

Abstract
Nucleic acid (NA)–sensing TLRs (NA-TLRs) promote the induction of anti-nuclear Abs in systemic lupus erythematosus. However, the extent to which other nonnuclear pathogenic autoantibody specificities that occur in lupus and independently in other autoimmune diseases depend on NA-TLRs, and which immune cells require NA-TLRs in systemic autoimmunity, remains to be determined. Using Unc93b13d lupus-prone mice that lack NA-TLR signaling, we found that all pathogenic nonnuclear auto-antibody specificities examined, even anti-RBC, required NA-TLRs. Furthermore, we document that NA-TLRs in B cells were required for the development of antichromatin and rheumatoid factor. These findings support a unifying NA-TLR–mediated mechanism of autoantibody production that has both pathophysiological and therapeutic implications for systemic lupus erythematosus and several other humoral-mediated autoimmune diseases. In particular, our findings suggest that targeting of NA-TLR signaling in B cells alone would be sufficient to specifically block production of a broad diversity of autoantibodies.
Systemic lupus erythematosus (SLE) is a heterogeneous autoimmune disease characterized by multiorgan involvement and high levels of circulating autoantibodies, most commonly to nuclear Ags. Importantly, substantial evidence supports a critical role for the endolysosome-restricted nucleic acid (NA)–sensing subset of TLRs (NA-TLRs) in the production of such anti-nuclear autoantibodies and in the pathophysiology of lupus (1). Accordingly, overexpression of the ssRNA-binding TLR7 exacerbated disease in susceptible strains and could even induce lupus in nonautoimmune mice (2–6), whereas absence of most or all NA-TLR signaling in lupus-prone mice deficient in MyD88 (7) or Unc93b1 (3d mutation) (8) reduced most clinical manifestations and mortality. Further dissection of the NA-TLRs suggested that TLR7 and, to a lesser extent, the DNA-binding TLR9 are most critical for lupus induction (9–14). Notably, deletion of these TLRs inhibited autoantibodies to self-Ags containing their corresponding ligands: anti-ribonucleoprotein (RNP) was inhibited with TLR7 deficiency, and anti-dsDNA or chromatin was inhibited with TLR9 deficiency (7, 9, 13).
Although the relationship of NA-TLRs to nuclear and RNP autoantibodies is well documented, SLE is also associated with a wider array of autoantibodies that include specificities with less clear connections to NAs, several of which are associated with diseases that can occur independent of lupus (15, 16). These include anti–β2-gp1 (GP1) and anti-cardiolipin in the anti-phospholipid syndrome, anti-myeloperoxidase (MPO) in certain vasculitides, and anti-RBCs, such as those against band 3 or glycophorin A, in autoimmune hemolytic anemia (17–19). In models of antiphospholipid syndrome and hemolytic anemia, studies have shown increased autoantibody production due to TLR7 duplication (Yaa mutation), suggesting NA-TLRs might affect most lupus autoantibody specificities (20, 21). However, it is not known to what extent non-NA–targeted autoantibodies are dependent on NA-TLRs or if they share a common production mechanism with anti-nuclear Abs (ANAs) and anti-RNP.
NA-TLRs are postulated to promote lupus by both nonspecific activation of the innate immune system and specific induction of autoreactive B cells. In the former, activation of the endosomal NA-TLRs can occur after engulfment of NA-containing immune complexes via FcγRIIa-mediated endocytosis in plasmacytoid dendritic cells (pDCs), conventional DCs (cDCs), and neutrophils (1, 22). Such activated pDCs and cDCs could potentially enhance lupus through the production of proinflammatory and immunostimulating factors, particularly type I IFNs and BAFF, and could also act as potent APCs for self-Ags, whereas such activation of neutrophils has been shown in vitro to cause cell death and the release of neutrophil extracellular traps that activate pDCs (1, 22). In contrast, more specific activation of autoreactive B cells recognizing self-antigenic cargoes containing NAs is postulated to occur following receptor endocytosis and release of NAs into the endosomal compartment (1, 23). Such NA-TLR–mediated activation of self-reactive B cells has been suggested to play a role in both central and peripheral tolerance as well as amplification of autoantibody responses (1, 6, 9, 24–27). These studies provide insights into potential individual NA-TLR–dependent mechanisms, but their contribution as a whole to the pathophysiology of SLE has not been directly examined.
Unc93b13d (3d) mice have a mutation (H412R) that blocks UNC93B1-mediated trafficking of endosomal TLRs from the endoplasmic reticulum to the endosome, which abolishes endosomal TLR signaling, including all NA-TLRs (28, 29). In this study, we used 3d lupus-prone mice to determine the role of NA-TLRs in the development of nonnuclear lupus-related autoanti-body specificities and cryoglobulins, the effects of complete NA-TLR deficiency on clinical manifestations, and finally the impact of cell-intrinsic NA-TLR expression on pDCs, cDCs, and B cell activation and expansion in lupus. The findings delineate specific and critical roles of NA-TLRs in autoantibody responses and broaden understanding of their significance in SLE pathogenesis.
Materials and Methods
Mice
MRL-Faslpr Unc93b13d (3d) and NZB-3d mice were generated by marker-assisted congenic breeding to C57BL/6 (B6)-3d mice as previously described (30). MRL-Faslpr and MRL-Faslpr 3d/WT (Het) mice had concordant phenotypes and were analyzed together as wild-type (WT)/Het. Data for MRL-Faslpr 3d mice were from female and littermate controls from N4–N7 generations except for survival, which compared N10 generation mice. NZB-3d mice were N6–N8, and littermate WT/Het controls had similar severity of autoimmune hemolytic anemia as parental NZB mice. Mice were bred at The Scripps Research Institute vivariums. Procedures were approved by The Scripps Institutional Animal Care and Use Committee.
