

Abstract
Purpose
To determine the frequency, pathology and clinical relevance of amyloid deposited in corneas of CHED.
Methods
Clinical and histopathological case series.
Results
Amyloid subepithelial deposition was found in 5 (6.6%) corneal buttons of 75 patients with histopathologically confirmed CHED diagnosis. Clinical findings included history of parental consanguinity, poor vision (ranging from counting fingers from one foot to 3/200), corneal edema, and central whitish subepithelial corneal nodules in all the five cases and positive family history in 4 of 5 cases. The patients underwent PKP at a mean age of 15 years (range 3–22 years). Histological findings included attenuated endothelium (6/6) thickened Descemet’s membrane (6/6), stromal edema (2/6), and subepithelial amyloid deposits (6/6). All patients improved from vision point of view. To date, no recurrence of the amyloid has been seen in the grafts.
Conclusion
Considering the consanguinity, family history, early onset, and bilaterality, this study supports our hypothesis that the amyloid is primary in nature in our patients and indicates a new subtype of autosomal recessive CHED that require further chemical and genetic analysis. This subtype has the same prognosis for PKP as all CHED patients, if not better.
Keywords: Subepithelial amyloidosis, Congenital hereditary endothelial dystrophy, Amyloid deposits
1. Introduction
Congenital hereditary endothelial dystrophy (CHED) presents at or shortly after birth with bilateral corneal edema. This corneal disorder can be inherited in an autosomal dominant or recessive form. The pathology of CHED is attributed to endothelial cells degeneration during gestation (Maumenee, 1960; Pearce et al., 1969; Kenyon and Antine, 1971; Judisch and Maumenee, 1978).
Cornea is a known ocular site for amyloid deposition either as primary or secondary pathology. Primary corneal amyloid deposition (amyloid AL) can be seen in lattice corneal dystrophy, polymorphic amyloid degeneration (PAD), and gelatinous drop-like corneal dystrophy. Secondary corneal amyloid deposition (amyloid AA) is most frequently associated with local eye disease such as uveitis, ocular trauma, CDK, ROP, keratoconus, trichiasis, and trachoma (Nagataki et al., 1972; Stern et al., 1988; Hidayat and Risco, 1989; Aso and Wakakura, 2000; Matta et al., 1991; Hill et al., 1990).
Two recent studies addressed the association of corneal amyloid deposition with congenital hereditary amyloid dystrophy (Mahmood and Teichmann, 2000; Vemuganti et al., 2002). Amyloid deposition was postulated to be possibly primary in nature in association with CHED by Mahmood and Teichmann (2000). While Vemuganti et al. concluded that this association was secondary in nature (Vemuganti et al., 2002).
In this study we evaluated the clinical and histopathological findings in five patients with CHED associated with subepithelial amyloid deposition.
2. Materials and methods
Corneal buttons pathology reports of 86 patients with CHED who underwent PKP from 1983 to 2006 at King Khalid eye specialist hospital (KKESH) were reviewed for presence or absence of amyloid. Histopathology slides of corneal buttons reported to have amyloid deposits as well as histopathology slides unclearly or insufficiently reported were reviewed by a single pathologist at KKESH to determine the presence of deposits and details of the amyloid distribution. Out of 86 operated CHED patients at KKESH, 75 patients had histopathology slides available from one or both eyes. Therefore, 11 patients were excluded. Out of the 75 patients, five patients only had amyloid deposition in association with CHED.
The corresponding clinical and demographic data of all CHED patients with corneal amyloid deposition were reviewed for clinico-pathologic correlation and analysis. We identified five patients (six corneal buttons) with subepithelial amyloid deposits. Corneal buttons from relatives of those five patients who had PKP for CHED at KKESH were also reviewed for the presence or absence of amyloid and were all negative.
All five patients (eight eyes) underwent PKP while they were receiving general anesthesia. The trephine size was 6.75 mm in one eye, and 7 mm in seven eyes in the recipient cornea, although the donor corneal trephine size was larger by 0.25 mm in four eyes, and 0.5 mm in the other four eyes. The sutures were continuous in two eyes, interrupted in five eyes, and combined in one eye. Sutures were removed completely in a mean time of 13.5 months (range, 3–44 months). Patients were followed up for a mean of 112 months (range, 10–264 months).
Paraffin sections were prepared from six corneal buttons after overnight 10% formalin fixation, stained with Hematoxylin and Eosin (H&E), Periodic Acid-Schiff (PAS) and Congo red stains. Two corneal buttons from two different patients (patients 4 and 5) were submitted only for electron microscopy studies and no histopathology slides were prepared.
