

Abstract
The distal convoluted tubule (DCT) plays a central role in blood pressure and potassium homeostasis, as evidenced by diseases that occur when its function is modified. The paper by van der Lubbe and colleagues in this issue of Kidney International makes clear that angiotensin II itself increases the activity and abundance of the thiazide-sensitive Na-Cl cotransporter (NCC), independent of changes in circulating aldosterone. This commentary provides additional perspective on that work.
These are exciting times for the distal convoluted tubule (DCT), or at least for those who study it. During the golden age of micropuncture, this short nephron segment was studied widely. Later, however, attention shifted to other nephron segments, owing to ease of study and the belief that NaCl transport along the DCT is determined ‘in large part by delivered load’, with only ‘equivocal’ evidence for regulatory modulation1. New molecular tools and techniques, coupled with exciting insights into genetic hypertension and salt wasting, however, now identify the DCT as a key site for regulated NaCl transport. As with any field that is moving rapidly, however, emerging results often raise confusing questions. Our understanding of DCT transport remains inchoate, but the paper by van der Lubbe and colleagues (page XXX) helps to bring some clarity.
During the past fifteen years, evidence has accumulated that aldosterone increases sodium reabsorption along the DCT2. More recently, arginine vasopressin (AVP) has also been shown to enhance sodium reabsorption along this segment3. Aldosterone and AVP have long been known to stimulate Na transport along the cortical collecting duct, by acting on the epithelial Na channel, ENaC; AVP also increases water permeability of this segment (via aquaporin-2), where both mineralocorticoid (MR) and vasopressin type 2 receptors (V2R) are expressed. Yet DCT cells also expresses MR4 and V2R5. These receptors probably mediate direct hormonal effects in DCT, as aldosterone increases the activity2 and abundance6 of the thiazide-sensitive Na-Cl cotransporter (NCC), as does AVP3, 7, 8.
The dominant NaCl transport pathway of the DCT is NCC. To transport NaCl, NCC must move (‘traffic’) to, and be inserted into, the apical plasma membrane; it is also phosphorylated along its amino terminal cytoplasmic domain, enhancing activity (see Figure 1). WNKs are intracellular kinases that modulate NCC activity by altering both trafficking and phosphorylation. WNK4 reduces NCC movement to the apical membrane9 from sites where it is synthesized (endoplasmic reticulum) and processed (golgi apparatus), at least in part, by targeting it to lysosomes, where it can be degraded10, 11; the effects of WNK4 may be modulated by angiotensin II (see below). In contrast, WNK3 increases NCC abundance and activity12-14. Thus, some WNKs are predominantly inhibitory, while others are predominantly stimulatory, at least with respect to NCC. Little is known about how NCC is removed from the apical membrane, although the process does not appear to involve clathrin-mediated endocytosis 11, 15.
Figure 1.

Simplified scheme of regulation of thiazide-sensitive Na-Cl cotransporter (NCC) regulation. NCC is synthesized and then glycosylated (green fork) within the golgi apparatus (not shown, for clarity). The NCC then moves to and into the apical membrane, where it exists as a dimer. To be full active, NCC undergoes phosphorylation along its amino terminal cytoplasmic domain, mediated largely by SPAK, thereby permitting NaCl transport. Little is know about mechanisms of removal from the membrane. Arginine vasopressin (AVP), aldosterone (Aldo), and angiotensin II (Ang II) all stimulate NCC activity. Trafficking may be a rapid effect, modulated predominantly by AVP and Ang II. Phosphorylation may occur within the membrane, and is enhanced by all three factors.
As noted, NCC is also activated by phosphorylation (Figure 1). Phosphorylation activates NCC without changing its membrane abundance, at least when it is expressed heterologously in Xenopus oocytes 16. The major kinase that phosphorylates and activates NCC appears to be SPAK17, 18. SPAK, which is expressed along the distal nephron19, can itself be phosphorylated and activated by WNK kinases, so that WNK, SPAK, and NCC comprise a signaling pathway20. Nevertheless, although kinase domains of the several WNKS are homologous, all WNKs do not appear to have the same effects on NCC. As noted, WNK4 appears to act as an inhibitor of NCC9, 21, at least under some conditions22, whereas WNK1 phosphorylates SPAK to activate NCC18. In HeLa cells, WNK1, but not WNK4, activated SPAK and caused a large shift in electrophoretic mobility23; thus, details of how WNK kinases modulate NCC, remain confusing.