Pathology
Tissues were fixed in zinc formalin solution and sections stained with periodic acid-Schiff reagent and hematoxylin. The severity of glomerulonephritis (GN) was blindly graded on a 0–4 scale as previously described (31). To assess glomerular IgG deposits, 5-μm frozen kidney sections were stained with Alexa 488 goat anti-mouse IgG (Invitrogen), and then three to four areas per section were captured with a Zeiss AxioCam camera (Carl Zeiss) attached to a Nikon Eclipse E800 microscope (Nikon) using Axi-ovision AC software and background subtracted. Exposure time was uniform for all sections, and quantification of immunofluorescence was carried out using ImageJ software (National Institutes of Health) by measuring the fluorescence intensity per area of defined glomeruli (6–16 glomeruli/mouse). Skin sections were blindly graded on a 0–4 scale for lymphocytic infiltration, basement membrane changes, and follicle loss.
Proteinuria and serology
Urine was collected at 24 wk and proteinuria measured using a Bio-Rad protein assay (Bio-Rad). Cryoglobulins were precipitated at 4°C for 3 d, washed five times with cold PBS, and resuspended in 4 M urea prior to ELISA. Serum ANAs were detected on Hep2 slides (MBL Bion) at a 1:100 dilution using Alexa Fluor 488–conjugated anti-mouse IgG (Invitrogen) and scored on a 0–4 scale (8). ELISAs, performed as previously described (8), used capture and detection Abs purchased from Southern Biotechnology Associates, Jackson ImmunoResearch Laboratories, or Caltag Laboratories and Ags from Sigma-Aldrich (cardiolipin and calf thymus chromatin), SurModics (β2-GP1 and MPO), and Inova Diagnostics (Sm and RNP). Standard curves were generated using mouse reference sera (Bethyl Laboratories). For the detection of cross-reactive Abs, we used a competition ELISA (32) with 75 μg/ml chromatin as the competitor. IgG anti-RBCs were detected by direct Coomb test and flow cytometry as previously described (33).
ELISPOT
Splenocytes starting at 5 × 105 cells/ml were added at 2-fold serial dilutions to anti-IgG2aa–coated (clone 8.3; BD Pharmingen) Multiscreen-HTS IP plates (Millipore). After 18 h at 37°C, cells were lysed, and Ab-secreting cells detected using HRP-conjugated polyclonal goat anti-IgG2a (Southern Biotechnology Associates) and 3-amino-9-ethylcarbazole substrate.
Flow cytometry
Isolated splenocytes were blocked with anti-CD16/CD32 and stained with combinations of dye-conjugated Abs to CD90 (G7), CD11c (N418), H2-IA/ IE (M5/114.15.2), CD19 (6D5), CD138 (281-2), CD21 (7G6), CD23 (B3B4), CD4 (GK1.5), CD3 (145-2C11), CD40 (1C10), CD93 (AA4.1), CD43 (S7), IgD (11-26C.2a), IgM (RMM-1), B220 (RA3-6B2), Ter-119 (Ter-119), Gr-1 (RB6-8C5), CD11b (M1/70), Ly5a (A20), Ly5b (104), pDC Ag-1 (PDCA-1; ebio-927), F4/80 (BM8), CD86 (GL-1), Il-4 (11B-11), IFN-γ (XMG1.2), Il-17a (TC11-18H10.1), CD25 (PC-61), CD44 (IM-7), CD62L (MEL-14), and CD69 (H1-2F3) (BD Biosciences or BioLegend). Gating strategy: CD4+ T cells (CD90+CD4+), bone marrow B cells (CD3− Ter119−Gr-1−CD11b−B220+CD43+/−IgM+/−IgD+/−), marginal zone (MZ) B cells (CD19+CD138−CD21HiCD23−), follicular (FO) B cells (CD19+ CD138-CD21+CD23+), age-related B cells (CD19+CD138−CD11c+), plasma cells (PCs; CD19−CD138+), cDCs (CD90−CD19−CD11c+PDCA-1−), and pDCs (CD90−CD19−PDCA-1+). Data were acquired on an LSRII (BD Biosciences) and analyzed by FlowJo (Tree Star).
Bone marrow chimeras
B6-Faslpr or B6-Faslpr3d recipient mice were lethally irradiated at 1100 cGy and injected i.v. with 2 × 106 1:1 mixed bone marrow cells with either the combination of young (<3 mo old) WT Ly5a and 3d Ly5b or WT IgHa and 3d IgHb donors all of B6-Faslpr background. Mice were assessed by comparing allotypic serum Ab levels and cell populations in lymphoid organs 2–7 mo after bone marrow transfer. Differences in WT/3d stem cell ratios among individual recipients were corrected using the formula: normalized proportion (%) = 50 + (50 × (an − aT)/b), where an is the percent of WT or 3d in a cell population in mouse n (any individual mouse), aT is the percent of WT or 3d in the T cell population in mouse n, and b is (100 − aT) if an>aT or aT if an<aT. Normalized proportions using aT as the mean of thymic T cell populations, double-positive populations, or splenic T cells were concordant.
Statistical analysis
Unpaired or paired Student t test with Welch correction if unequal variance, log rank, and Mann–Whitney U test were used to compare groups. The p values <0.05 were considered significant.
Results
NA-TLRs are critical for some, but not all, lupus pathology in MRL- Faslpr mice
We previously demonstrated the significance of NA-TLRs in B6-Faslpr and BXSB mice (8). In this study, we examined the role of NA-TLRs on a wider spectrum of autoantibody specificities and additional disease manifestations by backcrossing the 3d mutation onto MRL-Faslpr mice. Strikingly in 3d mutants, proteinuria (Fig. 1A) was significantly reduced to essentially normal levels, and GN was minimal (Fig. 1A, 1B). Only faint areas of IgG glomerular deposits were present in 3d mice compared with intense deposits in Unc93b1WT/Het (WT/Het) littermate controls (Fig. 1C). Accordingly, absence of NA-TLR signaling dramatically prolonged survival, with all 3d mice surviving at 7 mo compared with only 8% of WT/Het controls (Fig. 1D). Thus, NA-TLRs are required for the development of GN.
