

Abstract
Mutations in the NLRP3 (CIAS1, Cryopyrin) gene are associated with cryopyrin-associated periodic syndromes (CAPS), auto inflammatory diseases characterized by excessive interleukin-1 (IL-1) production and neutrophilia in blood and tissues. Recent studies with gene-targeted mice expressing mutations homologous to those found in CAPS patients have advanced the understanding of NLRP3 associated auto inflammation. In this Viewpoint, we will discuss the mechanisms of NLRP3 inflammasome activation and its induction of Th17 cell dominant immunologic responses.
Keywords: inflammation, inflammasome, interleukin-1, Th17
Introduction
The understanding of various inflammasomes, particularly the NLRP3 inflammasome, has been greatly enhanced by the investigation of gene-targeted mice in which inflammasome components have been knocked out [1–5]. Such knock-out mice however, provide only limited insight into the function of the inflammasome in humans with auto inflammatory syndromes (i.e., patients with cryopyrin-associated periodic syndromes (CAPS)), since the latter are characterized by NLRP3 mutations causing inflammasome hyper activation rather than decreased function [6–8]. Recently, gene-targeted mice with such mutations of the Nlrp3 gene have been developed, and these mice do in fact express abnormalities associated with human auto inflammatory syndromes [9, 10]. These mice can be manipulated both environmentally and genetically in ways that are impossible in humans and thus offer a superb platform to investigate the mechanisms of excessive inflammasome activity and its immunological consequences.
Knock-In Mice with Nlrp3 Mutations
Mice with Nlrp3 mutations were developed independently by investigators in two laboratories. One group introduced a R258W mutation in the third exon of the Nlrp3 gene of C57BL/6 mice [9]. This corresponds to the R260W mutation frequently found in humans with the Muckle-Wells syndrome [7]. A second group introduced either an A350V or a L351P mutation in exon 3 of Nlrp3 in 129SvJ mice [10]. These mutations occur frequently in patients with Muckle-Wells syndrome and familial cold auto inflammatory syndrome (FCAS), respectively [10]. The targeting strategy used to obtain these strains required that the mice co-express Cre-recombinase to delete a neomycin cassette inserted in reverse orientation that when present causes gene silencing. This allowed studies of mice in which the Cre-recombinase was expressed under tissue-specific promoters and thus enabled tissue-specific expression of the mutated gene [10].
In studies to determine if the R258W mice exhibit the basic immunologic abnormality of patients with CAPS, bone marrow-derived macrophages (BMDM) and dendritic cells (BMDC) from these mice were stimulated with a TLR ligand (LPS) in the presence and absence of ATP, the latter an essential co-factor in NLRP3 inflammasome activation in wild type (WT) cells. It was shown that while cells from R258W mice were unable to produce IL-1β and IL-18 in the absence of stimulation, they produced large amounts of these cytokines upon LPS stimulation in the presence or absence of exogenous ATP. These cells therefore differed from WT cells in that the latter only exhibited IL-1β production upon LPS stimulation in the presence of ATP and thus were similar to cells of patients with CAPS. Interestingly, both WT and R258W cells produced equivalent amounts of other cytokines upon LPS stimulation. This suggested that the abnormality was limited to the NLRP3 inflammasome and that elevations in non-inflammasome cytokine production occurring during prolonged inflammation was due to secondary stimulation of cells by increased levels of IL-1β [6, 9]. In parallel studies of peritoneal macrophages and BMDC from the A350V and L351P knock-in (KI) mice, production of IL-1β in the absence of ATP was also found. In addition, it was shown that BMDC from L351P mice secreted IL-1β when incubated at 32 °C, as do CAPS patients with similar mutations. Thus, cold conditions seem to be an inflammasome activator in the presence of this mutation. Finally, cold-challenged dendritic cells from L351P KI mice exhibited spontaneous IL-1β secretion whereas A350V KI cells were more dependent on LPS priming; this may explain the greater neonatal mortality of the L351P KI mice as compared to A350 KI mice [10].