3. Results
Five patients (out of 75) were found to have subepithelial amyloid deposition (6.6%). The age of the five patients at the time of PKP ranged from 3 to 22 years (mean, 15 years); there were three females and two males. Four patients were all members of one Saudi family and one patient (patient 1) was Yemeni. The clinical features of these patients are summarized in Table 1. History of consanguinity was positive in all cases. The clinical diagnosis of CHED was made in all cases. Patient 1 had relatively good visual acuity in the left eye (20/100) for which PKP was not performed in that eye. Patient 2 was already grafted in the right eye 3 years earlier on presentation to KKESH and the patient had a pathology report documenting the diagnosis of CHED along with the presence of subepithelial amyloid deposits, but obtaining slides from original corneal button was not possible. Later on that right graft failed and we performed another PKP but we did not find any evidence of CHED recurrence nor amyloid deposition in that failed graft. Patients 4 and 5 entire left corneal buttons were submitted for Electron Microscopy studies which confirmed the clinical diagnosis of CHED as part of another ongoing prospective study concerning CHED but the gold stain used on those buttons precluded the possibility of amyloid identification by histopathology, nevertheless, both left eyes for both patient contained clinically the same white nodular subepithelial pathology proven to be amyloid in the contralateral eyes by histopathology (Figs. 1 and 2).
Table 1.
Clinical features of patients with CHED and amyloid corneal deposition.
| Case no. | Age (y)/sex | Symptom(s) | Duration of symptoms | Consanguinity | Siblings with CHED | Laterality | Preoperative VA | IOP | Nystagmus | Cornea | Graft recurrence | Last VA |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 22/M | DV/WE | 15 y | Yes | 2/2 | OD | CF: 2 f | 25 mmHg | No | Edema/haze/CWO | No | 20/100 |
| 2 | 16/M | DV | SB | Yes | 1/3 | OS | CF: 2 f | Normal | No | Edema/haze/CWO | No | 20/70 |
| 3 | 19/F | DV/WE | SB | Yes | 2/3 | OD | CF: 1 f | Normal | Yes | Edema/haze/CWO | No | 20/300 |
| ″ | ″ | ″ | ″ | ″ | ″ | OS | CF: 1 f | Normal | Yes | Edema/haze/CWO | No | 20/300 |
| 4 | 5/F | DV | SB | Yes | 2/3 | OD | 3/200 | Normal | Yes | Edema/haze/CWO | No | 20/30 |
| ″ | ″ | ″ | ″ | ″ | ″ | OS | 20/300 | Normal | Yes | Edema/haze/CWO | No | 20/125 |
| 5 | 3/F | WE | SB | Yes | 3/4 | OD | FF | Normal | No | Edema/haze/CWO | No | 20/160 |
| ″ | ″ | ″ | ″ | ″ | ″ | OS | FF | Normal | No | Edema/haze/CWO | No | 20/100 |
CF, counting fingers; CWO, central white opacities; DV, diminution of vision; FF, fixes and follows; IOP, intraocular pressure; SB, since birth; VA, visual acuity; WE, white eye.
Figure 1.
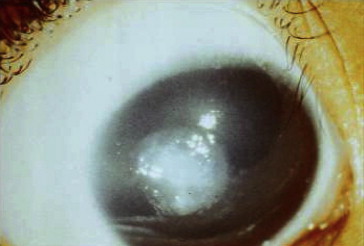
Patient 4, central grayish-white nodular subepithelial elevation in right eye.
Figure 2.
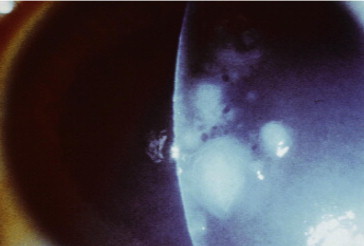
Patient 4, same focal nodules observed in the contralateral eye.
No graft rejection episodes were documented in any of the patients. Patient 3 had left graft failure 30 months after the primary graft that was not caused by neither rejection nor infection. This failed graft got infected 5 months later (treated elsewhere) and another PKP was performed 72 months after performing the primary graft. The secondary graft failed as well and a third PKP was performed 49 months later. The tertiary graft remains clear. Apart from patient 3 left graft, none of the remaining grafts (including patient 3 right graft) were infected nor labeled as failed. Vision in all eyes improved.
Histopathology features of these six corneal buttons are presented in Table 2. Amyloid deposits noted in six corneal buttons were subepithelial (patient 4 had even deeper amyloid deposits involving the mid-stroma) which appeared as amorphous plaque-like nodules (Figs. 3, 5–7) with characteristic birefringence under polarized light microscopy (Fig. 4). Bowman’s layer was either absent (in two patients) or interrupted (in four patients). The stromal edema (thickening) was evident in only two patients (patients #2 and 5) but without stromal scarring. Anterior or Mid-stromal neovascularization was evident in the first four patients. Stromal inflammation was seen in all but one patient (#4). Descemet’s membrane was thickened in all patients. The endothelium was attenuated in all patients as well.
Table 2.
