationic induced compaction/aggregation of DNA: condensation occurs when ∼90% of the DNA charge [as calculated by the Manning counterion condensation model (18)] is neutralized by counterions (19). This phenomenological rule has since then been established under a variety of conditions with a range of polycations (4).
Theoretically, DNA–DNA attraction caused by the presence of multivalent cations was investigated using analytical and simulation approaches (6,15,20). Analytical approaches include description of ion correlation effects pioneered by Oosawa (21), description of the DNA-multivalent ion system as a Wigner crystal (22,23) and modelling tight binding of cations to DNA (24). Simulations of the collapse and/or polyion–polyion attraction caused by the addition of multivalent were carried out using Monte Carlo (10,11,25–28) and molecular dynamics simulations (14,29–33).
So far, there is no comprehensive systematic study of the dependence of DNA condensation on the major factors that define the outcome of the process: the net charge of the cationic ligand, the concentration of monovalent salt (KCl or NaCl) and the DNA concentration. For the ‘simple’ condensation agents such as naturally occurring polyamines spermidine3+ (Spd3+) and spermine4+ (Spm4+) and hexamminecobalt(III) (CoHex3+), a large number of studies have been carried out (19,34–41). The corresponding dependencies for cationic ligands carrying positive charge +5 and higher have only been addressed in a few studies and remain enigmatic (39,42–44).
In this work we present a systematic account of DNA condensation, investigated for a broad range of charge of added cationic ligand (from Z = +3 to +31). We cover a variation of the concentration of DNA over almost two orders of magnitude and in a range of concentration of monovalent salt (KCl). The major result is the development of a simple universal thermodynamic model of DNA condensation by oligocationic ligands. The model gives a clear and quantitative relationship between concentration and charge of the ligand causing the DNA condensation, binding constant of the ligand–DNA interaction and concentrations of DNA and monovalent salt. It is able to predict the experimental behavior for a vast range of such conditions. As cationic ligands we use our recently designed ε-oligolysines and their branched derivatives (Figure 1) (45).
Chemical structures of linear ε-oligolysines and α–substituted derivatives of εK10. (L, Y and R represent amino-acid residue leucine, tyrosine and arginine, respectively).
MATERIALS AND METHODS
Materials
The plasmid pEGFP-N1 (4.7 kb) was a gift from Lu Yanning at Nanyang Technological University. The plasmid was amplified in Escherichia coli DH5α strain and isolated using the QIAGEN HiSpeed Plasmid Purification Giga Kit (Valencia, CA). DNA concentration was determined by Varian Cary 300 Bio UV-Visible (Varian Inc., Palo Alto, CA) or NanoDrop ND-1000 (NanoDrop Technologies Inc, Wilmington, DE) spectrophotometers using extinction coefficient 6600 M−1 cm−1 at 260 nm. The absorbance ratio (A260/A280) was >1.8, indicating absence of protein contamination. Stock solutions of DNA in 10 mM or 100 mM KCl were prepared from lyophilized DNA and stored at 4°C. DNA solutions for DLS, PA and ITC measurements were prepared by dilution of the stock DNA solution in KCl solutions of known molarity; pH of the DNA solutions was in the range 6.8–5.4 (solutions with higher CKCl had lower pH values).
Two types of cationic peptides were used: unsubstituted ε-oligo(l-lysines) of varying length and α-substituted ε-oligo(l-lysines) (Figure 1). The ε-oligolysines were synthesized using solid-phase peptide synthesis, purified and characterized as described (45). ε-Poly(l-lysine), εK31, with degree of polymerization 31 in the form of chloride salt was purchased from Chisso Corporation (Tokyo, Japan). Hexammine cobalt(III) chloride, Co(NH3)6Cl3; spermidine trihydrochloride, C7H19N3·3HCl; spermine tetrahydrochloride, C10H26N4·4HCl; potassium chloride and other salts were purchased from Sigma-Aldrich Inc. (St. Louis, MO) and used without purification. In all measurements we used KCl (Sigma-Aldrich Inc., St. Louis, MO) diluted to designed concentration from 3 M stock solution.
Dynamic light scattering (DLS)
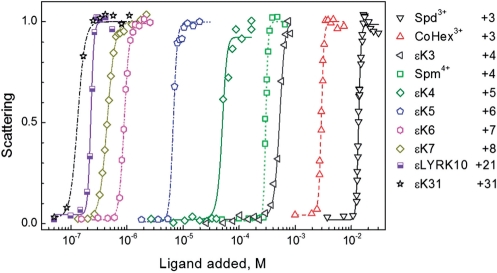
Dynamic light scattering (DLS) experiments were conducted using a Brookhaven 90plus particle size analyzer (Long Island, NY) as described in (45). The effectiveness in inducing DNA compaction has been quantified by the molar concentration of the ligand needed for a 50% increase in the static 90° light scattering intensity (EC50), see Figure 2. Values of EC50 were calculated from titration curves displaying the scattering intensity versus ligand concentration using sigmoidal fitting. The particle sizes (effective diameters assuming a lognormal distribution) were calculated applying standard theory using software provided by equipment manufacturer.
Light scattering of plasmid DNA solution in dependence on concentration of some cationic ligands with net-positive charge ranging from +3 to +31 (100 mM KCl and 5 µM in DNA phosphate groups). Points are measured values (normalized relative to maximal intensity of light scattering observed in each titration); curves are sigmoid fitting of the experimental data. Net-positive charge of the ligands is indicated in the graph.
Because of the vast number of EC50 values determined over a wide range of conditions with three methods, a systematic repetition of all measurements to determine standard deviation of the presented data was not possible, due to sample limitation. However, several randomly chosen individual EC50 points were repeated two times and on the basis of the scattering, the error is estimated at 3–10%. This estimate holds for EC50 determinations with all three methods used in this study.
Precipitation assay
Precipitation of the plasmid DNA was studied by UV absorption monitored assay using a NanoDrop ND-1000 UV/Vis spectrophotometer (NanoDrop Technologies Inc, Wilmington, DE). Solution of plasmid DNA (30 µl) of optical density A260 = 0.66 (DNA concentration 100 µM) with given KCl and ligand concentration were prepared by consecutive addition of the stock solutions (3 M KCl, plasmid DNA, and cationic ligand) to double distilled Millipore water in Eppendorf tubes, 15 s vortexing and equilibrating the mixtures for 20 min at room temperature followed by centrifugation 10 min × 11 000g in Eppendorf Centrifuge 5424 (Eppendorf AG, Hamburg Germany). Values of A260 were measured in supernatant by placing 2 µl in the NanoDrop spectrophotometer. The optical density of the supernatant was compared with the A260 absorbance of the two control solutions of DNA plus KCl (with and without spinning) in the absence of the ligand. Absorbance of the control solutions was equal within experimental uncertainty of the measurements (2–3% according to the NanoDrop specifications). Results of the precipitation assay were plotted as optical absorbance versus concentration of added ligand and fitted by sigmoidal function similar to the DLS titration data.
Isothermal titration calorimetry (ITC)
A VP-ITC microcalorimeter from MicroCal (Northampton, MA) was used at 25°C. The measurements were conducted as described earlier (45). The nonspecific interaction between cationic ligands and DNA is accompanied by very low heat effects with the need for relatively high DNA concentration (0.40 mM here) and great care in experimentation (46,47). Typically, heat effects associated with dilution of the ligands studied in this work were comparable with the heat capacity changes related to the ligand–DNA interaction (see Figure 1S, A in Supplementary Data). Titration peaks from the two titrations (with and without DNA) were integrated and the heats of ligand dilution were subtracted from the data obtained in the ligand–DNA titration. The Origin7.0 graphic and analysis program integrated in the VP-ITC calorimeter by VPViewer 2000 ITC software was used for data collection, analysis, and presentation. Examples of the raw ITC data, dependence of binding enthalpy on molar ratio of added ligand, and comparison of the calorimetry data with a fitting calculation based on a one-site-binding model are given in the Supplementary Figure 1S.
RESULTS
Light scattering
Figure 2 displays typical curves for the titration of the ε-oligolysine oligocationic ligands into solution of low concentrations of plasmid DNA (5 µM) and at fixed CKCl = 100 mM, including our data for the much studied (19,34–41) cations Spd3+, Spm4+ and CoHex3+. The transition of DNA from extended to compact form is accompanied by a steep increase of the scattered light. We will call these measurements ‘DLS’, since scattering in all cases was obtained from a dynamic light scattering (DLS) experiments. The capacity of the ligand to induce DNA condensation can be quantified by the ligand concentration needed for a 50% increase of light scattering intensity, EC50, applying a sigmoidal fitting of the experimental titration curves (lines in Figure 2). Spermidine3+ is the least efficient condensing agent with EC50, in the range of 10–20 mM Spd3+. The other ligand with the same positive charge, cobalt(III) CoHex3+ displays higher efficiency. εK3 (Z = +4) shows higher DNA condensation competence than CoHex3+ but somewhat lower than the other tetracationic ligands, Spm4+. In the presence of 100 mM KCl, the efficiency of the εK4 (Z = +5) is about five times higher than that of Spm4+. ε-Oligolysines demonstrate slightly weaker DNA condensation potential than polyamines of similar charge. An increase of charge of the cationic ligand from Z = +4 (Spm4+ and εK3) to Z = +6 (εK5) leads to a pronounced decrease of EC50. Further increase of the ligand charge from Z = +8 to Z = +31 is accompanied by a gradual increase of the effectiveness of cationic ligand.