Nlrp3 Knock-In Mice Have a Hyper-Active Inflammasome
The mechanism of ATP co-activation of the NLRP3 inflammasome was studied in the R258W KI mice. Previous work has shown that this ATP function is an extra-cellular activity that involves activation of a membrane receptor, P2X7R [11]. Upon stimulation by ATP, P2X7R interacts with a membrane-bound channel protein pannexin-1 (Panx1), after which the Panx1 forms a large transmembrane channel [12]. Thus ATP may be acting to allow inflammasome-activating TLR ligands (or other inflammasome activators) to enter the cell. Support for this idea comes from the fact that down-regulation of Panx1 or inhibition of its binding to P2X7R by an inhibitory peptide, 10Panx1, down-regulates LPS in the presence of ATP induction of NLRP3 inflammasome activity [13]. Another proposed mechanism is based on the fact that the ATP interaction with P2X7R leads to K+ efflux; thus ATP may be acting to cause an intra-cellular cation change necessary for inflammasome activation [14, 15]. This idea is supported by the fact that inhibition of K+ efflux by increased extra-cellular K+ concentrations suppresses NLRP3 inflammasome activation [16, 17]. When reconciling these two mechanisms one should note that inhibition of K+ efflux does not affect Panx1 channel formation and that, conversely, 10Panx1 peptide inhibition of Panx1 mediated pore formation does not inhibit potassium efflux [12, 18]. Thus it is possible that channel formation and potassium efflux are independent functions of the P2X7R/Panx1 complex that are both necessary for NLRP3 inflammasome activation.
In initial studies to determine why ATP is not necessary for inflammasome activation in R258W knock-in mice, it was found that the lack of ATP dependence occurred in spite of inhibition of K+ efflux. Therefore, the mutation did not cause a defect in the intracellular cation balance. In addition, there was no difference between KI cells and WT cells in their ability to generate endogenous extracellular ATP so the ATP independence was not the result of excessive ATP production from KI cells either [9].
Further insight into ATP function in R258W KI and WT cells came from studies of inflammasome activation (IL-1β release) in the presence of 10Panx1 peptide. We found that the presence of 10Panx1 decreased the inflammasome activity of WT cells by about 50% when added up to 4 hours prior to the ATP pulse but had no effect on KI cells. This indicated that WT cells were dependent on the rapid Panx1 channel formation whereas KI cells were not; however, residual inflammasome activation in WT cells in the presence of the Panx1 channel blockade was still dependent on the presence of ATP (perhaps acting via another cellular entry mechanism, depicted in Fig. 1 as the P2X7R/X channel). When 10Panx1 was added together with LPS (24 hours prior to the ATP pulse), even the inflammasome activation of KI cells was substantially inhibited. This indicated that Panx1-mediated entry also occurs in KI cells, albeit that this route of entry is not absolutely critical since inflammasome activation occurs at least partially in the absence of ATP (perhaps due to LPS entry via other cellular mechanism (indicated as channel X in Fig. 1) [9].
Figure 1.

NLRP3 mutations decrease the activation threshold of the inflammasome. In both WT and NLRP3 gene-targeted cells, mature IL-1β secretion needs pro-IL-1β synthesis dependent on TLR activation by pathogen associate molecular patterns (PAMPs) such as LPS. PAMPs also need to enter cells to induce production of the proposed direct ligand for NLRP3. WT NLRP3 needs a large quantity of ligand to be activated, which depends on sufficient entry of PAMPs through either the P2X7R/Panx1or an unknown “X” channel in association with P2X7R, requiring for both channels an exogenous ATP pulse and an accompanying K+ efflux. In contrast, mutated NLRP3 can be activated with much lower amounts of the proposed endogenous ligand, which can be generated by low concentrations of PAMPs entering cells via Panx1 and unknown “X” channels independently of exogenous ATP.
The above studies of Panx1 channel inhibition suggested that low amounts of TLR ligand are sufficient to activate the NLRP3 inflammasome in R258W cells. To verify this possibility, the concentration dependence of inflammasome activation in WT and KI cells (in the presence and absence of ATP) was determined. It was found that while inflammasome activation increased in both cell types with increasing LPS concentrations, WT cells required massive amounts of LPS (>1000 ng/ml) to activate the inflammasome in the absence of ATP, whereas KI cells required only minute amounts of LPS. It thus appears that KI cells do not require co-stimulation by ATP because the small amounts of TLR ligand that enter in the absence of ATP are sufficient to activate the altered inflammasome. Overall, these data are consistent with the concept previously suggested from studies of CAPS patients that NLRP3 mutations lead to changes in the conformation of the protein that, in turn, result in a reduced activation threshold and thus an inflammasome capable of responding to reduced amounts of TLR ligand or other activating factors [9, 19]. However, NLRP3 may not be able to directly bind to such a wide variety of ligands including PAMP and DAMPs, rather an endogenous activator induced by all these upstream stimuli may serve as the direct ligand for NLRP3 (Fig. 1). This concept has also been proposed independently by other researchers [20, 21].