Angiotensin II is another component of the renin/angiotensin/aldosterone system that stimulates Na transport along the DCT 24. This effect is also likely to be direct, owing to the presence of AT1 receptors along DCT 25. Genetic deletion of AT1a receptors reduces the abundance of NCC 26, and infusion of angiotensin II for 8 days increases the abundance and phosphorylation of NCC 27; thus, angiotensin II and aldosterone appear to have similar effects on NCC activity. Gamba and colleagues reported that angiotensin II relieved the inhibitory effect of WNK4 on NCC, in a SPAK-dependent manner 22.
Angiotensin II increases NCC activity, in part, by increasing the abundance of NCC at the apical plasma membrane. This effect occurs rapidly, with short-term angiotensin II infusions increasing the ratio of apical to sub-apical NCC 28. In cultured mpkDCT cells, angiotensin II also increases SPAK and NCC phosphorylation, suggesting that acute exposure to angiotensin II also activates the transporter allosterically27. Longer-term effects, induced by dietary NaCl restriction29 or angiotensin II infusions27 also stimulate NCC activity, increase NCC abundance, and increase its phosphorylation; in these situations, however, the effects may be direct, from AT1 receptor activation, or indirect, via aldosterone stimulation.
Talati and colleagues concluded, based on inhibitor studies, that long-term effects of angiotensin II on NCC are mediated by aldosterone27, and suggested therefore that aldosterone is the predominant NCC regulatory factor. T paper by van der Lubbe and colleagues in this issue of Kidney International (page XXX) shows clearly that angiotensin II itself increases NCC abundance and phosphorylation, even during chronic exposure; the authors used the definitive approach of performing adrenalectomy, and then infusing hormones chronically, to fix adrenal steroid concentrations. The results are clear; angiotensin II increases NCC abundance and phosphorylation even when serum aldosterone levels are fixed. Several additional points, derived from their data, however, deserve emphasis.
First, figure 2, redrawn from data in the paper, shows that aldosterone, but not angiotensin II, increased αENaC abundance substantially. This pattern of hormonal effect on ENaC contrasts with effects on NCC, in which both angiotensin II and aldosterone increase NCC abundance and phosphorylation. These results help to explain how aldosterone, a single hormone, can generate either NaCl retention or Na/K exchange, depending on the stimulatory signal (an effect termed the ‘aldosterone paradox’30). Thus, when aldosterone secretion is stimulated by angiotensin II (such as occurs when the extracellular fluid volume is depleted), Na reabsorption will be stimulated along much of the nephron (including the proximal and distal tubule by angiotensin II, and the distal tubule and collecting duct by aldosterone). These effects will restore extracellular fluid volume both because proximal segments reabsorb NaCl, and because Na delivery to the distal, K secretory sites, will be limited. In contrast, when aldosterone secretion is stimulated by hyperkalemia, in the absence of changes in angiotensin II, Na reabsorption will only be stimulated distally, favoring the exchange of Na for K. Although other mechanisms are likely to be involved, the patterns of angiotensin II and aldosterone effect on Na transport along the nephron certainly reflect physiologically adaptive processes.
Figure 2.
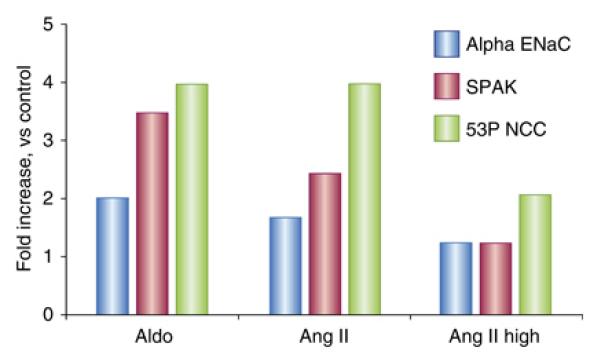
Redrawn from data in the paper by van der Lubbe. Bar plot shows the effects of aldosterone (Aldo), angiotensin II (Ang II) and pressor dose Ang II (Ang II High) on abundance of the alpha subunit of ENaC, SPAK, and phosphorylated NCC. Note that all three interventions increase phosphorylated NCC, whereas only aldosterone increases ENaC abundance. Please see text for more details.