FIGURE 1.

NA-TLRs are required for development of lupus nephritis in MRL-Faslpr mice. (A) Protein concentration in urine (left panel) and GN scores (right panel) from 7- to 8-mo-old 3d and WT/Het MRL-Faslpr. Urine protein in female B6 mice was 2.5 ± 0.1 mg/ml (n = 4). (B) Representative periodic acid-Schiff–stained kidney sections of 7- to 8-mo-old WT/Het and 3d mice. Glomerular enlargement, deposits (indicated by arrow), and proliferative changes in the mesangium of WT, but not in 3d kidneys, can be observed. (C) IgG immunofluorescence staining and quantification of frozen kidney sections. Fluorescent intensity was normalized to glomerular area. Seven- to 8-mo-old WT/Het and 3d (6–13 mice/group). (D) Kaplan-Meier survival plot of F10 WT and 3d MRL-Faslpr mice (6–11 mice/group). Data representative of three independent experiments for (A)–(C). Survival study was carried out once. +p < 0.05, ++ p < 0.01, +++p < 0.001.
In contrast, the incidence and severity of cutaneous lupus were not affected in 3d mutants, all of which developed skin disease by 7 mo with lesions similar to WT/Het MRL-Faslpr mice (Fig. 2A, 2B); immune infiltration, disruption, or thickening of the basement membrane and follicle loss were also similar between the two groups (Fig. 2C). Consistent with this, IgA anti-desmoglein 3 (Dsg3) levels, previously reported to correlate with skin disease in MRL-Faslpr mice (34), were not different (Fig. 2E), whereas IgG3 cryoglobulins, reported to be associated with skin disease in this model (35), were significantly reduced in 3d mice (Fig. 2F).
FIGURE 2.

Lupus-associated dermatitis in MRL- Faslpr mice is not significantly affected by absence of NA-TLRs. (A) Incidence of dermatitis in mice over time (9–11 mice/group). (B) Surface area of skin lesions at 7 mo (8–18 mice/group). (C) Blinded scores of immune infiltration, basement membrane changes, and follicle loss in skin sections (9–15 mice/ group). (D) Representative H&E sections of skin showing similar epidermal thickening and hyperkeratosis in 7- to 8-mo-old WT/Het and 3d skin compared with B6. IgA anti-Dsg3 levels (E) and IgG3 cryoglobulin levels (F) from 6-mo-old mice. Data representative of three independent experiments. +++p < 0.001.
Effect of NA-TLR signaling on hypergammaglobulinemia and ANAs in MRL-Faslpr mice
IgM concentrations were significantly lower in 3d mice compared with WT/Het littermate controls, reaching levels similar to non-autoimmune B6 mice (Fig. 3). Total IgG, although still higher than in B6 mice, was also significantly reduced in the 3d group due to lower concentrations of all IgG subclasses except for IgG1. Absence of NA-TLR signaling did not affect IgA concentrations, and IgE was higher in 3d mice compared with WT/Het littermate controls (Fig. 3).
FIGURE 3.
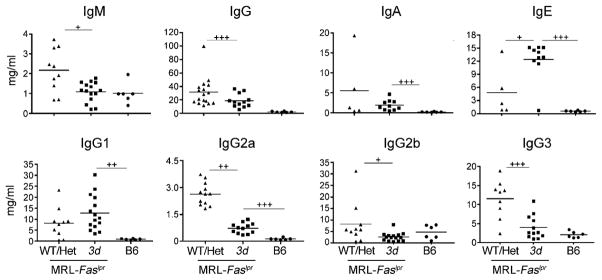
Total isotype and subclass Ig concentrations in 3d MRL-Faslpr mice. Ig isotype and IgG subclass levels in WT/Het and 3d MRL-Faslpr mice and B6 controls at 6 mo (6–17 mice/group). One of three independent experiments is shown. +p < 0.05, ++ p < 0.01, +++p < 0.001; p < 0.05 for WT/Het versus B6 for all Ig isotypes and IgG subclasses except IgG2b.
3d mice had significantly lower ANA scores, indicative of reduced autoantibodies to nuclear Ags (Fig. 4B). IgM and IgG autoantibodies to nuclear Ags, including chromatin, RNP, and Sm, were also uniformly reduced to the same level as B6 controls (Fig. 4C). Likewise, IgM rheumatoid factors (RFs) were almost undetectable (Fig. 4C). These findings underscore the central role of NA-TLRs in the production of autoantibodies to nuclear Ags.
FIGURE 4.