Clinical and General Immunologic Features of Mice Bearing NLRP3 Mutations
NLRP3 KI mice bearing an R258W mutation raised under pathogen free facility exhibit spontaneous clinical symptoms similar to those of the counterpart Muckle-Wells syndrome patients. These symptoms consist of poor linear growth, reduced reproductive capacity, impaired hair development and, in many animals, severe dermatitis affecting the ears, top of the head and tail base area occurring at 6–12 weeks of age that is associated with a deterioration of health. The skin lesions were clinically more severe than the urticaria-like skin disease seen in human CAPS and characterized by neutrophilic infiltration of the dermis and epidermis. Spleen and draining lymph nodes were enlarged in the KI mice and showed poorly developed follicles along with a diffuse infiltrate, again containing many neutrophils. However, these KI mice were free of lung, kidney or gut inflammation and the level of circulating inflammatory cytokines was normal [9].
The clinical features of mice bearing A350V and L351P mutations were qualitatively similar to those described for R258W mice, but were far more severe. These A350V/L351P KI mice had lifespan measured in days rather than weeks, and had more widespread skin inflammation and inflammatory infiltration (mainly neutrophilic) of many organs, including the joints, sinus, bone marrow and tongue. In addition, there was evidence of “necrotic degeneration” in the gut and kidney. Finally, in contrast to the R258W KI mice, the A350V/L351P KI mice exhibited elevated circulating levels of proinflammatory cytokines, most notably IL-1β and IL-6 as well as G-CSF and the neutrophil chemotactic factor, KC; in addition, there was a variable increase in Th2 (but not Th1) cytokines and no increase in IL-17. The more severe clinical disease and the presence of circulating inflammatory cytokines in these A350V/L351P KI mice as compared to R258W KI mice probably reflect an intrinsically more hyperactive NLRP3 inflammasome [10]. This may arise from the, as yet, undefined effects of the background strain on inflammasome activity, since R258W KI mice exhibit less severe disease when interbred with BALB/c mice. A similar factor could account for the variable penetrance of CAPS disease in humans.
The primary cells responsible for inducing disease in the R258W KI mice were shown to be hematopoietic cells, as bone marrow cells from R258W KI mice (but not from WT mice) transferred to irradiated WT recipients resulted in auto inflammation. In addition, inflamed KI mice resolved the inflammation upon irradiation followed by bone marrow transfer from WT donor mice ([9] and Meng and Strober, unpublished observation). A similar conclusion applies to A350V and L351P KI mice, since the disease observed in mice in which the mutation was limited to antigen-presenting cells (APCs) exhibited a similar phenotype to those mice in which the mutation occurred in all cells [10]. It should be noted, however, that other cells could also be contributing to disease manifestations [22, 23]. Furthermore, cells induced by APCs, such as T cells, could also be contributing to inflammation as indicated by the fact that in R258W KI mice inflammation exhibits a Th17 cell bias that may be shaping the overall response (see next section). Studies showing that L351P KI mice crossed with RAG1-deficient mice that do not have T cells have largely undiminished disease do not contradict this point, since it is possible that the hyper-robust inflammasome activity in these mice might not model the lesser degree of inflammation in humans with CAPS.
The Nature of Inflammation in NLRP3 mutated Mice
Detailed studies of the immune response underlying the inflammation in R258W KI mice have revealed important new insights into how a hyper-active NLRP3 inflammasome causes inflammation. In initial studies it was shown by RT-PCR examination of the spontaneously occurring skin lesions that the inflammation was associated with an increase in IL-17 family cytokines and factors, including IL-17A, IL-17F, IL-21, RORγt and IL-22 whereas, in contrast, the Th1 cytokine IFN-γ was only moderately elevated. In addition, other Th1 factors such as IL-12Rβ2 and T-bet were even decreased compared with levels in WT skin. Finally, the inflamed skin contained increased expression of a spectrum of pro-inflammatory cytokines including IL-12p40, IL-12p35, IL-1β, IL-6 and TNF-α. In further studies this bias toward a Th17-cell mediated inflammation was also observed in skin DTH responses in A350V/L351P KI mice as compared with WT mice [9].