Second, while NCC stimulation by either angiotensin II or aldosterone is associated with increases in SPAK abundance and SPAK phosphorylation, when animals received higher doses of angiotensin II, NCC appeared to be stimulated, even though SPAK (and phosphorylated SPAK) were at baseline levels; even though this effect did not quite reach statistical significance, it raises the possibility that other kinases can activate NCC.
Finally, while comparisons of protein abundance do not necessarily reflect changes in transporter activity, the ability of aldosterone to increase NCC abundance is quite impressive, in comparison with its ability to increase ENaC abundance. Many, if not most, introductory texts for medical and graduate students describe effects of aldosterone on ENaC, but omit effects on NCC 31. The accumulating data suggest that it is time to break old paradigms, and include NCC as a crucial aldosterone-regulated transport protein, when introducing students to the effects of adrenal steroids on the kidney.
Lest the current data are believed to clear all confusion, several questions remain. As noted, two groups7, 8 have shown that AVP increases trafficking and phosphorylation of NCC. In the study by van der Lubbe and colleagues, the abundance of aquaporin 2 was increased by both aldosterone and angiotensin II infusion. This suggests either that these peptides stimulated AVP secretion or that angiotensin II activated V2R directly; there is some evidence in support of the second model32. From a physiological standpoint, of course, the striking similarity of effects of aldosterone and AVP on distal transporters is hard to reconcile with effects on whole animal balance. Aldosterone and AVP stimulate both stimulate ENaC and NCC. Yet, hyperaldosteronism typically presents with hypertension, owing to sodium chloride retention, while the syndrome of inappropriate ADH secretion presents with hyponatremia, owing to effects on aquaporin 2, but without NaCl retention. This suggests either that the potency of stimulatory effects on Na transport, or the escape mechanisms that supervene, are different, or that other factors come into play. One possible factor is V1a receptors; most studies of AVP actions utilize the V2-receptor-specific agonist desmopressin (dDAVP). V1a-receptors, a second target of the native hormone arginine vasopressin, can increase natriuresis33.
Finally, the roles played by WNK kinases in modulating or mediating effects of angiotensin II and/or aldosterone remain intriguing, but are not fully elucidated. In view of the phenotype that results when WNK kinases are mutated, familial hyperkalemic hypertension (pseudohypoaldosteronism type II or Gordon syndrome), it seems clear that these kinases help to determine whether aldosterone is primarily kaliuretic or NaCl retentive. Yet changes in WNK4 were not observed in the experiments reported by van der Lubbe and colleagues, and data concerning WNK1 or WNK3 are not reported. WNK kinases may play a crucial role in determining NCC membrane abundance and states of phosphorylation, but the roles of the individual players, and their integration, remain poorly understood. Further, it seems likely that effects of WNK kinases, or at least WNK4, are modulated by circulating (or local) levels of angiotensin II22, as noted above. Much remains to be learned about the interactions between WNKs, SPAK, NCC, and the renin/angiotensin/aldosterone system. Yet, the possibility that small molecule WNK modulators might provide novel ways to ‘turn down’ the distal nephron means that this pathway is an attractive target for drug development.
Footnotes
“For now we see through a glass, darkly; but then face to face” (1 Corinthians 13)
References
- 1.Koeppen BM, Stanton BA. Sodium chloride transport: Distal Nephron. In: Seldin DW, Giebisch G, editors. The Kidney: Physiology and Pathophysiology. 2 edn Raven Press; New York: 1992. pp. 2003–2040. [Google Scholar]
- 2.Velazquez H, Bartiss A, Bernstein P, et al. Adrenal steroids stimulate thiazide-sensitive NaCl transport by rat renal distal tubules. The American journal of physiology. 1996;270:F211–219. doi: 10.1152/ajprenal.1996.270.1.F211. [DOI] [PubMed] [Google Scholar]