The dependencies of EC50 values on monovalent salt concentration (CKCl) are summarized in Figure 3; numerical values are given in the Supplementary Table 1S. The most noticeable feature for the ε-oligolysines with charge +5 ≤ Z ≤ +11, (εK4 to εK10) is the existence of two regimes: a salt-independent regime at low concentration of KCl and a salt-dependent one at high CKCl. Ligands with Z = +3 show only salt-dependent behaviour; above CKCl of 50 mM, EC50 for εK4 increases steeply with CKCl. For the ε-oligolysines εK5 to εK10, the transition from salt-independent to salt-dependent regime shifts to higher CKCl with increase of the ligand charge; EC50 for εK5 becomes salt-dependent above 50 mM KCl; whereas for εK10 a steep increase of EC50 is observed above CKCl of 200 mM. The ligand with the highest positive charge Z = +31, εK31, shows no salt dependence; EC50 gradually decreases with CKCl from 10 to 400 mM. For most of the ε-oligolysines, a switch between the two regimes is accompanied by small drop in EC50 and broadening of the titration curves.
KCl concentration dependence of midpoint (EC50) of DNA compaction by cationic ligands determined from the DLS titration data. (A) EC50 values in dependence on the KCl concentration and charge of the ligand. (B) Results for the substituted ε-oligolysines and εK10 and εK31. In (A) and (B), EC50 is expressed in mol/l of ligand; DNA concentration is constant and equal to 5 µM. Horizontal dashes below and above EC50 data points show respectively the EC20 and EC80 values for the cases when the titration curves display a broad transition. See text for details.
Figure 3B compares the EC50 values of the substituted εK10 homologues with different side chains: εLK10, εYK10 (Z = +11); εRK10, εYRK10 and εLYRK10 (Z = +21). Modification of εK10 by attaching a hydrophobic (leucine, εLK10) or aromatic (tyrosine, εYK10) amino acids without changing net positive charges of the peptide makes little influence on the ligand compaction capacity at low CKCl (10–20 mM). However, at higher CKCl, εK10, εLK10, and εYK10 behave differently: εLK10 demonstrates transition from salt-independent to the salt-dependent regime already at CKCl > 100 mM accompanied by a rather steep decrease in EC50 just before the switch between the regimes. EC50 for the εK10 becomes salt-dependent at CKCl > 200 mM, whereas EC50 of the εYK10 remains ‘flat’ at all KCl concentrations. The insertion of arginine in εK10 increases the positive charge of the peptide to Z = +21. All three arginine-containing compounds, εRK10, εYRK10 and εLYRK10, show only salt-independent behavior.
Variations of DNA particle size in dependence on DLS titration conditions
Dynamic light scattering measurements in DNA solutions are significantly complicated by the fact that the single-molecule collapse of the giant DNA polymer is accompanied by formation of multimolecular aggregates. DNA aggregation leads to the formation of large inhomogeneous particles and to DNA precipitation that are complex time-dependent functions of DNA molecular weight, DNA concentration, diffusion properties of the particle, kinetics of ligand–DNA interaction, and all other conditions of the DLS-monitored DNA titration. Due to the high molecular weight of the DNA, the concentration of DNA when exclusively monomolecular collapse can be observed is too low to be detected with DLS. For these reasons, particle-size information collected in the DLS measurements is of rather limited value although some experimental (9,48) and theoretical (8) studies have been performed earlier. In our titration experiments, we obtained size distributions and the onset of DNA condensation was accompanied by a sharp increase in light scattering signal with formation of particles having an average diameter of 200 nm (lowest observed values were 150 nm). This indicates that in the beginning of the DNA condensation, aggregates of many DNA molecules are formed (the diameter of a single-molecule plasmid DNA globule is estimated to be less than 30 nm).
Precipitation assay
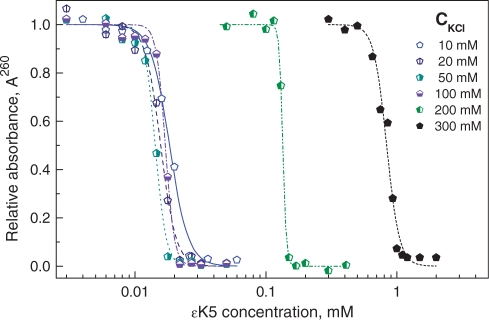
An example of a KCl concentration-dependence series of precipitation assays (DNA concentration CP = 100 µM) is shown in Figure 4 with data for εK5. Figure 5 summarizes the data for all cationic ligands (including, for comparison data for Spd3+, Spm4+ and CoHex3+); numerical values are given in the Supplementary Table 2S). The data of the PA (Figure 5) and DLS (Figure 3) methods generally show a qualitative similarity. There are, however, several small differences in the results. First, the values of EC50 in the salt-independent regime in Figure 5 (PA) are shifted to higher concentration as compared to Figure 3 (DLS). This change of EC50 is expected since the concentration of DNA in the precipitation assay was 100 µM compared with CP = 5 µM in the DLS titrations and more ligand will be needed to neutralize the negative charge of the DNA. Secondly, for the ligands of the same charge, in the precipitation assay data, the transition from the salt-independent to the salt-dependent regime is shifted to a region of higher salt concentrations. In addition, unlike the DLS data, even the lowest charged ε-oligolysine, ε K3, shows a salt-independent regime at CKCl < 20 mM. Furthermore, the PA data show that DNA condensation occurs in a more narrow range of ligand concentrations, compared to the width recorded by the DLS method.
Example of precipitation assay data showing optical absorbance A260 in supernatant of the plasmid DNA solution (CP = 100 µM) versus concentration of the added cationic ligand, εK5. Points are measured values (normalized relative to A260 measured in the DNA solution in the absence of εK5); curves are sigmoidal fitting of the experimental data.
Dependence of midpoint (EC50) of DNA condensation by cationic ligands determined from the precipitation assay data on KCl concentration. (A) Summary showing change of the EC50 values in dependence on the KCl concentration and charge of the ligand. (B) Results for the substituted ε-oligolysines. Data obtained for the unsubstituted εK10 and εK31 are also displayed for comparison. In (A) and (B), EC50 is expressed in mol/L of ligand; DNA concentration is constant and equal to 100 µM.
ITC
Measurements (DNA concentration 400 μM) were obtained at two concentrations of monovalent salt; CKCl = 10 and 100 mM (no data obtained for εK3 and εK4). An example of the raw ITC data is given in the Supplementary Figure 1S, A). The dependence of ligand–DNA interaction enthalpy (ΔH) on amount of the added ligand is given in Figure 6 and a typical curve of ΔH versus molar ratio of ligand is also given in Supplementary Data, Figure 1S, B. The dependence of ΔH on the amount of added ligand shows two stages of DNA–ligand interaction: (i). Initially the ligand binds to extended DNA in solution and after saturation; (ii). DNA condensation occurs accompanied by an additional heat peak. The enthalpy of the oligocation–DNA interaction in the first and in the second stage of interaction is positive (with some rare exceptions which will be discussed below). This implies that the driving force of the oligocation binding to DNA is the entropic gain which is the result of net release of the monovalent ions and water upon ligand binding (46,47,49,50). Formation of condensed DNA is reflected in the ITC output as increased heat capacity peaks (Supplementary Data, top panel in Supplementary Figure 1S,A) and as sharp peak or discontinuity in the ΔH versus molar ratio dependence (Figure 6). In discussion of the results, we will use two precisely defined quantities: The molar ratio corresponding to the peak of DNA condensation, Npeak (we assume this corresponds to the midpoint of DNA condensation with EC50 = Npeak × CP), and the molar-binding enthalpy corresponding to the initial plateau in the ΔH versus ligand/DNA molar ratio dependencies (see discussion about further analysis in Supplementary Data).
Results of ITC showing enthalpy at 298 K plotted versus ratio of added ligand charge to charge of the DNA. Panels (A) and (B) show the results obtained in 10 mM KCl; panels (C) and (D) present similar data determined in 100 mM KCl; DNA concentration 400 µM in all measurements. (A) and (C). Influence of degree of polymerization of ε-oligolysines on enthalpy of ligand–DNA interaction (data on εK7, εK9, and εK10 not shown). (B) and (D). Influence of side chain of the εK10 derivatives on enthalpy of ligand–DNA interaction (εLK10, εYK10, εYRK10 and unsubstituted εK10; data on εRK10 and εLYRK10 not shown).