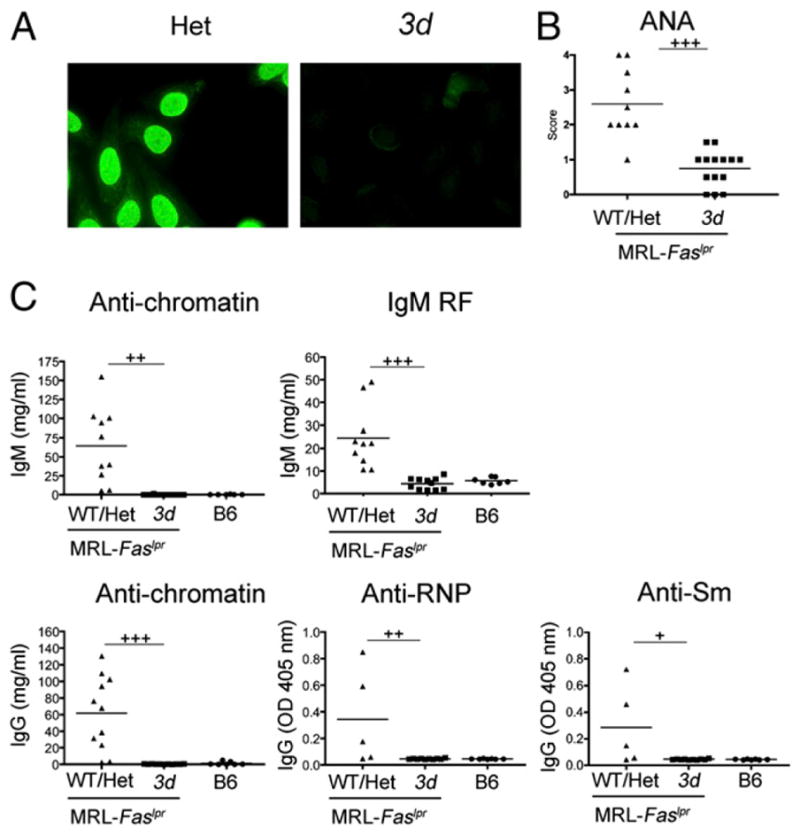
ANA specificities and IgM RF in MRL-Faslpr 3d mice. (A) Representative ANA staining from 6-mo-old Het and 3d MRL-Faslpr mice. (B) ANA scores for WT/Het and 3d MRL-Faslpr mice (10–14 mice/group). (C) IgM RF and IgM anti-chromatin (top panel) and IgG anti-chromatin, IgG anti-RNP, and IgG anti-Sm (bottom panel). Each symbol represents an individual mouse. Data representative of at least two independent experiments. +p < 0.05, ++p < 0.01, +++p < 0.001; p < 0.05 for WT/Het versus B6 for all autoantibodies in (C).
Effects of NA-TLRs on diverse autoantibody specificities in MRL-Faslpr mice
Next, to determine if NA-TLRs play a common role in Ab production in SLE and possibly other systemic autoimmune diseases, we examined the effect of NA-TLR deficiency on a wider spectrum of Ab specificities, specifically assessing responses to MPO, cardiolipin, β2-GP1, and Dsg3. Sera from MRL-Faslpr background mice showed greatly reduced IgM and IgG autoantibodies to cardiolipin, MPO, and β2-GP1 in 3d mutants compared with the WT/Het group but, in contrast to Abs to the nuclear Ags, levels of IgM anti–β2-GP1 and all IgG autoantibodies with these specificities remained significantly higher in 3d mutants than in B6 mice (Fig. 5A). IgG anti-Dsg3, similar to IgA anti-Dsg3, was unaffected by the absence of NA-TLR signaling.
FIGURE 5.

Nonnuclear autoantibody levels in 3d mice. (A) IgM anti-cardiolipin, –β2-GP1, and -MPO (top panel) and IgG anti-cardiolipin, –β2-GP1, -MPO, and -Dsg3 (bottom panel) in 6- to 7-mo-old WT/Het MRL-Faslpr, 3d MRL-Faslpr, and B6 mice. p < 0.01 for WT/Het versus B6 for all IgM and IgG autoantibodies. (B) Anti-RBC and splenomegaly in NZB WT/ Het and 3d mice. IgG direct Coomb test (n = 11–20 for WT/Het, 4–7 for 3d; p < 0.0001; left panel). Mean fluorescence intensities of IgG anti-RBCs (middle panel). Spleen weights from 10-mo-old mice (right panel). (C) Chromatin competition ELISA. Diluted sera from 6- to 7-mo-old WT/Het or 3d with IgG anti-cardiolipin, anti-MPO, or anti-chromatin specificity were added to ELISA plate wells with or without chromatin (6–17 mice/group). One of three independent experiments is shown. +p < 0.05, ++p < 0.01, +++p < 0.001.
To determine the effects of NA-TLRs on anti-RBC Abs, we generated and examined autoimmune hemolytic anemiaprone NZB mice congenic for the 3d mutation. Similar to other lupus-related nonnuclear autoantibodies, IgG anti-RBC production was partially, but significantly, inhibited in the NZB mice homozygous for 3d (Fig. 5B). Thus, although >50% of NZB 3d mice were Coomb positive by 10 mo, levels of IgG anti-RBCs and splenomegaly, even among the Coomb-positive mice, remained lower than in WT mice. Taken together, our data indicate that NA-TLR–mediated induction and/or amplification of autoantibodies is a common mechanism for most specificities in lupus, but the degree of dependence on NA-TLRs varies according to specificity.
Although evidence suggests that NA-TLR–dependent auto-antibodies in lupus largely develop because they can bind to NA-containing material (36), certain specificities such as anti-cardiolipin might also be induced indirectly because of known cross-reactivity (37). To assess the potential for this latter mechanism, we used a competitive ELISA to assess the relative amount of IgG anti-cardiolipin or anti-MPO in the sera of WT/Het (NA-TLR dependent) or 3d (NA-TLR independent) MRL-Faslpr mice that cross-reacted with chromatin. Indeed, anti-cardiolipin levels in WT/Het mice, but not 3d mutants, were significantly reduced in the presence of chromatin, whereas levels of anti-MPO, which does not exhibit cross reactivity with NAs, were not significantly affected by the addition of chromatin in either WT/Het or 3d sera (Fig. 5C).