Histopathological features of patients with CHED and amyloid deposits.
| Case no. /eye | Epithelium | Bowman’s layer | Stromal edema | Stromal scarring | Stromal vascularization | Stromal inflammation | Descemet’s membrane | Endothelium | Amyloid |
|---|---|---|---|---|---|---|---|---|---|
| 1/OD | Disrupted | Interrupted | Absent | Mid-stromal | Anterior | Anterior (perivascular) | Thickened | Marked attenuation, pigmented | Subepithelial |
| 2/OD | Disrupted | Interrupted, thickened | Present | Absent | Mid-stromal | Anterior and mid-stromal | Thickened | Marked attenuation | Subepithelial |
| 3/OD | Thinned | Absent | Absent | Anterior and mid-stromal | Mid-stromal | Diffuse | Thickened | Marked attenuation, pigmented | Subepithelial |
| 3/OS | Thinned | Absent | Absent | Full thickness | Mid-stromal | Perivascular | Thickened | Marked attenuation, pigmented | Subepithelial |
| 4/OD | Acanthosis, bullae | Interrupted | Absent | Anterior, mid-stromal | Absent | Absent | Thickened | Marked attenuation | Subepithelial, mid-stromal |
| 5/OD | Partially thickened | Interrupted | Present | Absent | Absent | Subepithelial | Thickened | Moderate attenuation | Subepithelial |
Figure 3.
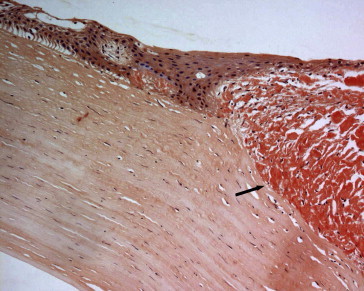
Patient 4, the corresponding corneal subepithelial amyloid nodule (arrow) reaching the mid-stroma of the right eye in the previous patient (Congo red stain; original magnification 200×).
Figure 5.
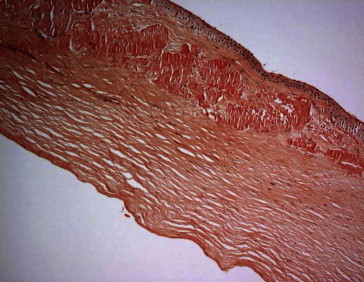
Patient 1, OD. Corneal subepithelial amyloid (Congo red stain; original magnification 100×).
Figure 6.
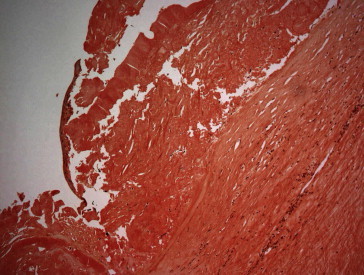
Patient 2, OD. Corneal subepithelial amyloid (Congo red stain; original magnification 100×).
Figure 7.
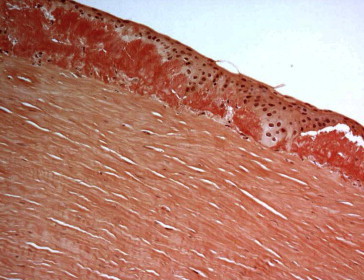
Patient 5, OD. Corneal subepithelial amyloid (Congo red stain; original magnification 200×).
Figure 4.

Patient 3, the subepithelial deposits exhibit birefringence with polarized light confirming the presence of amyloid (Congo red stain; original magnification 100×).
4. Discussion
Starch (amylum in Latin) was mistakenly thought to be the substance forming amyloid based on crude iodine-staining techniques, until it was finally resolved that it was rather a deposition of proteinaceous mass (Kyle, 2001). The classical, histopathological definition of amyloid is an extracellular, proteinaceous deposit exhibiting cross-beta structure due to mis-folding of unstable proteins. These deposits are generally identified by apple-green birefringence when stained with Congo red and seen under polarized light. They often recruit various sugars and other components such as serum Amyloid P component, resulting in complex, and sometimes inhomogeneous structures (Sipe and Cohen, 2000).
There are at least two types of amyloid. Type A (known also as AA) is a non-immunoglobulin protein of unknown origin. Type B amyloid has been shown to be identical to a fragment of light chain of immunoglobulin. Amyloid deposits are associated with a structural protein known as P or AP (Spark et al., 1978). The deposition of amyloid in various body tissues results in amyloidosis and the reason for it is unknown. It may be a disorder of protein metabolism or hypersensitivity, an abnormality of reticular endothelial system, the result of chronic immunologic reaction, or a combination of these defects (Scheinberg and Cathcart, 1974).
Amyloidosis is a heterogeneous group of disorders in which fibrillar hyaline proteins including amyloid P protein (AP), prealbumin or transthyretin (AF), immunoglobulin light chains (AL), and acute phase reactants (AA) are deposited in a variety of target tissue (Blodi and Apple, 1979).