In additional studies designed to elucidate the origin of the above Th17 cytokine bias, it was first demonstrated that anti-CD3/CD28-stimulated WT CD4+ T cells cultured under Th17 cell conditions (TGF-β + IL-6) produced substantially increased amounts of IL-17, but substantially less IFN-γ, when co-cultured with CD11b+ cells from R258W KI mice but not WT mice. It was then shown that culture of T cells from IL-1R1 deficient mice which cannot respond to IL-1β, exhibited substantially less IL-17 bias than WT T cells when co-cultured with R258W CD11b+ cells. Similar results were obtained when T cells were co-cultured with supernatants of R258W KI APC. Taken together, these findings indicate that the KI APCs act on differentiating CD4+ T cells to favor Th17 cell differentiation via IL-1β, providing that the T cells have undergone initial Th17− inductive steps. It should be noted, however, that since there was residual Th17 cell bias in the studies using IL-1R1−/− cells, other factors secreted by APCs from R258W KI mice may also play a role in inducing Th17 cell differentiation [9].
Parallel studies of T cell differentiation directed by APCs from A350V and L351P KI mice were conducted with antigen-specific T cells. It was found that these APCs exhibited a normal capacity to induce T cells to differentiate into any type of T cell lineage under subset-specific conditions, and exhibited only a modest bias toward IL-17 under neutral conditions. This result was consistent with the fact that skin inflammation in these mice did not show a IL-17 cytokine bias. This discrepancy may be due to the fact that these in vitro studies were not conducted under conditions that allowed initial Th17 cell induction and thus did not assess IL-1β effects at an appropriate phase of T cell differentiation [9, 10].
The mechanism underlying the Th17 cell bias in the inflammasome-associated inflammation noted above for R258W KI mice is not fully understood. Previous studies have shown that IL-1β together with TNF-α can augment TGF-β/IL-6-induced Th17 cell differentiation and that in fact IL-6 induces IL-1R1 expression on T cells [24, 25]. In addition, IL-1β has been shown to upregulate factors that induce/enhance IL-17 transcription such as RORγt and IRF-4 [24, 26]; however, the molecular mechanism underlying this upregulation is not known. As for the fact that the inflammasome-associated inflammation is marked by decreased IFN-γ as well as increased IL-17 production, it may be due to the fact that IL-1β down regulates IL-6-induced STAT-1 activation and thereby inhibits T-bet transcription [27]. Additionally, it was observed that the inflamed tissue of the KI mice exhibited decreased IL-12Rβ2 expression and that treatment of mice with anti-IL-1R1 reversed this effect. Thus IL-1β may inhibit IL-12p70 induction of STAT-4 activation, the essential initial step in Th1 cell development [28].
Given the well known propensity of IL-17 to induce a neutrophil-rich inflammation [29, 30], the Th17 cell bias inherent in inflammasome activity may be a major reason why neutrophils are a major component of auto inflammation in CAPS. In addition, since neutrophils express NLRP3 [4], these cells may augment IL-1β production in inflamed lesions and thereby provide positive feedback for inflammasome activity (Fig. 2).
Figure 2.