NA-TLR signaling promotes B cell and DC activation in lupus
To investigate the role of NA-TLRs on immune cells in lupus, we first compared spleen cell populations in 3d and WT MRL-Faslpr mice. Notably, the development of splenomegaly in 3d mutants was delayed, but eventually became as severe as in the WT/Het group, suggesting the expansion of splenocytes was primarily dependent on Fas deficiency (Fig. 6A and not shown). Indeed, other than a reduction in the DN (CD4−CD8−) T cell subset, the numbers and percentages of most spleen cell populations, including PCs, B cells, CD4+ and CD8+ T cells, and DCs, were similar in the two groups (Fig. 6B). There was also no significant skewing of CD4+ T cells that secrete IFN-γ, IL-4, or IL-17 (Fig. 6C). However, although the total number of B (CD19+) cells were unaffected in 3d mice, there were significant shifts in the proportion of B cell subsets with a marked increase in the FO (CD19+ CD138−CD21−CD23+) subset and a major decrease in the recently described age-associated B cell (ABCs; CD19+CD138− CD21−CD23−CD11c+) (38, 39) population (Fig. 6D, 6E). There was also a lower percentage of cDCs (CD11c+) expressing high levels of CD40 in 3d mice consistent with reduced activation in the absence of NA-TLR signaling, although no differences in MHC class II (MHC II) or CD86 expression were detected (Fig. 6F and not shown). Likewise, levels of the activation-associated CD69 and the activation- and regulatory T cell–associated CD25 in WT and 3d mice were similar in CD4+ and CD8+ T cells, as was expression of CD86, CD40, and MHC II on F4/80+ macrophages (not shown). These results suggest that NA-TLRs primarily affect certain B cell populations, particularly the ABC subset, and have a modest effect on the activation of cDCs.
FIGURE 6.

The effect of NA-TLR deficiency on immune cells in lupus mice. (A) Spleen weights and splenocyte numbers of WT/Het and 3d mice (11–19 mice/group). (B) Percentages and numbers of major spleen cell populations in MRL-Faslpr WT/Het (gray bars) and 3d mice (white bars). (C) Percentages and numbers of IFN-γ+, IL-4+, or IL-17+ CD4+ T cells. (D) Percentages and numbers of splenic B cell subsets: FO, MZ, and ABCs. (E) Representative flow cytometry plots of splenic CD19+CD138− B cell subsets defined by CD21 and CD23 in MRL-Faslpr WT and 3d mice at the indicated ages. ABC population is indicated by arrow to demonstrate age-related increase of this subset in WT mice. (F) Percentage of CD40hi DCs in WT/Het or 3d MRL-Faslpr mice. Data from 7-mo-old mice. For (B)–(F), four to seven mice per group. Mean ± SEMs are shown. Data representative of three experiments. +p < 0.05, ++p < 0.01, +++p < 0.001.
Cell-intrinsic effects of NA-TLR signaling
To more accurately parse the cell-intrinsic effects of NA-TLR signaling from secondary immunostimulation, we generated mixed bone marrow chimeras from 3d (Ly5b allotype) and WT (Ly5a allotype) B6-Faslpr mice and examined them at 2–6 mo posttransfer (Fig. 7). As expected, because T cells do not express NA-TLRs, the 3d mutation had no effect on the proportions of T cell subsets in the thymus (data not shown). In the bone marrow at 2 and 3 mo, the 3d mutation had little effect on Pro- (CD43+ B220lo) or Pre- (CD43− B220lo) B cell subsets; however, NA-TLR signaling gave WT cells a significant competitive advantage over 3d cells in the immature subset (B220hiIgM+IgD−), as reflected by similar differences in the mature (B220hiIgM+IgD+) B cell populations (Fig. 7A). In the spleen, greater expansion of total WT B cells affecting all subsets, including ABC, MZ, and FO, was evident at 3 and 6 mo postreconstitution (Fig. 7A). Strikingly, significant increases in the ABC and MZ WT populations were already present by 2 mo. Increases in WT PCs were also detected, although later than for WT B cells, at 3 mo in the bone marrow and at 6 mo in the spleen. Remarkably, NA-TLR–mediated expansion in the spleen could be estimated based on the difference in proportions of WT and 3d cells to constitute a striking 35% of the entire PC response. Relative expansion of WT cDCs was detected at 2 mo, but no further expansion was observed at later time points (Fig. 7A), and no significant differences in activation markers (CD86, MHC II, and CD40) were observed at any time point (data not shown). The WT pDC population exhibited a late downward shift at 6 mo (Fig. 7A), possibly reflective of greater activation and turnover (40).
FIGURE 7.

Cell-intrinsic NA-TLR signaling effects on immune cell subsets and autoantibodies in lupus. (A) Proportion (%) of WT cell populations in the bone marrow (BM) and spleen in 1:1 mixed bone marrow chimeras of WT (Ly5a):3d (Ly5b) B6-Faslpr donors at 2 (white bars) and 3 (gray bars) mo after reconstitution. Normalized percentage (see Materials and Methods) of Ly5a+ WT cells are shown. Mean ± SEM with error bars for line graphs in only one direction to avoid overlap. For spleen B cell subsets, p < 0.05–0.001 for all points except PCs at 2 and 3 mo and FO B cells at 2 mo. (B) Allotype-specific IgM RF (anti-IgG1), total IgM in 1:1 mixed bone marrow chimeras of 7-mo-old WT (IgHa):3d (IgHb) B6-Faslpr donors. IgG2a anti-chromatin, total IgG2a serum concentrations, and IgG2a ANA scores in 1:1 mixed bone marrow chimeras of 4.5-mo-old WT (IgHa):3d (IgHb) B6-Faslpr donors. Five to six mice per group. Similar data were obtained from WT and 3d recipients, and results represent combined data from both types of recipients. Data representative of two independent experiments. +p < 0.05, ++p < 0.01, +++p < 0.001.