To compare binding and condensation of DNA by the oligocations with different charges, values of ΔH were normalized by the charge of the ligand. The enthalpies of oligocation–DNA interaction in dependence of charge ratio (positive charge of added ligand/negative charge of DNA) are summarized in Figure 6 for two different KCl concentrations. ITC measurements carried out with Spd3+, CoHex3+ and Spm4+ are given in Supplementary Data (Figure 2S).
The positive enthalpy observed in the ITC data is a signature of ligand interaction with the DNA minor groove (49,50). For the data obtained in 10 mM KCl, heat effects of the initial ε-oligolysine–DNA binding remain similar to these at CKCl = 100 mM (Figure 6B). For Spm4+, Spd3+ and CoHex3+ at high salt, the value ΔH ≈ 0 indicates the occurrence of no or weak binding of these cations to DNA (data not shown). Our ITC results are in perfect agreement with the published data for Spd3+ and CoHex3+except slightly lower values of ΔH for CoHex3+ (46,47,51). The ITC result for Spm4+ and CKCl = 10 mM is in qualitative agreement with the data of Patel and Anchodoquy (52) determined at lower salt concentration (5 mM HEPES buffer, pH 6, no added salt).
The derivatives of εK10 demonstrate more variation in ΔH than the simple ε-oligolysines especially in low salt, CKCl = 10 mM (Figure 6B and D). εLK10 and εLYRK10 have more positive enthalpy than other ligands. These differences in ΔH might reflect different contributions of electrostatic, hydrophobic and specific interactions (like formation of hydrogen bond) in the binding of the ligand to DNA (47). Differences in binding enthalpy can influence DNA-condensation efficiency of the oligocations and will be discussed below.
For CKCl = 10 mM and ε-oligolysines εK5-εK8 and especially for the εK31, the contribution of DNA condensation to the total heat effect is larger (Figure 6A) than similar effects reported for CoHex3+ and polyamines [Supplementary Figure 2S and (46)]. At the higher monovalent salt concentration, CKCl = 100 mM (Figure 6C and D), all ε-oligolysines become more efficient, as compared to the situation in CKCl = 10 mM, with the condensation peak shifting towards lower ligand/DNA charge ratios. This reduction in Npeak is in agreement with the other DLS and PA results. The heat effects associated with both initial ligand–DNA interaction and with the DNA condensation are also reduced for the case of the arginine-containing derivatives of the εK10 (εRK10, εYRK10). At CKCl = 100 mM, DNA condensation by εRK10, εYRK10 is actually accompanied by a negative deflection in the dependence of ΔH on molar (charge) ligand/DNA ratio (Figure 6D). The observed reduced value of ΔH for εRK10, εLYRK10 and the negative enthalpy for εYRK10 might indicate that the arginine-containing ligands interact with the major groove of DNA during second stage of the ligand binding (49,50) and this interaction is salt-dependent.
Electrostatics is the major factor defining DNA condensation
We normalize the values of EC50, obtained by the DLS, PA and ITC methods using the concentration of ligand charge at the point of 50% DNA condensation, EZ50 = EC50·Z. Values of EZ50 in dependence on CKCl determined for ε-oligolysines obtained from all three methods are summarized in Figure 7 (for clarity, data for a selection of ε-oligolysines are displayed). Normalization of the EC50 value reduces the dispersion of the experimental data. The EZ50 values for the salt-independent regime clearly form three groups defined by the DNA concentration used in the different methods, 400 µM in the ITC, 100 µM in the PA and 5 µM in the DLS. In the salt-dependent regime, the values of the EZ50 for the same ligand obtained at different DNA concentrations in the DLS and in the PA methods become similar and are practically equal for εK5 and εK6 at the highest KCl concentration, CKCl = 400 mM. Still, there is a small and increasing difference between the EZ50 values obtained by the DLS and the PA methods for the ε-oligolysines from εK7 to εK10 (Figure 7A). Increase of the DNA concentration shifts the transition from the salt-independent to salt-dependent regime to higher CKCl values.
Dependence of EZ50 concentration on concentration monovalent salt (CKCl) obtained by DLS, PA and ITC methods. (A). EZ50 versus CKCl for the ε-oligolysivnes from εK4 to εK10 and εK31 (for clarity data on εK3, εK7, εK8, εK9 not shown); (B). EZ50 versus CKCl for the derivatives of εK10 (data on εRK10 not shown). Horizontal bars show constant DNA concentration in the solutions studied by correspondingly ITC, PA and DLS methods (from top to bottom).
Clearly, the charge of the ligand is the major factor defining its DNA condensation efficiency, which is illustrated by variation of the concentration of monovalent salt where condensation changes from being DNA-concentration to KCl-concentration dependent. However, modifications by addition of various amino acids to ε-oligolysine (or in general other chemical groups to the oligocation) can modulate this dependence. This is demonstrated in Figure 7B where the two ε-oligolysines with Z = +11, εLK10 and εYK10, show behavior different to each other and to that displayed by the simple ε-oligolysine of the same charge, εK10.
Explanation of the two regimes of salt dependence of EC50
The explanation of the existence of salt-independent and salt-dependent regimes in the log EC50 versus log CKCl curves is simple:
EC50 is the total concentration of the ligand (CtotL) at the critical point in solution where the free energies of extended DNA in solution and condensed DNA are equal. The total ligand concentration is the sum of the concentration of the ligand bound to the DNA and free in solution: , where CfreeL and CboundL refer to the conditions at the midpoint of DNA condensation. The value of CboundL is related to the critical degree of neutralization of DNA charge necessary to induce DNA condensation, due to bound cationic ligands (NLcrit) and to the total DNA concentration (CP) such that CfreeLCboundL = CPNLcrit/Z. The free ligand concentration,CfreeL is dependent on the DNA-binding affinity of the ligand: . Here, Kd ( = 1/Kb, binding constant) is the dissociation constant of the ligand–DNA complex, CfreeP = CP − ZCboundL is the concentration of free DNA; or more specifically, the concentration of DNA phosphate groups not neutralized by the oligocation. After a simple transformation the dependence of EC50 takes the form:
1
Equation (1) is general and based on thermodynamic-stoichiometric considerations of the DNA–ligand-binding equilibrium at the midpoint of condensation. The fraction of neutralized DNA phosphate charges caused by all bound cations present in solution, r (the notation used in Manning theory), is denoted Ncrit at the condensation point. Generally, , is the sum of the fractions of monovalent counterions, NMcrit and cationic ligands, NLcrit, bound per one phosphate group. At low concentration of monovalent salt, the contribution from LZ + is dominant as it has been shown by experimental measurements of the composition of ions in condensed DNA (53). Furthermore, theoretical calculations using CC or Poisson Boltzmann theories (54) as well as Monte Carlo simulations supports these observations (55) (see also the section describing the CC theory results in the Supplementary Data). Hence, it is assumed that the value of NLcrit is equal to Ncrit. This is a reasonable approximation in the low salt regime and in the high salt region the error due to this assumption has small effect on the behavior of the EC50 curves (see also text below and the section describing the CC theory results in the Supplementary Data).
In the salt-independent regime, DLS, PA and ITC results show that compaction of DNA occurs at an oligocation/DNA charge ratio below 1.0 (Figure 7) which indicates that the early observation by Wilson and Bloomfield that about 90% of the DNA charge must be neutralized to induce condensation holds (19). According to this concept, Ncrit is a constant with a universal value independent of conditions and we will assume that this phenomenological relation holds.
It is well established that oligocation–DNA interaction is strongly salt-dependent (56,57) with the dissociation (binding) constant showing a linear dependence in logarithmic coordinates, logKd versus logCsalt (KCl in the present work) (56–60):
2
or
2a
Here, Kd(1M) is the value of Kd extrapolated to CKCl = 1M, the term which accumulates all non-electrostatic effects of oligocation–DNA interactions (56,58–60); SK = dlogKd/dlogCKCl ≈ bZ is the slope of the logKd–logCsalt dependence; b is the thermodynamic degree of binding of the monovalent cation to the DNA polyion per negative phosphate charge. Estimates of b for double-stranded (ds) DNA and synthetic polynucleotides in interaction with α-oligolysines and α-oligoarginines give similar values of b that are close to unity (b ≈ 0.9) (57,59). Consequently, the slope SK is steep and sensitive to the value of Z. Combining Equations (1) and (2a) gives the relation for EC50:
3
The above result presents a simple and clear explanation of the observed two regimes in the dependencies of EC50 on monovalent salt concentration. Using the approximate value , Equation (1) takes a very simple form: EC50 = 0.9·[10·Kd + CP]/Z). Hence, one can illustrate the conditions defining DNA condensation either in the salt-independent or the salt-dependent regime:
For the low salt concentration when there is little competition between the ligand and monovalent cations, 10Kd < CP and EC50 is proportional to the DNA concentration and independent of monovalent salt (CKCl). For the high salt regime when there is a considerable competition between the ligand and the monovalent cations, Kd > CP/10 and EC50 is salt dependent and proportional to CbZKCI. The region of transition depends largely on the DNA concentration and the charge of the ligand such that high DNA concentration and/or high ligand charge preserves the salt-independent regime. This is precisely the general appearance of all the collected data as demonstrated by Figure 7. Equation (3) can be used to fit the experimental data with Kd(1M) and b (in SK = bZ, with Z given by ligand charge) as only independent variables. In the case of available experimental results for Kd(1M) and SK = bZ it can be used to compare with prediction on the basis of independent experimental data.