Amyloidosis can be classified into systemic and localized. Systemic amyloidosis can be primary or secondary. Primary systemic type usually involves the tongue (macroglossia), heart (cardiomyopathy), GIT (malabsorption), peripheral nerves (neuropathy including ptosis), kidney (nephrotic syndrome), ocular muscles (ophthalmoplegia), vitrouse, and cornea (Meretoja syndrome or lattice corneal dystrophy type II) (Meretoja, 1969, 1973). Secondary systemic amyloidosis is usually found in association with malignancies, tuberculosis, rheumatoid arthritis, syphilis, and other chronic inflammatory conditions and it is the most commonly encountered form McPherson (1966).
In primary amyloid deposition (AL) the amyloid fibrils consist of the variable portions of monoclonal kappa (κ) or lambda (λ) immunoglobulin light chains. In secondary amyloid deposition (AA) the amyloid fibrils consist of protein A, a non-immunoglobulin (Meretoja, 1969, 1973; McPherson, 1966).
Cornea is a frequent ocular site for amyloid deposition either as primary or secondary pathology. Primary corneal amyloid deposition (amyloid AL) can be seen in lattice dystrophy, polymorphic amyloid degeneration (PAD), and gelatinous drop-like corneal dystrophy. Secondary corneal amyloid deposition (amyloid AA) is most frequently associated with local eye disease such as uveitis, ocular trauma, climatic droplet keratopathy, retinopathy of prematurity, keratoconus, trichiasis, and trachoma (Stern et al., 1988; Hidayat and Risco, 1989; Aso and Wakakura, 2000; Matta et al., 1991; Hill et al., 1990; McPherson, 1966).
Polymorphic amyloid degeneration presents as corneal punctate and filamentous opacities that affect patients in their forth decade or older (Mannis et al., 1981; Woodward et al., 2007). Family studies failed to demonstrate heritability and therefore it is classified as degeneration rather than dystrophy (Mannis et al., 1981). The deposits are usually in the deeper layers associated with normal intervening stroma (Mannis et al., 1981). Although it is not a cause of visual dysfunction, this disorder (Mannis et al., 1981). It can be associated with posterior polymorphous corneal dystrophy (Molia et al., 1999) or posterior crocodile shagreen corneal degeneration (Woodward et al., 2007).
Lattice corneal dystrophy usually is an autosomal dominant condition, and it is one of the common stromal dystrophies. Like granular and Avellino dystrophy, the genetic defect of lattice dystrophy has been mapped to the BIG H3 gene on chromosome 5q. Examination of the cornea in the second to third decade of life will reveal stromal branching, refractile lattice lines representing amyloid protein with intervening haze, which are observed best in retroillumination. Lattice dystrophy can cause excessive corneal erosions and decreased visual acuity requiring a corneal transplant or phototherapeutic keratectomy (PTK). Recurrence after keratoplasty is a known complication (Chan et al., 1982).
Primary gelatinous drop-like corneal dystrophy (PGDD) is a rare corneal dystrophy, most probably autosomal recessive in nature and was first described in Japan in 1914 followed by other reports (Nagataki et al., 1972; Akiya et al., 1972; Santo et al., 1995). It usually presents with photophobia, foreign body sensation, and decreased vision in the second or third decades of life. PGDD has a high incidence of recurrence after keratoplasty (Santo et al., 1995).
Eyelid skin is another frequent site for amyloid deposition. Waxy, yellowish appearing small papules are typical. Conjunctival involvement is rather rare in the form of amyloid nodules, but of importance as it may mimic other forms of conjunctivitis, including trachoma (Blodi and Apple, 1979). Primary localized amyloid deposition in the orbit (mainly lacrimal gland and ocular muscles) is rare and can lead to proptosis (Knowles et al., 1975). Familial amyloidosis can affect the pupil in the form of segmental iris paralysis, pupillary dissociation with inequality, and heterochromia (Falls et al., 1955).
Amyloid deposits in the vitreous are known to occur in the systemic familial amyloidosis. Isolated vitreous deposits in the absence of a family history (primary non-familial amyloidosis of the vitreous) are extremely rare and may mimic other conditions including vitritis, lymphoma, endophthalmitis and old vitreous haemorrhage (Bitwas et al., 1992). Glaucoma occurring in association with vitreous amyloidosis is thought to result from transport of amyloid by the aqueous fluid and its deposition in the trabecular meshwork (Gregory et al., 1999).
Congenital hereditary endothelial dystrophy (CHED) was first described as “corneitis interstitialis in utero” in 1893 by Laurence. Initially classified as an intrauterine interstitial keratitis, then stromal dystrophy (Pearce et al., 1969). In 1960, Maumenee was the first to describe CHED as primary corneal endothelial dysfunction (Maumenee, 1960). In 1971, the name Congenital hereditary endothelial dystrophy was suggested by Kenyon and Antine (1971). The inheritance of the Autosomal-dominant (AD) type (CHED1) has been linked to chromosome 20, near posterior polymorphous dystrophy (PPMD) locus, while the Autosomal-recessive (AR) type (CHED2) is not linked to this chromosome, thus indicating a genetically distinct entity (Al-Rajhi, 2000; Kanis et al., 1999; Shah et al., 2008).