To further demonstrate the significance of B cell–intrinsic NA-TLR signaling, we generated mixed bone marrow chimeras of B6-Faslpr mice that were either 3d/IgHb or WT/IgHa and measured allotype-specific IgM RF, anti-IgG2a anti-chromatin, and IgG2a ANA responses (Fig. 7B). Strikingly, autoantibodies were derived almost exclusively from WT donor cells, with minimal levels of IgMb RF and anti-chromatin IgG2ab, despite similar allotype total IgM and IgG2ab. IgG2a ANA scores also indicated that virtually all nuclear autoantibodies were from WT donor cells; IgG2aa (WT) ANA was positive in 9 out of 12, whereas IgG2ab (3d) ANA was positive in 1 out of 12 chimeras (p < 0.0003). These findings definitively underscore the central role of B cell–intrinsic NA-TLR signaling in loss of tolerance and production of autoanti-bodies.
Discussion
In this study, we used the 3d mutation that selectively abolishes NA-TLR signaling to probe the specific role of NA-TLRs in lupus pathogenesis and document two main findings. First, NA-TLRs are critical for the production of all pathogenic autoantibody specificities in the MRL-Faslpr and NZB lupus models affecting both nuclear and nonnuclear self-Ags. Second, B cell–intrinsic NA-TLR engagement is required for optimal production of auto-antibodies in B6-Faslpr mice. These and other results provide new insights into the disease-shaping role of NA-TLRs in SLE and have implications for treatment.
Although inferred from previous studies, our findings in MRL-Faslpr and NZB mice now document that autoantibody specificities associated with autoimmune pathology, including ANAs, anti-MPO, anti–β2-GP1, anti-cardiolipin, and anti-RBCs, are highly dependent on NA-TLRs, providing direct evidence for a unifying pathogenic basis for autoantibody production in lupus and possibly other autoimmune diseases such as p-antineutrophil cytoplasmic antibody–associated vasculitides, anti–phospholipid syndrome, and autoimmune hemolytic anemia. These findings suggest that most autoantibodies associated with lupus bind to NAs or antigenic cargoes containing a sufficient amount of NAs to stimulate NA-TLRs upon endolysosome uptake (1). Indeed, MPO binds to DNA in neutrophil extracellular traps, and both cardiolipin and β2-GP1 can be detected on the surface of apoptotic cells; Dsg3, however, for which autoantibodies were not dependent on NA-TLRs, is not associated with NAs (41–44). Our findings also support the possibility that cross reactivity to nuclear material might enhance the production of some autoantibody specificities, such as anti-cardiolipin. Furthermore, because circulating RBCs have extruded both nuclei and mitochondria and therefore lack DNA, the dependence of anti-RBC on NA-TLRs implies that cellular RNA, which in RBCs includes both ribosomal and mRNA, is sufficient to activate NA-TLRs and induce auto-antibodies to surface Ags that do not themselves directly bind to NAs. It is also possible that cytoplasmic membrane–associated DNA attached to the plasma membrane could contribute to NA-TLR stimulation if present in mature RBCs (45). Abs to mouse endogenous retroviral gp70 proteins are associated with the development of lupus and are dependent on NA-TLRs, specifically TLR7 (46), similar to our findings with NA–associated SLE autoantibodies. In this case, because gp70 does not bind to NAs, it was postulated that in lupus strains, the generation of replication-competent mouse polytrophic retroviruses that contain RNA might be the triggering factor for the anti-gp70. Additionally, in B6. Nba2 TLR9-deficient congenic mice that develop more severe disease than the lupus-prone B6. Nba2 congenic because of increased TLR7 signaling, the additional deletion of TLR7 reduced anti-DNA to levels below B6. Nba2 mice, and also, to a lesser extent, other autoantibodies, including anti-histones, anti-RNA–related Ags, anti-glomerular Ags, and anti-cytochrome, to the same levels found in B6. Nba2 mice (13). These findings also support the dependence of nonnuclear autoantibody production in lupus on NA-TLRs.
Another interesting observation was that lupus autoantibodies in our study fell into three general groups based on the requirement for NA-TLRs. The first group, which primarily includes autoantibodies to nuclear Ags, was exquisitely dependent on NA-TLRs with the same levels in 3d as in nonautoimmune B6 mice. Included in this category are IgM RFs, which, by recognizing Fc regions of ANAs, were previously shown to bind indirectly to NA-containing immune complexes (36). The second group, which includes auto-antibodies to diverse nonnuclear targets such as MPO, RBCs, β2-GP1, and cardiolipin, was greatly amplified by NA-TLRs, but not completely dependent, as the 3d mutant MRL-Faslpr mice had significantly higher concentrations of these autoantibodies than B6 mice. Finally, the third group represented by anti-Dsg3 was not affected by the absence of NA-TLRs. Taken together, the data suggest NA-TLRs might be critical for both breaking tolerance and amplifying B cell responses to nuclear Ags, whereas loss of tolerance to nonnuclear Ags is more likely to be independent of NA-TLR signaling. The latter is consistent with previous congenic mouse studies showing that increased TLR7 signaling caused by the Yaa mutation induced ANAs and magnified other existing autoantibody specificities in various lupus-prone and non-autoimmune strains, but did not give rise to new pathologic autoantibody specificities (2). Possible reasons for the differences in requirements for NA-TLRs in tolerance might be related to the amount or accessibility of NAs in antigenic targets and the need for additional genetic or environmental factors.
Lack of NA-TLRs prevented the development of significant GN in MRL-Faslpr mice similar to MyD88-deficient or TLR7/TLR9–double-deficient MRL-Faslpr mice (7). Nonetheless, we detected low levels of glomerular IgG deposits, possibly related to the residual low levels of nonnuclear autoantibodies and the presence of IgG3 cryoglobulins. Potentially, NA-TLR deficiency might prevent GN by reducing pathogenic autoantibody production and by locally impairing effector cell recruitment/activation in kidneys (1, 47).