Interpretation of the experimental data for the ε-oligolysines
Figure 8 shows results of fitting Equation (3) to the experimental data for the ε-oligolysines εK5 to εK10 (Figure 8A–C) and for the substituted εLK10 (Figure 8D). We used the user-defined fitting service of the graphical software package Origin8.0 (OriginLab Corp., Northamton, MA) for fitting the values SK = bZ and Kd(1M) of Equation (3) to the experimental data at various fixed values of Ncrit. At low KCl concentrations in the salt-independent regime CP ≫ Kd and Equation (3) gives a good estimation for the value of Ncrit: Ncrit ≈ EC50·Z/CP. Variation of Ncrit in the range 0.85–0.92 with fitting values of SK and Kd(1M) produces good agreement between experimental and calculated values of EC50 in the whole range of KCl concentration. The value Ncrit = 0.88 gives the best fit to the experimental data. The fitting of Equation (3) to the experimental data for the ε-oligolysines eK6–eK10 results in SK = 0.85Z and Kd(1M) = 0.22 M; the lower charged ε-oligolysines give following fitting parameters: eK5, SK = 0.85Z, Kd(1 M) = 0.33 M; eK4, SK = 0.8Z, Kd(1 M) = 0.33 M; eK3, SK = 0.6Z, Kd(1 M) = 0.08 M (Ncrit = 0.88 for all fittings; for eK3 fitting is of poor quality since only PA data can be used).
Comparison of the experimental dependencies of EC50 on CKCl (data of DLS and PA shown as points with thin lines) to the calculated values using Equation (3) (displayed as smooth thick lines). (A–C) Results obtained for ε-oligolysine homologues from εK5 to εK10. Theoretical curves were drawn as follows: (A) fixed value Ncrit = 0.88 and the best fit values SK = 0.85Z and Kd(1 M) = 0.33 (eK5), 0.22 M (eK10) are used. Theoretical curves in (B) and (C) were built without fitting using the values SK = 0.85Z and Kd(1M) = 0.22 M; (B). Data for εK6 and εK9; (C) data for εK7 and εK8. (D) Comparison of the results calculated with Equation (3) with experimental results of the DLS and PA methods of εK10 and εLK10. Best fit is for Kd(1M) = 18 M and SK = 0.85Z with the fixed value Ncrit = 0.88; the data for εK10 are the same as in (A). See text for details.
Data on DNA condensation by εK3 and εK4 are shown in Figure 9. For εK3 and εK4 (Figure 9) these values give good fitting of all the experimental data obtained at the higher DNA concentration (PA method) and for the high salt data from the lower DNA concentration (DLS method), but not for the DLS data at low KCl concentration. Comparing the oligocations with increasing charge, from εK5 to εK10, there is a systematic shift to higher concentration of KCl where the EC50 values from the PA data (high DNA concentration) converge with those from the DLS (low DNA concentration) which remarkably is borne out by the theoretical curves.
Comparison of the experimental dependencies (points with thin lines) with prediction based on Equation (3) (shown as smooth thick lines) for data on DNA condensation by the ε-oligolysines εK3 and εK4. DLS and PA data are shown respectively as hollow and solid points. The parameters of Equation (3) are the same as in Figure 8A. See text and Supplementary Figure 4S and Table 4S) for more detail.
In Figure 8D, experimental and theoretically fitted results for the εLK10 and εK10 ε-oligolysines are compared. These ligands carry the same charge Z = +11, show similar EC50 values in the salt-independent regime (for the PA method) but εLK10 demonstrates a shift to the salt-dependent regime for CKCl > 100 mM, which is noticeably lower than the same transition for εK10 (CKCl > 200 mM). In addition, the DLS data for εLK10 display larger downward shift at CKCl = 50 – 100 mM than the case of εK10. The best fit of the experimental data for the εLK10 and Ncrit = 0.88 is obtained at b = 0.85 and Kd(1M) = 18 M. Kd(1M) value of εLK10 is about 80 times larger than the same parameter of εK10. This corresponds to a difference in free energy of ligand–DNA binding of about +2.6 kcal/mol. Thus, the difference in CKCl where the shift from salt-independent to salt-dependent regime occurs is caused by a higher unfavorable non-electrostatic (positive) enthalpy of the εLK10–DNA interaction compared to the enthalpy value of εK10. Remarkably, this picture is confirmed by the independent ITC results, demonstrating more positive ΔH values for εLK10 (Figure 6B and D).
The ε-oligolysines from εK6 to εK10 show a uniform dependence of the binding constant on KCl concentration with values of Kd(1M) in the range 0.18–0.25 M when Ncrit varies from 0.90 to 0.85 and the SK value is equal to 0.85Z. Depending on the choice , the best fitting is achieved for Kd(1M) = 0.22 and Ncrit = 0.88 or Kd(1M) = 0.18 when Ncrit = 0.90. Our fitted values of SK = bZ (which gives b = 0.85) for the ε-oligolysines are in agreement with the parameters available in the literature for α-oligolysines (58,61) and α-oligoarginines (59), which are in the range of 0.82–0.95 (62) with the value centered at 0.88. The values of Kd(1M) = 0.18–0.25 M for the ε-oligolysines indicates a small favorable non-electrostatic contribution to the ligand–DNA binding. The literature Kd(1M) values for α-oligolysines and α-oligoarginines often show a varying behavior (58,61). However, a thorough study of α-oligolysine binding to single stranded polynucleotides gave very similar results of the extrapolated log Kd(1M) values for a range of ligand charges (58,63). In any case, the conclusion about ε-oligolysine–DNA binding is the same as for the other unstructured basic peptides: their non-specific interaction with double stranded DNA is dominated by electrostatic forces, defined by the net ligand charge and monovalent salt concentration, with the non-electrostatic contribution being relatively small in the range of physiological salt concentrations (56,57).
Fitting Equation (3) to the experimental data for tri- and tetracationic ligands
Figure 9 shows fitting of Equation (3) to experimental data of the tetra- and pentacationic ε-oligolysines. For εK3 and εK4, with the exception of the data at low CKCl and low DNA concentration (DLS method), all the experimental results demonstrate good agreement with theoretical curves obtained using the fitted parameters calculated for the ε-oligolysines with charge Z > +5: Kd(1M) = 0.22 and SK = 0.85Z (see Figure 8A–C).
Comparing experimental values of tetracationic Spm4+ and εK3, one can see that spermine4+ is slightly more efficient condensation agent than εK3.
For the cations Z = +3 − +5, the experimental EC50 values at low DNA concentration at low CKCl are larger than expected from the theoretical estimations. The difference for the tetravalent oligolysine, εK3, is significant. A similar discrepancy is observed for the other ligands with charges Z = +3 and +4 (Supplementary Data, Figure 4S). The reduced efficiency of the ligands with charge +3 and +4 observed in the DLS measurements might be related to some unaccounted effect caused by the very low concentration of DNA typically applied in the DLS studies (in the present work, CP = 5 µM). At the same time, for the ε-oligolysines with higher positive charge Z > +5 no such effect is observed and Equation (3) gives consistent and universal fitting of all experimental data for both PA and DLS data (see above). Additional analysis and experimental study is needed to resolve this issue.