Congenital hereditary endothelial dystrophy is characterized by diffuse, non-inflammatory corneal opacities with edema. The disease is bilateral and tends to be symmetric. Marked impairment of vision is characteristic. CHED typically presents at birth or in early infancy as a common cause of childhood corneal opacification.
The two forms of CHED have different clinical characteristics (Judisch and Maumenee, 1978). Children with dominant CHED have clear corneas at birth with clouding first noted during the first or second year of life then slowly progresses over 5–10 years. Photophobia and epiphora are common and may be the presenting signs of the disease. Nystagmus is uncommon and vision tends to be better (in the range of 20/40 to 20/400) than in recessive CHED. Some authors have suggested that CHEDI is more appropriately termed infantile hereditary endothelial dystrophy, given its clinical characteristics (Judisch and Maumenee, 1978; Al-Rajhi, 2000). In contrast, corneal clouding is present at birth or within the neonatal period in recessive CHED. Corneal opacification is dense at the time of diagnosis, and does not tend to progress. There is no associated photophobia or epiphora. Nystagmus is invariably present, presumably as a result of severe corneal opacification at an early age (Judisch and Maumenee, 1978).
CHED has been associated with congenital glaucoma. Abnormalities in the neural crest could theoretically cause both entities in a single patient. Because both conditions can present with corneal opacification and edema, accurate diagnosis may be difficult and false elevations of intraocular pressure (IOP) caused by stromal edema can compound this problem. Other clinical characteristics must often be considered to distinguish the two diseases. For example, progressively enlarging corneal diameter is more characteristic of congenital glaucoma and corneal edema from congenital glaucoma should resolve after the IOP is lowered (Mullaney et al., 1995).
Histopathologic changes in CHED are concentrated in the endothelium and Descemet’s membrane. The endothelial cells of the peripheral cornea in CHED have a relatively normal appearance. The endothelium becomes attenuated in the midperiphery and is completely absent from the central cornea. In the transition zone, cells are irregularly shaped, with pleomorphism and polymegathism. The normal hexagonal pattern is lost. By ultrastructure studies, endothelial organelles are abnormal, including dilated mitochondria. Descemet’s membrane may be either thickened (Chan et al., 1982) or thinned (Kirkness et al., 1987) in CHED, possibly related to the degree and timing of endothelial dysfunction (Moller-Pedersen, 1997). Thinned or attenuated Descemet’s membrane may be the sequel of endothelial dysfunction in utero, so that only the fetal anterior banded zone is produced. Thickened Descemet’s membrane, on the other hand, is the result of persistent dystrophic or dysfunctional endothelium that secretes a reactive posterior collagenous layer or an exaggerated, but structurally normal, posterior non-banded layer (Moller-Pedersen, 1997). Finally the corneal endothelial permeability is significantly increased, as is expected with a dysfunctional endothelial barrier (Chan et al., 1982). Progressive stromal and epithelial edema occur later accompanied by secondary structural changes. The normal appearance of the anterior banded zone of Descemet’s membrane suggests that the endothelium is most likely functionally normal up to the fifth month in utero (Chan et al., 1982; Moller-Pedersen, 1997). Degeneration starting with the central cornea occurs thereafter which is believed to arise from an abnormality in the terminal differentiation of neural crest cells (Mullaney et al., 1995).
Penetrating keratoplasty is currently the best option for visual rehabilitation and remains the most acceptable form of management with good results (Vemuganti et al., 2002). Corneal transplantation for CHED has a better prognosis than do other pediatric indications because these eyes typically lack corneal neovascularization, inflammation, and concomitant intraocular pathology (Dana et al., 1995). One series have demonstrated a 90% graft survival rate in CHED with a mean follow-up of approximately 3 years (Sajjadi et al., 1995). The autosomal dominant type is associated with relatively better vision due to later onset and lack of nystagmus (Kenyon and Antine, 1971; Judisch and Maumenee, 1978). In Saudi Arabia, a previous study has shown the graft survival rate to be higher in delayed-onset CHED (96%) than in CHED present at birth (56%) (Al-Rajhi and Wagoner, 1997). Pediatric penetrating keratoplasty poses greater technical challenges and, in general, is less successful than corneal transplantation in adults (Frueh and Brown, 1997). Effective visual rehabilitation in patients with CHED is time-consuming for parents, child, and surgeon. Aggressive amblyopia management is critical for optimal visual recovery (Sajjadi et al., 1995; Frueh and Brown, 1997).