- 3.Ecelbarger CA, Kim G-H, Terris J, et al. Vasopressin-mediated regulation of epithelial sodium channel abundance in rat kidney. AmJPhysiol Renal Physiol. 2000;279:F46–F53. doi: 10.1152/ajprenal.2000.279.1.F46. [DOI] [PubMed] [Google Scholar]
- 4.Bostanjoglo M, Reeves WB, Reilly RF, et al. 11Beta-hydroxysteroid dehydrogenase, mineralocorticoid receptor, and thiazide-sensitive Na-Cl cotransporter expression by distal tubules. J Am Soc Nephrol. 1998;9:1347–1358. doi: 10.1681/ASN.V981347. [DOI] [PubMed] [Google Scholar]
- 5.Mutig K, Paliege A, Kahl T, et al. Vasopressin V2 receptor expression along rat, mouse, and human renal epithelia with focus on TAL. American journal of physiology. 2007;293:F1166–1177. doi: 10.1152/ajprenal.00196.2007. [DOI] [PubMed] [Google Scholar]
- 6.Kim GH, Masilamani S, Turner R, et al. The thiazide-sensitive Na-Cl cotransporter is an aldosterone-induced protein. Proceedings of the National Academy of Sciences of the United States of America. 1998 Nov 24;95:14552–14557. doi: 10.1073/pnas.95.24.14552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Pedersen NB, Hofmeister MV, Rosenbaek LL, et al. Vasopressin induces phosphorylation of the thiazide-sensitive sodium chloride cotransporter in the distal convoluted tubule. Kidney international. 2010 doi: 10.1038/ki.2010.130. [DOI] [PubMed] [Google Scholar]
- 8.Mutig K, Saritas T, Uchida S, et al. Short-term stimulation of the thiazide-sensitive Na+-Cl− cotransporter by vasopressin involves phosphorylation and membrane translocation. American journal of physiology. 2010;298:F502–509. doi: 10.1152/ajprenal.00476.2009. [DOI] [PubMed] [Google Scholar]
- 9.Yang CL, Angell J, Mitchell R, et al. WNK kinases regulate thiazide-sensitive Na-Cl cotransport. J Clin Invest. 2003;111:1039–1045. doi: 10.1172/JCI17443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Subramanya AR, Liu J, Ellison DH, et al. WNK4 Diverts the Thiazide-sensitive NaCl Cotransporter to the Lysosome and Stimulates AP-3 Interaction. The Journal of biological chemistry. 2009;284:18471–18480. doi: 10.1074/jbc.M109.008185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cai H, Cebotaru V, Wang YH, et al. WNK4 kinase regulates surface expression of the human sodium chloride cotransporter in mammalian cells. Kidney Int. 2006;69:2162–2170. doi: 10.1038/sj.ki.5000333. [DOI] [PubMed] [Google Scholar]
- 12.Glover M, Zuber AM, O’Shaughnessy KM. Renal and brain isoforms of WNK3 have opposite effects on NCCT expression. J Am Soc Nephrol. 2009;20:1314–1322. doi: 10.1681/ASN.2008050542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yang CL, Zhu X, Ellison DH. The thiazide-sensitive Na-Cl cotransporter is regulated by a WNK kinase signaling complex. The Journal of clinical investigation. 2007;117:3403–3411. doi: 10.1172/JCI32033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Rinehart J, Kahle KT, de Los Heros P, et al. WNK3 kinase is a positive regulator of NKCC2 and NCC, renal cation-Cl− cotransporters required for normal blood pressure homeostasis. Proc Natl Acad Sci U S A. 2005;102:16777–16782. doi: 10.1073/pnas.0508303102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Golbang AP, Cope G, Hamad A, et al. Regulation of the Expression of the Na/Cl cotransporter (NCCT) by WNK4 and WNK1: evidence that accelerated dynamin-dependent endocytosis is not involved. Am J Physiol Renal Physiol. 2006;291:F1369–1376. doi: 10.1152/ajprenal.00468.2005. [DOI] [PubMed] [Google Scholar]
- 16.Pacheco-Alvarez D, Cristobal PS, Meade P, et al. The Na+:Cl− cotransporter is activated and phosphorylated at the amino-terminal domain upon intracellular chloride depletion. J Biol Chem. 2006;281:28755–28763. doi: 10.1074/jbc.M603773200. [DOI] [PubMed] [Google Scholar]
- 17.Moriguchi T, Urushiyama S, Hisamoto N, et al. WNK1 regulates phosphorylation of cation-chloride-coupled cotransporters via the STE20-related kinases, SPAK and OSR1. The Journal of biological chemistry. 2005;280:42685–42693. doi: 10.1074/jbc.M510042200. [DOI] [PubMed] [Google Scholar]