In contrast to the reduction in GN, we detected no effect of NA-TLRs on cutaneous disease despite detailed examination of clinical and pathological features. This is similar to findings in TLR7/ TLR9–double-deficient MRL-Faslpr mice, but contrasts with those in MyD88-deficient MRL-Faslpr mice, which exhibited a reduction in incidence presumably because of additional signaling pathways mediated by MyD88 (9, 48). Although previous studies have associated IgG3 cryoglobulinemia and RFs with skin disease in the MRL-Faslpr mice (35), our finding of unabated cutaneous disease in 3d mice despite reduced cryoglobulins and RF suggests that NA-TLR–dependent cryoglobulin specificities or cryoglobulins themselves are not required. In contrast, cutaneous lupus induced by tape stripping in (NZB × NZW)F1, which is both dependent on pDCs and associated with a persistent type I IFN signature, was curtailed by treatment with a bifunctional TLR7/9 inhibitor (49). We also found no effect of the 3d mutation on the production of IgA anti-Dsg3, consistent with the presence of skin disease. It should be mentioned that although anti-Dsg3 Abs were detectable at significantly higher concentrations than in nonautoimmune mice or mice without skin disease, there was no evidence of a pemphigus vulgaris–like bullous disease caused by pathologic Abs to Dsg3 (44). Thus, the anti-Dsg3 associated with cutaneous lupus in the MRL-Faslpr model might be a consequence of epitope spreading secondary to skin inflammation rather than being pathogenic.
Our findings clearly identify B cells as the immune cell type most affected by NA-TLRs in the Faslpr model. This was shown in mixed BM chimeras in which early and sustained changes in WT B cell subsets preceded detectable shifts in WT pDCs, and only transient early expansions in WT cDCs were detected. Furthermore, based on the absence of IgG2a anti-chromatin, greatly reduced incidence of ANAs, and only marginal IgM RF production by 3d B cells in chimeric mice despite substantial production by WT B cells, we conclude that intrinsic NA-TLR engagement in B cells has a major influence on the production of autoantibodies to lupus Ags found in complex with NA material. This broad principle is supported by recent studies showing that normalization of TLR7 in B cells in a TLR7 transgenic model of lupus reduces some, but not all, RNA-associated autoantibodies (6) and the requirement of TLR9 expression in B cells for anti-chromatin autoantibodies (27). These studies, however, are limited to ANAs and, although likely, the extent to which this conclusion applies to Abs to nonnuclear Ags remains to be documented. Interestingly, in our chimeras, the effects of NA-TLR expression on B cells were already detectable at the immature B cell stage, suggesting that shaping of the autoimmune repertoire occurs even prior to full maturation. We also could estimate that a remarkably large (~20–30%) proportion of the PC population in the spleen and bone marrow were induced by NA-TLRs.
Another notable finding in the mixed bone marrow chimeras was a significant reduction in NA-TLR–expressing (WT) pDCs in the spleen, consistent with greater activation of these cells (40) and supporting recent findings demonstrating that pDCs are essential for lupus pathogenesis (50). WT cDCs were increased compared with 3d at the earliest 2 mo time point, but not at later time points despite presumably higher levels of immune complexes and previous evidence that cDCs can be activated by NA-containing immune complexes (51, 52). Moreover, we did not find significant differences in the activation profiles of WT and 3d cDCs in the mixed bone marrow chimeras, although increased activation of cDCs in WT compared with 3d MRL-Faslpr mice was detected. Taken together, this suggests a more limited role for NA-containing immune complexes in the activation of cDCs later in disease, which could be also mediated by type I and type II IFNs or by TLR2-mediated stimulation of cDCs by HMGB1-containing nucleosomes from apoptotic cells (53).
Previous models of autoantibody pathogenesis in lupus have proposed a multifaceted process that includes the initial activation of self-reactive B cells and the subsequent activation of pDCs and cDCs by immune complexes containing NAs, resulting in a self-perpetuating amplification of the autoimmune response (1). Our data support this overall scheme, but indicate the presence of NA-TLRs in B cells is necessary to drive the initial autoimmune response and to promote the activation and escape of tolerance of self-reactive B cells recognizing a broad range of NA-containing materials. We were also able to detect modest changes in cDCs and pDCs associated with intrinsic expression of NA-TLRs during the development of lupus, but not major shifts in activation, as might be anticipated. Nevertheless, deletion of CD11c+ cells, including cDCs, pDCs, Langerhan’s cells, and possibly ABC B cells, was shown to substantially reduce lymphocyte expansion and glomerulonephritis in MRL-Faslpr mice (54), indicating the importance of one or a combination of these cell types in disease pathogenesis. Further studies will be needed to determine the extent to which NA-TLR expression in these cell populations is required.
The initial characterization of the 3d mutation revealed a partial, but definite, defect in Ag presentation that might have influenced our results (28). However, we previously found that 3d did not reduce T-dependent humoral immune responses in mice immunized with NA-free adjuvant (8). Moreover, UNC93B1-deficient cell lines showed no defects in MHC class I or MHC II molecules (29) or class II Ag presentation (55), there was no defect in CD8 T cell responses in UNC93B1-deficient mice infected with murine CMV (56), and in vitro activation of CD8 T cells by WT and 3d APCs was reportedly equivalent (57). Thus, it seems unlikely that the partial reduction in Ag presentation in 3d mice is responsible for our findings. The 3d mutation also blocks trafficking of several other endosomal TLRs, including TLR8, for which the natural ligand in mice is not known (58), TLR11 and 12, which recognize profilin-like proteins from Toxoplasma gondii (59, 60), and TLR13, which recognizes bacterial rRNA (61, 62). TLR8 deficiency leads to lupus-like autoimmunity due to compensatory increased expression of TLR7 (12), and TLR11–13 do not recognize mammalian NAs and are not expressed in humans. Thus, it is unlikely that the lack of signaling from these endosomal TLRs accounts for our findings.