In our current study we are presenting the clinical and histologic features of the association of CHED and Amyloid corneal deposition. The clinical diagnosis of CHED was established in all cases, and this was confirmed by histologic examination which demonstrated attenuated endothelium and thickened Descemet’s membrane. White superficial nodular corneal opacities were an interesting clinical finding and were found to be amyloid deposits in all cases on histopathology. These deposits resembled clinically the amyloid deposits seen in primary gelatinous drop-like corneal dystrophy (PGDD), which is a rare corneal dystrophy, most probably autosomal recessive in nature. PGDD was first described in Japan in 1914 followed by other reports (Al-Rajhi, 2000; Solomon et al., 2008; Akiya et al., 1972). It usually presents with photophobia, foreign body sensation, and decreased vision in the second or third decades of life. PGDD has a high incidence of recurrence after keratoplasty (Akiya et al., 1972). The amyloid deposition in PGDD is subepithelial as in our patients, nevertheless, none of our patients had any recurrence after keratoplasty which indicates a separate pathology.
An article from India described another five patients with CHED and amyloid deposition. The authors attributed amyloid depositions to degenerative changes and stromal inflammation and they classified amyloid as secondary in nature based on immunohistochemical findings. None of their patients were related and the youngest patient was 8 years old at the time of PKP (Mahmood and Teichmann, 2000). Despite the phenotypic and histologic resemblance to the patients in our study (especially patient 1) we assume the amyloid type in our patients is primary rather than secondary (especially patients 2–5) for several reasons. First, we observed these deposits in four related patients with history of consanguinity in which case, inheritance is considered a major role in explaining the amyloid deposition. Second, the size and configuration of amyloid deposits is too large to accumulate as secondary amyloid over such a short time, since our patients were very young at the time of surgery (patient 4 was 5 years old and patient 5 was only 3 years). Third, known local causes of secondary amyloid deposition like spheroidal degeneration, trichiasis, or trachoma were not observed in any of the patients. Fourth, amyloid typing based on immunohistochemical testing may be inconclusive or even misleading, therefore unreliable (Al-Rajhi and Wagoner, 1997; Satoskar et al., 2007). Immunohistochemical amyloid identification utilizes immunoglobulin light chains immunofluorescence staining which is presumed to identify amyloid AL (primary amyloid) fibrils through positive reaction to commercially available anti-light chain antibodies which lack specificity by cross-reaction with amyloid AA (secondary amyloid) fibrils (Satoskar et al., 2007). To obtain an accurate amyloid typing, chemical analysis using tandem mass spectrometry is necessary (Al-Rajhi and Wagoner, 1997). This method of testing, which is based on proteomics technologies, will allow direct molecular identification of the amyloid protein and has the advantage of less tissue needed for identification compared to other means of testing (Al-Rajhi and Wagoner, 1997; Satoskar et al., 2007). Unfortunately, this is a new technology and no laboratory in Saudi Arabia has the capability of conducting such testing, so running these test on our samples was not possible.
The prognosis of penetrating keratoplasty in CHED associated with amyloid deposits is good with no recurrence of amyloid as observed in our patients as well as previously reported cases in the literature (Hill et al., 1990; Mahmood and Teichmann, 2000). The amyloid deposition in association with CHED dose not seem to increase future graft rejection, infection nor failure.
In summary, we have described five cases of CHED associated with presumably primary subepithelial amyloid deposition. This finding is not as rare to be associated with CHED as previously described. We believe that this finding indicates a new subtype of autosomal recessive CHED, however it requires further chemical and genetic analysis.
References
- Akiya S., Ito K., Matsui M. Gelatinous drop-like dystrophy of the cornea: light and electron microscopy study of superficial stromal lesion. Jpn. J. Ophthalmol. 1972;26:815–826. [Google Scholar]