DISCUSSION
ITC results explain discrete (all-or-nothing) mechanism of DNA condensation
A number of studies report that condensation of DNA due to the addition of condensing agent occurs as a sharp all-or-nothing transition and intermediate states of DNA do not exist. Direct observation of DNA molecules by fluorescent microscopy revealed that the DNA exists in solution either as an extended molecule or as a compact globule (65,66). ITC data shows that DNA condensation is the second stage of the oligocation–DNA interaction which follows the first stage of ligand binding to DNA and it occurs after sufficient neutralization of DNA by the ligand has taken place. Calculation of thermodynamic quantities for the two stages of oligocation DNA interaction on the basis of ITC titration curves revealed that the binding constant of ligand interaction in condensed DNA was larger than the Kb for interaction of the oligocation with extended DNA (46,51). A recent combined single molecule magnetic tweezer and osmotic stress measurements of the DNA condensation induced by Spd3+, Spm4+ and the hexacationic analog of spermine, sp6+, demonstrated that the ratio of attractive to repulsive free energy is about two, providing a strong driving force towards DNA condensation upon addition of oligocationic ligand (67). Our ITC data are in qualitative and quantitative agreement with the literature. Therefore, the onset of DNA condensation creates new population of binding sites (inside condensed DNA) with lower free energy. Consequently, at the onset of DNA compaction, redistribution of oligocations between collapsed and extended DNA occurs. Compacted DNA forms by depletion of ligands from the extended DNA until the free energies of the extended and compacted DNA are equal. This depletion of the soluble DNA with cationic ligand might explain the observation that DNA molecules in solution repel each other even above the critical concentration of the condensing agent (68).
Comparison with literature data
Dependence of EC50 values on concentration of monovalent salt (NaCl) is reported in literature for a number of oligocations with charge varied from +3 to +6. The most studied cations are CoHex3+ and Spd3+ (19,46). Using light scattering, Thomas and co-workers determined the salt (NaCl) dependence of EC50 by a number of oligocations with Z = +4, polyamines (37–39) and for α-tetralysine (43). The existence of two regimes in the NaCl dependence for penta- and hexamine, with different slopes for these polylysines, was noted and attributed to a difference in binding affinity to DNA. Comparison of the DLS, PA and ITC results of the present work with literature data for tri- and tetravalent cations is presented in the Supplementary Figure 3SA and with a number of penta- and hexacationic polyamines in Supplementary Figure 3SB. The comparison shows that our results are in general agreement with most literature data. Values of EC50 determined by ITC at CKCl = 10 mM are in perfect agreement with similar data reported for CoHex3+ (46,51) and Spd3+ (46) in 10 mM NaCl. Similar increase of the critical concentration of Spm4+ (40,41) or X salmon protamine (Z = +21) (44) with increase of CNaCl was observed by Livolant and colleagues. These authors noted the effects of NaCl and appreciated the importance of such factors as DNA concentration, DNA neutralizing counterions and counterion competition for DNA condensation.
One of the major results of our study is a derivation of Equation (3) which provides a universal description of the oligocation-induced DNA condensation and establishes a clear connection between DNA condensation and salt-dependent binding of the ligands to DNA. The linear logarithmic salt dependence of dissociation (binding) constant of the ligand–DNA interaction [Equation (2)] is well justified, not only on the basis of experimental data. The relation dlogKd/dlogCKCl = SK ≈ bZ follows from both Manning counterion condensation theory and the Poisson-Boltzmann cylindrical cell model, under limiting law conditions (62). The limiting law value of b is 0.88 in both of these approaches, in very good agreement with the fitted values obtained in our model. However, such theories cannot be applied to theoretically predict the onset of attraction in condensation experiments since they always predict repulsion between rod-like polyelectrolytes (6). Therefore, the assumption that Ncrit = 0.88 – 0.90 has to be based on the wide range of experimental observations that have documented this behavior and remains an empirical relation. In order to understand the origin of the attractive interactions that lead to condensation, when this critical degree of DNA charge neutralization has been met by the addition of oligocations, theoretical models beyond mean field theories must be applied (14,32,69).
The model also assumes that at the condensation point, the fraction of DNA charge neutralized by bound counterions is dominated by oligocations (). This approximation is certainly not generally valid. The inaccuracy of this assumption becomes larger in the range of high salt concentration. However, it has a small effect on the dependence of EC50 on CKCl, which is steep in this region. Even a significant substitution of LZ+ for K+ with decrease in NLcrit (say, from 0.90 to 0.60; see theoretical Supplementary Figure 8S) does not produce a great influence on the general appearance of the EC50 versus CKCl curve. In fact, the presence of this assumption in Equation (3) highlights the competition from monovalent ions for binding to DNA as the most likely origin of the small decrease in EC50 at low to intermediate CKCl, observed mainly for ligands of higher charge.
The present study was carried out on plasmid (circular) DNA of relatively low molecular weight. We do not expect that for linear DNA of different (larger) length, the major qualitative features of the dependencies reported here would be different. Bloomfield and co-workers (46) reported that ITC-monitored condensation of plasmid DNA in circular and linear forms produced absolutely identical results. A recent study by Toma et al. (44) reports little difference between condensation of short (about 146 bp) and long (48.5 kb) DNA polyions by protamines from salmon sperm (average Z = +21) in conditions corresponding to the salt-dependent regime (CNaCl from 0.4 to 1.5 M). It might still be possible that for DNA of different molecular weight there will be some difference in ranges where the salt-independent and salt dependent regimes are observed. Additional studies may be necessary to address the issue of how molecular weight of the DNA might influence dependencies of EC50 on CP, CKCl and Z.
Variation of EC50 in the salt-independent regime
In the salt-independent regime, the logEC50–logCKCl dependencies of the ε-oligolysines (in particular those with Z > +4) show a gradual and small decrease of EC50 with increase in KCl concentration (Figures 3, 5 and 7). In addition, just before the transition to the salt-dependent regime an additional ‘plunge’ of EC50 is often observed. Equation (3) is not able to account for the decrease of EC50 in the salt-independent regime. There are two (not mutually exclusive) mechanisms that can explain why an increase in CKCl may facilitate DNA condensation in the salt-independent regime: First, increase of salt introduces competition between mono- and multivalent cations which is neglected in Equation (3) due to the assumption . This causes a decrease in EC50 (see below discussion on application of Manning counterion condensation theory). Secondly, the flexible ε-oligolysines can form DNA–DNA bridges and the bridging may be facilitated by an increase of CKCl. We applied the counterion condensation model in an attempt to estimate the dependence of total degree of DNA charge neutralization on the concentration of monovalent salt due to binding of two cations with different valency. Briefly, this analysis gives the following result (Supplementary Data): showed the best agreement with the experimental EC50 values in the salt-independent regime for the ε-oligolysines with Z from +3 to +10 (Supplementary Figures 6S and 8S). However, the counterion condensation theory fails to predict the following major trends in the experimental data: (i) The magnitude of the slope of EC50 versus CKCl in the salt-dependent regime for all values of Ncrit and Z; (ii) the CC theory shows poor prophecy of the range of monovalent salt concentration where a transition from salt-dependent to salt-independent regime occurs. Clearly, more detailed theoretical models are needed to explain these principal features of the experimental dependencies. At the same time, we found that the CC theory captures the slight decrease of EC50 in the salt-independent regime, which is neglected in Equation (3) (due to the assumption ). This slight drop of the EC50 is caused by substitution of the LZ+ ligands by K+ upon increase of the salt concentration and is dependent on the choice of Ncrit.
CONCLUSIONS
We have proposed a simple and universal model for the dependence of oligocation ligand concentration needed to induce DNA condensation (EC50) on the DNA (CP) and monovalent salt (Csalt) concentrations. The DNA condensation is well described for a range of conditions, by a model combining the established and uniform salt dependence of the oligocation–DNA dissociation constant with the necessity to neutralize about 90% of the DNA charge. These two assumptions, together with the approximation that the fraction of DNA charge neutralized by bound counterions is dominated by oligocations (), leads to Equation (3). The virtue of this phenomenological relation is that it relates the DNA condensation behavior to its dependence on the experimental conditions in a very clear and simple way. Equation (3) connects two well-studied phenomena of DNA polyelectrolyte solution behavior, namely DNA oligocation-induced condensation and the salt-dependent DNA oligocation binding in a novel way. The appearance of one salt dependent and one salt-independent regime in this condensation follows naturally within this model. This model gives a qualitatively correct prediction of the salt and DNA concentration dependent ε-oligolysine induced DNA condensation. For the simple cations Spd3+, Spm4+ and CoHex3+, the description is good in the high salt regime as well as in the low salt regime at higher DNA concentrations (PA data), while the prediction fails in the low-salt and low-DNA concentration limit. To our knowledge, this is the first study reporting a direct and simple connection between oligocation binding to DNA (Kd value) and DNA condensation.
Implications to in vivo conditions
The dynamic units of the histone–DNA interaction in chromatin are the histone tails with net positive charge from +8 to +14 and average charge density of one lysine or arginine per every three amino acids (70). The ε-oligolysines studied in this work can be considered as a simplified model of the histone tails with respect to positive charge and charge density. To a certain extent, the ε-oligolysines with high positive charge, Z = +31 (εK31) and Z = +21 (εRK110, εYRK10, εLYRK10) may serve as a simplified model for the nonspecific interaction between DNA and basic proteins of similar net positive charge (e.g. histones).