- 18.Richardson C, Rafiqi FH, Karlsson HK, et al. Activation of the thiazidesensitive Na+-Cl− cotransporter by the WNK-regulated kinases SPAK and OSR1. Journal of cell science. 2008;121:675–684. doi: 10.1242/jcs.025312. [DOI] [PubMed] [Google Scholar]
- 19.Reiche J, Theilig F, Rafiqi FH, et al. SORLA/SORL1 functionally interacts with SPAK to control renal activation of Na+-K+-Cl− cotransporter 2. Mol Cell Biol. 2010 doi: 10.1128/MCB.01560-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Richardson C, Alessi DR. The regulation of salt transport and blood pressure by the WNK-SPAK/OSR1 signalling pathway. Journal of cell science. 2008;121:3293–3304. doi: 10.1242/jcs.029223. [DOI] [PubMed] [Google Scholar]
- 21.Wilson FH, Kahle KT, Sabath E, et al. Molecular pathogenesis of inherited hypertension with hyperkalemia: the Na-Cl cotransporter is inhibited by wild-type but not mutant WNK4. Proc Natl Acad Sci U S A. 2003;100:680–684. doi: 10.1073/pnas.242735399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.San-Cristobal P, Pacheco-Alvarez D, Richardson C, et al. Angiotensin II signaling increases activity of the renal Na-Cl cotransporter through a WNK4-SPAK-dependent pathway. Proceedings of the National Academy of Sciences of the United States of America. 2009;106:4384–4389. doi: 10.1073/pnas.0813238106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Anselmo AN, Earnest S, Chen W, et al. WNK1 and OSR1 regulate the Na+, K+, 2Cl− cotransporter in HeLa cells. Proceedings of the National Academy of Sciences of the United States of America. 2006;103:10883–10888. doi: 10.1073/pnas.0604607103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wang T, Giebisch G. Effects of angiotensin II on electrolyte transport in the early and late distal tubule in rat kidney. AmJPhysiolRenal,Fluid Electrolyte Physiol. 1996;271:F143–F149. doi: 10.1152/ajprenal.1996.271.1.F143. [DOI] [PubMed] [Google Scholar]
- 25.Mujais SK, Kauffman S, Katz AI. Angiotensin II binding sites in individual segments of the rat nephron. The Journal of clinical investigation. 1986;77:315–318. doi: 10.1172/JCI112293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Brooks HL, Allred AJ, Beutler KT, et al. Targeted proteomic profiling of renal Na(+) transporter and channel abundances in angiotensin II type 1a receptor knockout mice. Hypertension. 2002 Feb;39:470–473. doi: 10.1161/hy02t2.102959. [DOI] [PubMed] [Google Scholar]
- 27.Talati G, Ohta A, Rai T, et al. Effect of angiotensin II on the WNK-OSR1/SPAK-NCC phosphorylation cascade in cultured mpkDCT cells and in vivo mouse kidney. Biochemical and biophysical research communications. 2010 doi: 10.1016/j.bbrc.2010.02.096. [DOI] [PubMed] [Google Scholar]
- 28.Sandberg MB, Riquier AD, Pihakaski-Maunsbach K, et al. ANG II provokes acute trafficking of distal tubule Na+-Cl cotransporter to apical membrane. American journal of physiology. 2007;293:F662–669. doi: 10.1152/ajprenal.00064.2007. [DOI] [PubMed] [Google Scholar]
- 29.Ellison DH, Velazquez H, Wright FS. Adaptation of the distal convoluted tubule of the rat. Structural and functional effects of dietary salt intake and chronic diuretic infusion. The Journal of clinical investigation. 1989;83:113–126. doi: 10.1172/JCI113847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Halperin ML, Kamel KS. Dynamic interactions between integrative physiology and molecular medicine: the key to understand the mechanism of action of aldosterone in the kidney. Can J Physiol Pharmacol. 2000;78:587–594. [PubMed] [Google Scholar]
- 31.Briggs JP, Kriz W, Schnermann JB. Overview of kidney function and structure. In: Greenberg A, editor. Primer on Kidney Diseases. 5th edn Saunders; Philadelphia: 2009. pp. 2–18. [Google Scholar]
- 32.Jensen AM, Bae EH, Fenton RA, et al. Angiotensin II regulates V2 receptor and pAQP2 during ureteral obstruction. American journal of physiology. 2009;296:F127–134. doi: 10.1152/ajprenal.90479.2008. [DOI] [PubMed] [Google Scholar]
- 33.Perucca J, Bichet DG, Bardoux P, et al. Sodium excretion in response to vasopressin and selective vasopressin receptor antagonists. J Am Soc Nephrol. 2008;19:1721–1731. doi: 10.1681/ASN.2008010021. [DOI] [PMC free article] [PubMed] [Google Scholar]