- Al-Rajhi A.A. Congenital hereditary endothelial dystrophy: current concepts and management. Saudi J. Ophthalmol. 2000;14:101–114. [Google Scholar]
- Al-Rajhi A.A., Wagoner M.D. Penetrating keratoplasty in congenital hereditary endothelial dystrophy. Ophthalmology. 1997;104:956–961. doi: 10.1016/s0161-6420(97)30200-0. [DOI] [PubMed] [Google Scholar]
- Aso K., Wakakura M. Corneal amyloidosis complicated by trichiasis. Immunohistochemical identification of amyloid light chain protein. Jpn. J. Ophthalmol. 2000;44:191. doi: 10.1016/s0021-5155(99)00210-5. [DOI] [PubMed] [Google Scholar]
- Bitwas J., Badrinath S.S., Rao N.A. Primary nonfamilial amyloidosis of the vitreous. A light microscopic and ultrastructural study. Retina. 1992;12(3):251–253. doi: 10.1097/00006982-199212030-00010. [DOI] [PubMed] [Google Scholar]
- Blodi F.C., Apple D.J. Localized conjunctival amyloidosis. Am. J. Ophthalmol. 1979;88:346–450. doi: 10.1016/0002-9394(79)90631-7. [DOI] [PubMed] [Google Scholar]
- Chan C.C., Green W.R., Barraquer J. Similarities between posterior polymorphous and congenital hereditary endothelial dystrophies: a study of 14 buttons of 11 cases. Cornea. 1982;1:155–172. [Google Scholar]
- Dana M.R., Moyes A.L., Gomes J.A., Rosheim K.M., Schaumberg D.A., Laibson P.R., Holland E.J., Sugar A., Sugar J. The indications for and outcome in pediatric keraroplasty: a multicenter study. Ophthalmology. 1995;102:1129–1138. doi: 10.1016/s0161-6420(95)30900-1. [DOI] [PubMed] [Google Scholar]
- Falls H.F. Ocular manifestations of hereditary primary systemic amyloidosis. Arch. Ophthalmol. 1955;54:66. doi: 10.1001/archopht.1955.00930020666004. [DOI] [PubMed] [Google Scholar]
- Frueh B.E., Brown S.I. Transplantation of congenitally opaque corneas. Br. J. Ophthalmol. 1997;83:115–119. doi: 10.1136/bjo.81.12.1064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gregory A., Nelson M.D., Deepak P. Ocular amyloidosis and secondary glaucoma. Ophthalmology. 1999;106(7):1363–1366. doi: 10.1016/S0161-6420(99)00726-5. [DOI] [PubMed] [Google Scholar]
- Hidayat A.A., Risco J.M. Amyloidosis of corneal stroma in patients with trachoma. A clinicopathologic study of 62 cases. Ophthalmology. 1989;96:1203–1211. doi: 10.1016/s0161-6420(89)32765-5. [DOI] [PubMed] [Google Scholar]
- Hill J.C., Maske R., Bowen R.M. Secondary localized amyloidosis of the cornea caused by tertiary syphilis. Cornea. 1990;9:98–101. [PubMed] [Google Scholar]
- Judisch G.F., Maumenee I.H. Clinical differentiation of recessive congenital hereditary endothelial dystrophy and dominant hereditary endothelial dystrophy. Am. J. Ophthalmol. 1978;85:606–612. doi: 10.1016/s0002-9394(14)77091-6. [DOI] [PubMed] [Google Scholar]
- Kanis A.B., Al-Rajhi A.A., Taylor C.M., Mathers W.D., Folberg R.Y., Nishimura D.Y., Sheffield V.C., Stone E.M. Exclusion of AR-CHED from the chromosome 20 region containing the PPMD and AD-CHED loci. Ophthalmic Genet. 1999;20:243–249. doi: 10.1076/opge.20.4.243.2273. [DOI] [PubMed] [Google Scholar]
- Kenyon K.R., Antine B. The pathogenesis of congenital hereditary endothelial dystrophy of the cornea. Am. J. Ophthalmol. 1971;72:787–795. doi: 10.1016/0002-9394(71)90019-5. [DOI] [PubMed] [Google Scholar]
- Kirkness C.M., McCartney A., Rice N.S.C. Congenital hereditary corneal edema of Maumenee: its clinical features, management, and pathology. Br. J. Ophthalmol. 1987;71:130–144. doi: 10.1136/bjo.71.2.130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knowles D.M. Amyloidosis of the orbit and adnexa. Surv. Ophthalmol. 1975;19:367. [PubMed] [Google Scholar]
- Kyle R.A. Amyloidosis: a convoluted story. Br. J. Haematol. 2001;114:529–538. doi: 10.1046/j.1365-2141.2001.02999.x. [DOI] [PubMed] [Google Scholar]
- Mahmood M.A., Teichmann K.D. Corneal amyloidosis associated with congenital hereditary endothelial dystrophy. Cornea. 2000;19:570–573. doi: 10.1097/00003226-200007000-00035. [DOI] [PubMed] [Google Scholar]
- Mannis M.J., Krachmer J.H., Rodrigues M.M., Pardos G.J. Polymorphic amyloid degeneration of the cornea: a clinical and histopathologic study. Arch. Ophthalmol. 1981;99:1217–1223. doi: 10.1001/archopht.1981.03930020091008. [DOI] [PubMed] [Google Scholar]