The concentration of the DNA in the cell nucleus is very high (71,72). Recent fluorescent measurements give average concentration of 116 µM of nucleosomes (CP = 46.4 mM) with the highest concentration 260 µM of nucleosomes in heterochromatin (CP = 104 mM) (73). Extrapolating the EZ50 versus Csalt dependencies to such high DNA concentrations (Figure 7), it is clear that under physiological concentration of salt (K+ in the range 50–300 mM), condensation of DNA should always proceed in the salt-independent regime for all natural oligocations (histones, protamines, basic domains of the nuclear proteins, polyamines, etc.). At physiological concentration of salt, the critical degree of DNA neutralization needed to induce DNA condensation would be somewhat lower, in the range Nneut = 0.5 – 0.7, than the higher values Nneut = 0.7 – 0.95 observed at low salt concentration (CKCl = 10–20 mM). In addition, other factors such as the presence of Mg2+, polyamines as well as the crowded environment of the cell cytoplasm, would facilitate DNA condensation. Therefore, it is reasonable to suggest that the conditions of DNA in the nucleus are very close to the borderline separating the extended and collapsed DNA phases. Since DNA in the nucleus is in the salt-independent regime of condensation, the principal factor regulating DNA condensation should be any addition/removal of cationic (or any other) components facilitating the DNA collapse. As a result, there exist powerful ‘brakes’ for overproduction of the basic components (polyamines, protamines, histones), such that any excess of these components would result in DNA condensation blocking access to DNA for the transcription/replication machinery. On the other hand, insufficient amount of DNA condensing agents will lead to DNA expansion that might be damaging and harmful in the crowded space of the cell nucleus (74). The tight stoichiometry between DNA and the histones has recently been illustrated by the in vivo observation of strong correlation between the amount of the core histones, linker histones and nucleosome repeat length in the chromatin (75,76).
Counter-intuitively, the presence of a certain excess of DNA-condensing agents in chromatin may also facilitate access to DNA of the proteins responsible for replication and transcription. From the ITC observation that DNA condensation proceeds with higher affinity than the preceding non-specific (charge neutralizing) interaction, it follows than when the amount of condensing agent exceeds the critical value, the onset of DNA condensation triggers a redistribution of the ligand between the two phases of DNA. Thus, in the region of coexistence between the condensed and extended DNA states, the soluble phase of DNA is depleted of cationic ligands. The remaining uncondensed DNA binds less ligand than under the conditions before the onset of condensation. For the cell nucleus it means that increase in cationic components may lead to DNA condensation (formation of heterochromatin). This will be accompanied by a depletion of oligocations for the rest of chromatin, thus making the euchromatin more open and accessible for transcription. Such an unexpected consequence of excessive concentration of the condensing agents in the chromatin might explain the observation of higher concentration of the polyamines in the eukaryotic cells with high metabolic activity (e.g. in cancer cells) (77).
Our study demonstrates that the electrostatic mechanism is a universal one and salt-dependence of binding constant (and free energy) of ligand–DNA interaction is expected to be similar for the ligand–DNA interaction in solution and in condensed state. The apparent independence of EC50 on salt concentration simply indicates that the DNA condensation is in salt-independent regime and that dissociation constant of ligand–DNA complex is very low so that Kd ≪ CP in Equation (3). The net positive charge of the histone octamer forming the basic unit of eukaryotic chromatin, the nucleosome core particle, is about +147 (70). Therefore, from the salt dependence of oligocation–DNA-binding constant one can predict that the total electrostatic binding energy of the histone octamer to the nucleosomal DNA (the sum of binging energies of the all basic domains in the octamer) must be significant: [assuming Z = +147, Csalt = 0.15M, b = 0.9 and lnKd(1M) ≈ 0]. This estimation demonstrates that the NCP and chromatin in general is an extremely stable polycation–polyanion complex, which challenges the view of a marginal stability of chromatin (78). This evaluation for the electrostatic free energy of chromatin formation is a rather low estimate. Some additional favorable contributions to this term might originate from the non-stoichiometric charge ratio between DNA and the histones (70,79,80). Notably, favorable electrostatic free energy greatly exceeds the unfavorable contributions to the formation of the NCP [mostly due to energy cost of the DNA bending (81,82)] that was confirmed by early experimental observation (83) and by theoretical analysis based on the counterion condensation model (84). More detailed presentation of the arguments in support of a high stability of chromatin originating from nonspecific histone–DNA interaction as well as the consequence of the high stability of chromatin for its statics and dynamics and for the conditions for the protein machines operating on the chromatin template, is discussed in our earlier work (70,80,85).
Singapore Ministry of Education University Research Committee (URC) Tier grant T206B3207 to LN. A*STAR grant (05/1/22/19/414) and Lee Wee Nam foundation (post doctoral funding to KDE) to JPT. Funding for open access charge: Singapore Ministry of Education URC Tier 2 grant T206B3207.
Conflict of interest statement. None declared.
ACKNOWLEDGEMENTS
The authors thank Dr Dandan Huang for help in experimental work.
REFERENCES
1.Mintzer M.A., Simanek E.E. Nonviral vectors for gene delivery. Chem. Rev. 2009;109:259–302. doi: 10.1021/cr800409e. [DOI] [PubMed] [Google Scholar]
2.Mann A., Thakur G., Shukla V., Ganguli M. Peptides in DNA delivery: current insights and future directions. Drug Discov. Today. 2008;13:152–160. doi: 10.1016/j.drudis.2007.11.008. [DOI] [PubMed] [Google Scholar]
4.Bloomfield V.A. DNA condensation by multivalent cations. Biopolymers. 1997;44:269–282. doi: 10.1002/(SICI)1097-0282(1997)44:3<269::AID-BIP6>3.0.CO;2-T. [DOI] [PubMed] [Google Scholar]
5.Vijayanathan V., Thomas T., Thomas T.J. DNA nanoparticles and development of DNA delivery vehicles for gene therapy. Biochemistry. 2002;41:14085–14094. doi: 10.1021/bi0203987. [DOI] [PubMed] [Google Scholar]
6.Nordenskiöld L., Lyubartsev A.P., Korolev N. In: DNA Interactions with Polymers and Surfactants. Dias R.S., Lindman B., editors. London: John Wiley & Sons, Inc.; 2008. pp. 209–237. [Google Scholar]
7.Iwaki T., Saito T., Yoshikawa K. How are small ions involved in the compaction of DNA molecules? Colloids Surf. B Biointerfaces. 2007;56:126–133. doi: 10.1016/j.colsurfb.2006.10.040. [DOI] [PubMed] [Google Scholar]
8.Post C.B., Zimm B.H. Theory of DNA condensation: collapse versus aggregation. Biopolymers. 1982;21:2123–2137. doi: 10.1002/bip.360211104. [DOI] [PubMed] [Google Scholar]
9.Post C.B., Zimm B.H. Light-scattering study of DNA condensation: competition between collapse and aggregation. Biopolymers. 1982;21:2139–2160. doi: 10.1002/bip.360211105. [DOI] [PubMed] [Google Scholar]
10.Guldbrand L., Nilsson L.G., Nordenskiöld L. A Monte Carlo simulation study of electrostatic forces between hexagonally packed DNA double helices. J. Chem. Phys. 1986;85:6686–6698. [Google Scholar]
11.Lyubartsev A.P., Nordenskiöld L. Monte Carlo simulation study of ion distribution and osmotic pressure in hexagonally oriented DNA. J. Phys. Chem. 1995;99:10373–10382. [Google Scholar]