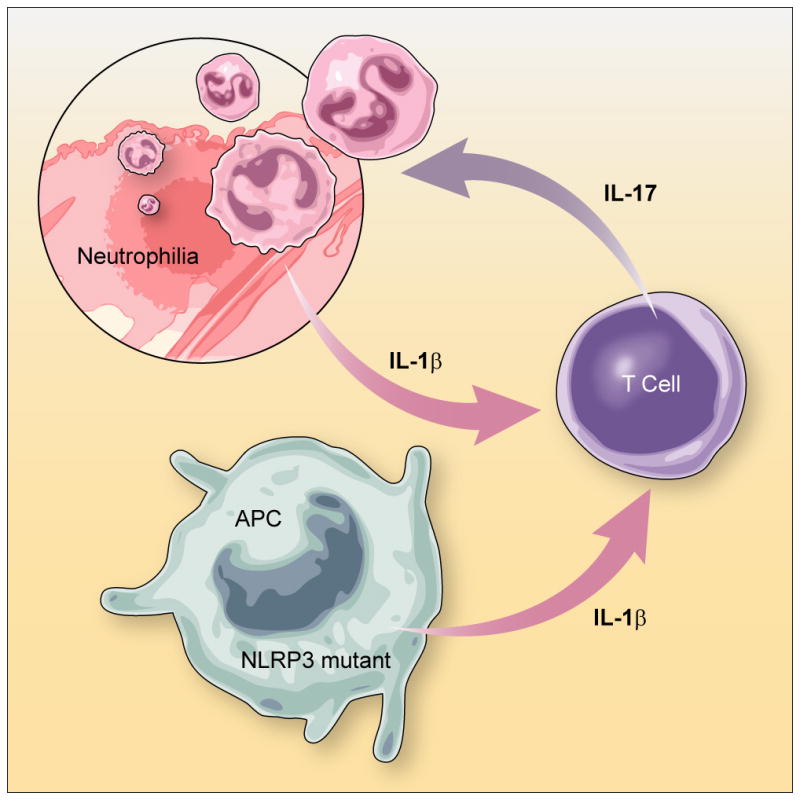
Excessive IL-1β production from NLRP3 mutated APCs induces T cell production of IL-17 and consequently neutrophilic inflammation in tissues. Antigen presenting cells (APCs) from NLRP3 mutated mice secrete elevated amounts of IL-1β due to hyperactive inflammasome activation, possibly triggered by minor trauma. Subsequently, IL-1β enhances IL-17 production from helper T cells. The Th17 induces neutrophil infiltration and results in tissue damage. Positive feed back also operates since neutrophils also produce IL-1β due to NLRP3 inflammasome hyper activation and thus amplify the inflammation.
Treatment of Inflammation in NLRP3 Knock-In Mice
Administration of anti-IL-1R1 to R258W KI mice results in nearly complete reversal of skin inflammation [9]. This result parallels findings in humans with CAPS who usually manifest striking clinical improvement upon treatment with IL-1β blocking agents [31–33], and supports the view that IL-1β is the main, if not the sole, basis of the inflammation. In contrast, administration of an IL-1β blocking agent (mIL-1 Trap) to A350V and L351P KI mice resulted in virtually no improvement in the inflammatory state, even though IL-1R1−/− mice bearing these mutations do not show inflammation [10]. This outcome could be the result of the intense NLRP3 inflammasome activity in these mice that leads to effects such as cell necrosis that are not easily reversed by an exogenous agent that neutralizes IL-1β [34, 35].
Given the Th17 cell bias of the inflammation in R258W KI mice, the effect of administration of anti-IL-17A was also assessed. Interestingly, this agent was also effective in ameliorating inflammation, despite the fact that it does not block the inflammatory effect of IL-17F, an IL-17 isotype also elevated in lesions [9]. These studies suggest that if humans with CAPS can also be shown to have inflammations with a Th17 cell bias it may be possible to control CAPS with anti-IL-17 as well as IL-1β inhibitory agents.
Conclusion
It is evident from the studies described above that mice carrying mutations of the Nlrp3 gene have already yielded valuable new insights into the immunopathology associated with CAPS, including a possible new treatment approach. Further studies in which these mice are used to elucidate the role of the NLRP3 inflammasome in various types of organ-specific inflammation hold the promise of defining the role of the inflammasome in a host of inflammatory conditions.
Acknowledgments
This research was supported [inpart] by the Intramural Research Program of the NIH, NIAID, DIR, LHD.
This work was supported by the Division of Intramural Research, National Institute of Allergy and Infectious Diseases, National Institutes of Health. We apologize to those authors whose work could not be cited due to space limitations.
Footnotes
Conflict of interest
The authors declare no financial or commercial conflict of interest.