- Matta C.S., Tabara K.F., Cameron J.A., Hidayat A.A., Al-Raghi A.A. Climatic droplet keratopathy with corneal amyloidosis. Ophthalmology. 1991;98:192–195. doi: 10.1016/s0161-6420(91)32317-0. [DOI] [PubMed] [Google Scholar]
- Maumenee A.E. Congenital hereditary corneal dystrophy. Am. J. Ophthalmol. 1960;50:1114–1124. doi: 10.1016/0002-9394(60)90998-3. [DOI] [PubMed] [Google Scholar]
- McPherson S.D. Corneal amyloidosis. Am. J. Ophthalmol. 1966;62:1025–1033. doi: 10.1016/0002-9394(66)92549-9. [DOI] [PubMed] [Google Scholar]
- Meretoja J. Familial systemic paramyloidosis with lattice dystrophy of the cornea, progressive cranial neuropathy, skin changes and various internal symptoms. A previously unrecognized heritable syndrome. Ann. Clin. Res. 1969;1(4):314–324. [PubMed] [Google Scholar]
- Meretoja J. Genetic aspects of familial amyloidosis with corneal lattice dystrophy and cranial neuropathy. Clin. Genet. 1973;4(3):173–185. doi: 10.1111/j.1399-0004.1973.tb01140.x. [DOI] [PubMed] [Google Scholar]
- Molia L.M., Lanier J.D., Font R.L. Posterior polymorphous dystrophy associated with posterior amyloid degeneration of the cornea. Am. J. Ophthalmol. 1999;127:86–88. doi: 10.1016/s0002-9394(98)00276-1. [DOI] [PubMed] [Google Scholar]
- Moller-Pedersen T. A comparative study of human corneal kerratocytes and endothelial cell density during aging. Cornea. 1997;16:333–338. [PubMed] [Google Scholar]
- Mullaney P.B., Risco J.M., Teichmann K. Congenital hereditary endothelial dystrophy associated with glaucoma. Ophthalmology. 1995;102:186–192. doi: 10.1016/s0161-6420(95)31037-8. [DOI] [PubMed] [Google Scholar]
- Nagataki S., Tanishima T., Sakimoto T. A case of primary gelatinous drop-like corneal dystrophy. Jpn. J. Ophthalmol. 1972;16:107–116. [Google Scholar]
- Pearce W.G., Tripathi R.C., Mrgan G. Congenital endothelial corneal dystrophy. Clinical pathological, and genetic study. Br. J. Ophthalmol. 1969;53:577–591. doi: 10.1136/bjo.53.9.577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sajjadi H., Javadi M.A., Hemmati R., Mirdeghan A., Parvin M., Nassiri N. Results of penetrating keratoplasty in CHED: congenital hereditary endothelial dystrophy. Cornea. 1995;14:18–25. [PubMed] [Google Scholar]
- Santo R.M., Yamaguchi T., Kanai A., Okisaka S., Nakajima A. Clinical and histopathologic feature of corneal dystrophies in Japan. Ophthalmology. 1995;102:557–567. doi: 10.1016/s0161-6420(95)30982-7. [DOI] [PubMed] [Google Scholar]
- Satoskar A.A., Burdge K., Cowden D.J., Nadasdy G.M., Herbert L.A., Nadasdy T. Typing of amyloidosis in renal biopsies: diagnostic pitfalls. Arch. Pathol. Lab. Med. 2007;131:917–922. doi: 10.5858/2007-131-917-TOAIRB. [DOI] [PubMed] [Google Scholar]
- Scheinberg M.A., Cathcart E.S. Casein-induced experimental amyloidosis III. Responses to mitogens, allogenic cells and graft versus host reaction in the murine model. Immunology. 1974;27:953. [PMC free article] [PubMed] [Google Scholar]
- Shah S.S., Al-Rajhi A.A., Brandt J.D., Mannis M.J., Roos B., Sheffield V.C., Syed N.A., Stone E.M., Fingert J.H. Mutation in the SLC4A11 gene associated with autosomal recessive congenital hereditary endothelial dystrophy in a large Saudi family. Ophthalmic Genet. 2008;28:41–45. doi: 10.1080/13816810701850033. [DOI] [PubMed] [Google Scholar]
- Sipe J.D., Cohen A.S. Review: history of amyloid Fibril. J. Struct. Biol. 2000;130:88–89. doi: 10.1006/jsbi.2000.4221. [DOI] [PubMed] [Google Scholar]
- Solomon A., Murphy C.L., Westermark P. Unreliability of immunohistochemistry for typing amyloid deposits. Arch. Pathol. Lab. Med. 2008;132:14–15. doi: 10.5858/2008-132-14b-IR. [DOI] [PubMed] [Google Scholar]
- Spark E.D. The identification of amyloid P-component (protein AP) in normal cultured human fibroblasts. Lab. Invest. 1978;38:556. [PubMed] [Google Scholar]
- Stern G.A., Knapp A., Hood C.l. Corneal amyloidosis associated with keratoconus. Ophthalmology. 1988;95:52–55. doi: 10.1016/s0161-6420(88)33225-2. [DOI] [PubMed] [Google Scholar]
- Vemuganti G.K., Sridhar M.S., Edward D.P. Subepithelial amyloid deposits in congenital hereditary endothelial dystrophy. Cornea. 2002;21:524–529. doi: 10.1097/00003226-200207000-00017. [DOI] [PubMed] [Google Scholar]
- Woodward M., Randleman J.B., Larson P.M. In vivo confocal microscopy of polymorphic amyloid degeneration and posterior crocodile shagreen. Cornea. 2007;26:98–101. doi: 10.1097/01.ico.0000240103.47508.c4. [DOI] [PubMed] [Google Scholar]