13.Tang J.X., Janmey P.A., Lyubartsev A.P., Nordenskiöld L. Metal ion-induced lateral aggregation of filamentous viruses fd and M13. Biophys. J. 2002;83:566–581. doi: 10.1016/S0006-3495(02)75192-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
14.Korolev N., Lyubartsev A.P., Nordenskiöld L. Computer modeling demonstrates that electrostatic attraction of nucleosomal DNA is mediated by histone tails. Biophys. J. 2006;90:4305–4316. doi: 10.1529/biophysj.105.080226. [DOI] [PMC free article] [PubMed] [Google Scholar]
15.Podgornik R., Licer M. Polyelectrolyte bridging interactions between charged macromolecules. Curr. Opin. Colloid Interface Sci. 2006;11:273–279. [Google Scholar]
16.Podgornik R., Saslow W.M. Long-range many-body polyelectrolyte bridging interactions. J. Chem. Phys. 2005;122:204902. doi: 10.1063/1.1908870. [DOI] [PubMed] [Google Scholar]
17.Podgornik R. Two-body polyelectrolyte-mediated bridging interactions. J. Chem. Phys. 2003;118:11286–11296. [Google Scholar]
18.Manning G.S. The molecular theory of polyelectrolyte solutions with application of the electrostatic properties of polynucleotides. Q. Rev. Biophys. 1978;11:179–246. doi: 10.1017/s0033583500002031. [DOI] [PubMed] [Google Scholar]
19.Wilson R.W., Bloomfield V.A. Counterion-induced condensation of deoxyribonucleic acid. A light-scattering study. Biochemistry. 1979;18:2192–2196. doi: 10.1021/bi00578a009. [DOI] [PubMed] [Google Scholar]
20.Lyubartsev A.P., Nordenskiöld L. In: Handbook of Polyelectrolytes and their Applications v. 3. Tripathy S.K., Kumar J., Nalwa H.S., editors. Los Angeles: American Scientific Publishers; 2002. pp. 309–326. [Google Scholar]
21.Oosawa F. Interaction between parallel rodlike macroions. Biopolymers. 1968;6:1633–1647. [Google Scholar]
22.Nguyen T.T., Grosberg A.Y., Shklovskii B.I. Screening of a charged particle by multivalent counterions in salty water: Strong charge inversion. J. Chem. Phys. 2000;113:1110–1125. [Google Scholar]
23.Nguyen T.T., Rouzina I., Shklovskii B.I. Reentrant condensation of DNA induced by multivalent counterions. J. Chem. Phys. 2000;112:2562–2568. [Google Scholar]
24.Tan Z.-J., Chen S.-J. Nucleic acid helix stability: effects of salt concentration, cation valence and size, and chain length. Biophys. J. 2006;90:1175–1190. doi: 10.1529/biophysj.105.070904. [DOI] [PMC free article] [PubMed] [Google Scholar]
25.Nilsson L.G., Guldbrand L., Nordenskiöld L. Evaluation of the electrostatic osmotic pressure in an infinite system of hexagonally oriented DNA molecules. A Monte Carlo simulation study. Mol. Phys. 1991;72:177–192. [Google Scholar]
26.Lyubartsev A.P., Nordenskiöld L. Monte Carlo simulation study of DNA polyelectrolyte properties in the presence of multivalent polyamine ions. J. Phys. Chem. B. 1997;101:4335–4342. [Google Scholar]
28.Mel'nikov S.M., Khan M.O., Lindman B., Jönsson B. Phase behavior of single DNA in mixed solvents. J. Am. Chem. Soc. 1999;121:1130–1136. [Google Scholar]
29.Allahyarov E., Löwen H. Effective interaction between helical biomolecules. Phys. Rev. E. 2000;62:5542–5556. doi: 10.1103/physreve.62.5542. [DOI] [PubMed] [Google Scholar]
30.Allahyarov E., Gompper G., Löwen H. Attraction between DNA molecules mediated by multivalent ions. Phys. Rev. E. 2004;69:041904. doi: 10.1103/PhysRevE.69.041904. [DOI] [PubMed] [Google Scholar]
31.Luan B., Aksimentiev A. DNA attraction in monovalent and divalent electrolytes. J. Am. Chem. Soc. 2008;130:15754–15755. doi: 10.1021/ja804802u. [DOI] [PMC free article] [PubMed] [Google Scholar]
32.Dai L., Mu Y., Nordenskiöld L., van der Maarel J.R.C. Molecular dynamics simulation of multivalent-ion mediated DNA attraction between DNA molecules. Phys. Rev. Lett. 2008;100:118301. doi: 10.1103/PhysRevLett.100.118301. [DOI] [PubMed] [Google Scholar]
33.Korolev N., Nordenskiöld L. H4 histone tail mediated DNA-DNA interaction and effects on DNA structure, flexibility and counterion binding. A molecular dynamics study. Biopolymers. 2007;86:409–423. doi: 10.1002/bip.20749. [DOI] [PubMed] [Google Scholar]
34.Widom J., Baldwin R.L. Cation-induced toroidal condensation of DNA studies with Co3+(NH3)6. J. Mol. Biol. 1980;144:431–453. doi: 10.1016/0022-2836(80)90330-7. [DOI] [PubMed] [Google Scholar]
35.Widom J., Baldwin R.L. Monomolecular condensation of λ-DNA induced by cobalt hexammine. Biopolymers. 1983;22:1595–1620. doi: 10.1002/bip.360220612. [DOI] [PubMed] [Google Scholar]
36.Plum G.E., Arscott P.G., Bloomfield V.A. Condensation of DNA by trivalent cations. 2. Effects of cation structure. Biopolymers. 1990;30:631–643. doi: 10.1002/bip.360300515. [DOI] [PubMed] [Google Scholar]
37.Saminathan M., Antony T., Shirahata A., Sigal L.H., Thomas T., Thomas T.J. Ionic and structural specificity effects of natural and synthetic polyamines on the aggregation and resolubilization of single-, double-, and triple-stranded DNA. Biochemistry. 1999;38:3821–3830. doi: 10.1021/bi9825753. [DOI] [PubMed] [Google Scholar]
38.Vijayanathan V., Thomas T., Shirahata A., Thomas T.J. DNA condensation by polyamines: a laser light scattering study of structural effects. Biochemistry. 2001;40:13644–13651. doi: 10.1021/bi010993t. [DOI] [PubMed] [Google Scholar]
39.Vijayanathan V., Thomas T., Antony T., Shirahata A., Thomas T.J. Formation of DNA nanoparticles in the presence of novel polyamine analogues: a laser light scattering and atomic force microscopic study. Nucleic Acids Res. 2004;32:127–134. doi: 10.1093/nar/gkg936. [DOI] [PMC free article] [PubMed] [Google Scholar]
40.Raspaud E., Olvera de la Cruz M., Sikorav J.-L., Livolant F. Precipitation of DNA by polyamines: a polyelectrolyte behavior. Biophys. J. 1998;74:381–393. doi: 10.1016/S0006-3495(98)77795-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
41.Raspaud E., Chaperon I., Leforestier A., Livolant F. Spermine-induced aggregation of DNA, nucleosome, and chromatin. Biophys. J. 1999;77:1547–1555. doi: 10.1016/S0006-3495(99)77002-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
42.Vijayanathan V., Lyall J., Thomas T., Shirahata A., Thomas T.J. Ionic, structural, and temperature effects on DNA nanoparticles formed by natural and synthetic polyamines. Biomacromolecules. 2005;6:1097–1103. doi: 10.1021/bm0493234. [DOI] [PubMed] [Google Scholar]
43.Nayvelt I., Thomas T., Thomas T.J. Mechanistic differences in DNA nanoparticle formation in the presence of oligolysines and poly-L-lysine. Biomacromolecules. 2007;8:477–484. doi: 10.1021/bm0605863. [DOI] [PMC free article] [PubMed] [Google Scholar]
44.Toma A.C., de Frutos M., Livolant F., Raspaud E. DNA condensed by protamine: A “short” or “long” polycation behavior. Biomacromolecules. 2009;10:2129–2134. doi: 10.1021/bm900275s. [DOI] [PubMed] [Google Scholar]
45.Huang D., Korolev N., Eom K.D., Tam J.P., Nordenskiöld L. Design and biophysical characterization of novel polycationic ε-peptides for DNA compaction and delivery. Biomacromolecules. 2008;9:321–330. doi: 10.1021/bm700882g. [DOI] [PubMed] [Google Scholar]
46.Matulis D., Rouzina I., Bloomfield V.A. Thermodynamics of DNA binding and condensation: isothermal titration calorimetry and electrostatic mechanism. J. Mol. Biol. 2000;296:1053–1063. doi: 10.1006/jmbi.1999.3470. [DOI] [PubMed] [Google Scholar]
47.Matulis D., Rouzina I., Bloomfield V.A. Thermodynamics of cationic lipid to DNA and DNA condensation: roles of electrostatics and hydrophobicity. J. Am. Chem. Soc. 2002;124:7331–7342. doi: 10.1021/ja0124055. [DOI] [PubMed] [Google Scholar]
48.Arscott P.G., Ma C., Wenner J.A., Bloomfield V.A. DNA condensation by cobalt hexammine(III) in alcohol-water mixtures: dielectric constant and other solvent effects. Biopolymers. 1995;36:345–364. doi: 10.1002/bip.360360309. [DOI] [PubMed] [Google Scholar]