References
- 1.Martinon F, et al. Nature. 2006;440:237–241. doi: 10.1038/nature04516. [DOI] [PubMed] [Google Scholar]
- 2.Mariathasan S, et al. Nature. 2006;440:228–232. doi: 10.1038/nature04515. [DOI] [PubMed] [Google Scholar]
- 3.Kanneganti TD, et al. Nature. 2006;440:233–236. doi: 10.1038/nature04517. [DOI] [PubMed] [Google Scholar]
- 4.Sutterwala FS, et al. Immunity. 2006;24:317–327. doi: 10.1016/j.immuni.2006.02.004. [DOI] [PubMed] [Google Scholar]
- 5.Mariathasan S, et al. Nature. 2004;430:213–218. doi: 10.1038/nature02664. [DOI] [PubMed] [Google Scholar]
- 6.Gattorno M, et al. Arthritis Rheum. 2007;56:3138–3148. doi: 10.1002/art.22842. [DOI] [PubMed] [Google Scholar]
- 7.Neven B, et al. Blood. 2004;103:2809–2815. doi: 10.1182/blood-2003-07-2531. [DOI] [PubMed] [Google Scholar]
- 8.Galeazzi M, et al. Clin Exp Rheumatol. 2006;24:S79–85. [PubMed] [Google Scholar]
- 9.Meng G, et al. Immunity. 2009;30:860–874. doi: 10.1016/j.immuni.2009.04.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Brydges SD, et al. Immunity. 2009;30:875–887. doi: 10.1016/j.immuni.2009.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ferrari D, et al. J Immunol. 2006;176:3877–3883. doi: 10.4049/jimmunol.176.7.3877. [DOI] [PubMed] [Google Scholar]
- 12.Pelegrin P, Surprenant A. Embo J. 2006;25:5071–5082. doi: 10.1038/sj.emboj.7601378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kanneganti TD, et al. Immunity. 2007;26:433–443. doi: 10.1016/j.immuni.2007.03.008. [DOI] [PubMed] [Google Scholar]
- 14.Walev I, et al. Embo J. 1995;14:1607–1614. doi: 10.1002/j.1460-2075.1995.tb07149.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Perregaux D, Gabel CA. J Biol Chem. 1994;269:15195–15203. [PubMed] [Google Scholar]
- 16.Sutterwala FS, et al. J Endotoxin Res. 2006;12:251–256. doi: 10.1177/09680519060120040701. [DOI] [PubMed] [Google Scholar]
- 17.Petrilli V, et al. Cell Death Differ. 2007;14:1583–1589. doi: 10.1038/sj.cdd.4402195. [DOI] [PubMed] [Google Scholar]
- 18.Pelegrin P, Surprenant A. J Biol Chem. 2007;282:2386–2394. doi: 10.1074/jbc.M610351200. [DOI] [PubMed] [Google Scholar]
- 19.Aksentijevich I, et al. Arthritis Rheum. 2007;56:1273–1285. doi: 10.1002/art.22491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hornung V, et al. Nat Immunol. 2008;9:847–856. doi: 10.1038/ni.1631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Allen IC, et al. Immunity. 2009;30:556–565. doi: 10.1016/j.immuni.2009.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Nakamura Y, et al. J Exp Med. 2009;206:1037–1046. doi: 10.1084/jem.20082179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Watanabe H, et al. J Invest Dermatol. 2007;127:1956–1963. doi: 10.1038/sj.jid.5700819. [DOI] [PubMed] [Google Scholar]
- 24.Chung Y, et al. Immunity. 2009;30:576–587. doi: 10.1016/j.immuni.2009.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Veldhoen M, et al. Immunity. 2006;24:179–189. doi: 10.1016/j.immuni.2006.01.001. [DOI] [PubMed] [Google Scholar]
- 26.Brustle A, et al. Nat Immunol. 2007;8:958–966. doi: 10.1038/ni1500. [DOI] [PubMed] [Google Scholar]
- 27.Shen X, et al. Biochem J. 2000;352(Pt 3):913–919. [PMC free article] [PubMed] [Google Scholar]
- 28.Nishikomori R, et al. J Immunol. 2002;169:4388–4398. doi: 10.4049/jimmunol.169.8.4388. [DOI] [PubMed] [Google Scholar]
- 29.Steinman L. J Exp Med. 2008;205:1517–1522. doi: 10.1084/jem.20072066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kroenke MA, et al. J Exp Med. 2008;205:1535–1541. doi: 10.1084/jem.20080159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lachmann HJ, et al. N Engl J Med. 2009;360:2416–2425. doi: 10.1056/NEJMoa0810787. [DOI] [PubMed] [Google Scholar]
- 32.Goldbach-Mansky R, et al. N Engl J Med. 2006;355:581–592. doi: 10.1056/NEJMoa055137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hoffman HM, et al. Lancet. 2004;364:1779–1785. doi: 10.1016/S0140-6736(04)17401-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Fujisawa A, et al. Blood. 2007;109:2903–2911. doi: 10.1182/blood-2006-07-033597. [DOI] [PubMed] [Google Scholar]
- 35.Willingham SB, et al. Cell Host Microbe. 2007;2:147–159. doi: 10.1016/j.chom.2007.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]