50.Privalov P.L., Dragan A.I., Crane-Robinson C., Breslauer K.J., Remeta D.P., Minetti C.A. What drives proteins into the major or minor grooves of DNA? J. Mol. Biol. 2007;365:1–9. doi: 10.1016/j.jmb.2006.09.059. [DOI] [PMC free article] [PubMed] [Google Scholar]
51.Kim W., Yamasaki Y., Kataoka K. Development of a fitting model suitable for the isothermal titration calorimetric curve of DNA with cationic ligands. J. Phys. Chem. B. 2006;110:10919–10925. doi: 10.1021/jp057554e. [DOI] [PubMed] [Google Scholar]
52.Patel M.M., Anchodoquy T.J. Contribution of hydrophobicity to thermodynamics of ligand-DNA binding and DNA collapse. Biophys. J. 2005;88:2089–2103. doi: 10.1529/biophysj.104.052100. [DOI] [PMC free article] [PubMed] [Google Scholar]
53.Korolev N., Lyubartsev A.P., Rupprecht A., Nordenskiöld L. Competitive substitution of hexammine cobalt(III) for Na+ and K+ ions in oriented DNA fibers. Biopolymers. 2001;58:268–278. doi: 10.1002/1097-0282(200103)58:3<268::AID-BIP1004>3.0.CO;2-8. [DOI] [PubMed] [Google Scholar]
54.Rouzina I., Bloomfield V.A. Competitive electrostatic binding of charged ligands to polyelectrolytes: practical approach using the non-linear Poisson-Boltzmann equation. Biophys. Chem. 1997;64:139–155. doi: 10.1016/s0301-4622(96)02231-4. [DOI] [PubMed] [Google Scholar]
55.Korolev N., Lyubartsev A.P., Nordenskiöld L., Laaksonen A. Spermine: an “invisible” component in the crystals of B-DNA. A grand canonical Monte Carlo and molecular dynamics simulation study. J. Mol. Biol. 2001;308:907–917. doi: 10.1006/jmbi.2001.4642. [DOI] [PubMed] [Google Scholar]
56.Record M.T., Zhang W., Anderson C.F. Analysis of effects of salts and uncharged solutes on protein and nucleic acid equilibria and processes: a practical guide to recognizing and interpreting polyelectrolyte effects, Hofmeister effects, and osmotic effects of salts. Adv. Protein Chem. 1998;51:281–353. doi: 10.1016/s0065-3233(08)60655-5. [DOI] [PubMed] [Google Scholar]
58.Mascotti D.P., Lohman T.M. Thermodynamics of single-stranded RNA and DNA interactions with oligolysines containing tryptophan. Effects of base composition. Biochemistry. 1993;32:10568–10579. doi: 10.1021/bi00091a006. [DOI] [PubMed] [Google Scholar]
59.Mascotti D.P., Lohman T.M. Thermodynamics of oligoarginines binding to RNA and DNA. Biochemistry. 1997;36:7272–7279. doi: 10.1021/bi970272n. [DOI] [PubMed] [Google Scholar]
60.Mascotti D.P., Lohman T.M. Thermodynamic extent of counterion release upon binding oligolysines to single-stranded nucleic acids. Proc. Natl Acad. Sci. USA. 1990;87:3142–3146. doi: 10.1073/pnas.87.8.3142. [DOI] [PMC free article] [PubMed] [Google Scholar]
61.Record M.T., Lohman T.M., de Haseth P. Ion effects on ligand-nucleic acid interactions. J. Mol. Biol. 1976;107:145–158. doi: 10.1016/s0022-2836(76)80023-x. [DOI] [PubMed] [Google Scholar]
62.Anderson C.F., Record M.T. Polyelectrolyte theories and their applications to DNA. Annu. Rev. Phys. Chem. 1982;33:191–222. [Google Scholar]
64.Braunlin W.H., Strick T.J., Record M.T. Equilibrium dialysis studies of polyamine binding to DNA. Biopolymers. 1982;21:1301–1314. doi: 10.1002/bip.360210704. [DOI] [PubMed] [Google Scholar]
65.Takahashi M., Yoshikawa K., Vasilevskaya V.V., Khokhlov A.R. Discrete coil-globule transition of single duplex DNAs induced by polyamines. J. Phys. Chem. B. 1997;101:9396–9401. [Google Scholar]
66.Murayama H., Yoshikawa K. Thermodynamics of the collapsing phase transition in a single duplex DNA molecule. J. Phys. Chem. B. 1999;103:10517–10523. [Google Scholar]
67.Todd B.A., Parsegian V.A., Shirahata A., Thomas T.J., Rau D.C. Attractive forces between cation condensed DNA double helices. Biophys. J. 2008;94:4775–4782. doi: 10.1529/biophysj.107.127332. [DOI] [PMC free article] [PubMed] [Google Scholar]
68.Qiu X., Andresen K., Lamb J.S., Kwok L.W., Pollack L. Abrupt transition from a free, repulsive to a condensed, attractive DNA phase, induced by multivalent polyamine cations. Phys. Rev. Lett. 2008;101:228101. doi: 10.1103/PhysRevLett.101.228101. [DOI] [PMC free article] [PubMed] [Google Scholar]
69.Yang Y., Lyubartsev A.P., Korolev N., Nordenskiöld L. Computer modeling reveals that modifications of the histone tail charges define salt-dependent interaction of the nucleosome core particles. Biophys. J. 2009;96:2082–2094. doi: 10.1016/j.bpj.2008.10.073. [DOI] [PMC free article] [PubMed] [Google Scholar]
70.Korolev N., Vorontsova O.V., Nordenskiöld L. Physicochemical analysis of electrostatic foundation for DNA-protein interactions in chromatin transformations. Prog. Biophys. Mol. Biol. 2007;95:23–49. doi: 10.1016/j.pbiomolbio.2006.11.003. [DOI] [PubMed] [Google Scholar]
71.Daban J.-R. High concentration of DNA in condensed chromatin. Biochem. Cell Biol. 2003;81:91–99. doi: 10.1139/o03-037. [DOI] [PubMed] [Google Scholar]
72.Cano S., Caravaca J.M., Martin M., Daban J.-R. Highly compact folding of chromatin induced by cellular cation concentration. Evidence from atomic force microscopy studies in aqueous solution. Eur. Biophys. J. 2006;35:495–501. doi: 10.1007/s00249-006-0057-7. [DOI] [PubMed] [Google Scholar]
73.Weidemann T., Wachsmuth M., Knoch T., Muller G., Waldeck W., Langowski J. Counting nucleosomes in living cells with a combination of fluorescent correlation spectroscopy and confocal imaging. J. Mol. Biol. 2003;334:229–240. doi: 10.1016/j.jmb.2003.08.063. [DOI] [PubMed] [Google Scholar]
74.Mazumder A., Roopa T., Basu A., Mahadevan L., Shivashankar G.V. Dynamics of chromatin decondensation reveals the structural integrity of a mechanically prestressed nucleus. Biophys. J. 2008;95:3028–3035. doi: 10.1529/biophysj.108.132274. [DOI] [PMC free article] [PubMed] [Google Scholar]
75.Fan Y., Nikitina T., Zhao J., Fleury T.J., Bhattacharyya R., Bouhassira E.E., Stein A., Woodcock C.L., Skoultchi A.I. Histone H1 depletion in mammals alters global chromatin structure but causes specific changes in gene regulation. Cell. 2005;123:1199–1212. doi: 10.1016/j.cell.2005.10.028. [DOI] [PubMed] [Google Scholar]
76.Woodcock C.L., Skoultchi A.I., Fan Y. Role of linker histone in chromatin structure and function: H1 stoichiometry and nucleosome repeat length. Chromosome Res. 2006;14:17–25. doi: 10.1007/s10577-005-1024-3. [DOI] [PubMed] [Google Scholar]
77.Thomas T., Thomas J. Polyamines in cell growth and cell death: molecular mechanisms and therapeutic applications. Cell. Mol. Life Sci. 2001;58:244–258. doi: 10.1007/PL00000852. [DOI] [PMC free article] [PubMed] [Google Scholar]
78.van Holde K.E. New York: Springer-Verlag; 1988. Chromatin. [Google Scholar]
79.Sivolob A.V., Khrapunov S.N. Binding of positively charged ligands to DNA. Effects of ionic strength, ligand charge and size. Mol. Biol. 1988;22:414–422. [PubMed] [Google Scholar]
80.Korolev N., Lyubartsev A.P., Laaksonen A. Electrostatic background of chromatin fiber stretching. J. Biomol. Struct. Dyn. 2004;22:215–226. doi: 10.1080/07391102.2004.10506997. [DOI] [PubMed] [Google Scholar]
81.Schiessel H. The physics of chromatin. J. Phys. Condens. Mat. 2003;15:R699–R774. doi: 10.1088/0953-8984/27/6/060301. [DOI] [PubMed] [Google Scholar]
82.Flaus A., Owen-Hughes T. Mechanisms for nucleosome mobilization. Biopolymers. 2003;68:563–578. doi: 10.1002/bip.10323. [DOI] [PubMed] [Google Scholar]
83.Walker I.O. Differential dissociation of histone tails from core chromatin. Biochemistry. 1984;23:5622–5628. doi: 10.1021/bi00318a037. [DOI] [PubMed] [Google Scholar]
84.Marky N.L., Manning G.S. A theory of DNA dissociation from the nucleosome. J. Mol. Biol. 1995;254:50–61. doi: 10.1006/jmbi.1995.0598. [DOI] [PubMed] [Google Scholar]
85.Korolev N., Nordenskiöld L. Polyelectrolyte properties of nucleosome disassembly and assembly reactions. Recent Res. Dev. Biophys. Chem. 2001;2:103–124. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.