Finally, our report that NA-TLRs are required for all the examined autoantibodies associated with lupus pathology and that NA-TLR expression in B cells is essential suggests that specific inhibition of NA-TLRs in B cells would be sufficient to treat SLE. Furthermore, combined with our previous finding of essentially normal humoral response to nonnuclear Ags in NA-TLR–deficient 3d mice (8) and the fact that selective blocking of NA-TLRs in B cells would spare the innate immune system, this approach could provide a step toward more specific treatment of autoimmune disease with less compromise of the overall immune response necessary for combating pathogens and malignancy.
Acknowledgments
This work was supported by National Institutes of Health Research Grants AR53731, AR39555, and ES14847; National Institutes of Health Training Grant T32 A1007244-26; and Arthritis Foundation Fellowship Grant 5479. This is publication 21907-IMM from the Department of Immunology and Microbial Science, The Scripps Research Institute.
We thank Dr. D. Barnette for analyzing skin pathology and M. Kat Occhipinti for editorial assistance.
Abbreviations used in this article
- ABC
age-associated B cell
- ANA
anti-nuclear Ab
- cDC
conventional dendritic cell
- Dsg3
desmoglein 3
- FO
follicular
- GN
glomerulonephritis
- MPO
myeloperoxidase
- MZ
marginal zone
- NA
nucleic acid
- NA-TLR
nucleic acid–sensing TLR
- PC
plasma cell
- pDC
plasmacytoid dendritic cell
- PDCA-1
plasmacytoid dendritic cell Ag-1
- RF
rheumatoid factor
- RNP
ribonucleoprotein
- SLE
systemic lupus erythematosus
- WT
wild-type
Footnotes
Disclosures
The authors have no financial conflicts of interest.
References
- 1.Theofilopoulos AN, Gonzalez-Quintial R, Lawson BR, Koh YT, Stern ME, Kono DH, Beutler B, Baccala R. Sensors of the innate immune system: their link to rheumatic diseases. Nat Rev Rheumatol. 2010;6:146–156. doi: 10.1038/nrrheum.2009.278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Izui S, Ibnou-Zekri N, Fossati-Jimack L, Iwamoto M. Lessons from BXSB and related mouse models. Int Rev Immunol. 2000;19:447–472. doi: 10.3109/08830180009055507. [DOI] [PubMed] [Google Scholar]
- 3.Pisitkun P, Deane JA, Difilippantonio MJ, Tarasenko T, Satterthwaite AB, Bolland S. Autoreactive B cell responses to RNA-related antigens due to TLR7 gene duplication. Science. 2006;312:1669–1672. doi: 10.1126/science.1124978. [DOI] [PubMed] [Google Scholar]
- 4.Deane JA, Pisitkun P, Barrett RS, Feigenbaum L, Town T, Ward JM, Flavell RA, Bolland S. Control of toll-like receptor 7 expression is essential to restrict autoimmunity and dendritic cell proliferation. Immunity. 2007;27:801–810. doi: 10.1016/j.immuni.2007.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Subramanian S, Tus K, Li QZ, Wang A, Tian XH, Zhou J, Liang C, Bartov G, McDaniel LD, Zhou XJ, et al. A Tlr7 translocation accelerates systemic autoimmunity in murine lupus. Proc Natl Acad Sci USA. 2006;103:9970–9975. doi: 10.1073/pnas.0603912103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hwang SH, Lee H, Yamamoto M, Jones LA, Dayalan J, Hopkins R, Zhou XJ, Yarovinsky F, Connolly JE, Curotto de Lafaille MA, et al. B cell TLR7 expression drives anti-RNA autoantibody production and exacerbates disease in systemic lupus erythematosus-prone mice. J Immunol. 2012;189:5786–5796. doi: 10.4049/jimmunol.1202195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Christensen SR, Shupe J, Nickerson K, Kashgarian M, Flavell RA, Shlomchik MJ. Toll-like receptor 7 and TLR9 dictate autoantibody specificity and have opposing inflammatory and regulatory roles in a murine model of lupus. Immunity. 2006;25:417–428. doi: 10.1016/j.immuni.2006.07.013. [DOI] [PubMed] [Google Scholar]
- 8.Kono DH, Haraldsson MK, Lawson BR, Pollard KM, Koh YT, Du X, Arnold CN, Baccala R, Silverman GJ, Beutler BA, Theofilopoulos AN. Endosomal TLR signaling is required for anti-nucleic acid and rheumatoid factor autoantibodies in lupus. Proc Natl Acad Sci USA. 2009;106:12061–12066. doi: 10.1073/pnas.0905441106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Nickerson KM, Christensen SR, Shupe J, Kashgarian M, Kim D, Elkon K, Shlomchik MJ. TLR9 regulates TLR7- and MyD88-dependent autoantibody production and disease in a murine model of lupus. J Immunol. 2010;184:1840–1848. doi: 10.4049/jimmunol.0902592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Barrat FJ, Meeker T, Chan JH, Guiducci C, Coffman RL. Treatment of lupus-prone mice with a dual inhibitor of TLR7 and TLR9 leads to reduction of autoantibody production and amelioration of disease symptoms. Eur J Immunol. 2007;37:3582–3586. doi: 10.1002/eji.200737815. [DOI] [PubMed] [Google Scholar]
- 11.Pawar RD, Ramanjaneyulu A, Kulkarni OP, Lech M, Segerer S, Anders HJ. Inhibition of Toll-like receptor-7 (TLR-7) or TLR-7 plus TLR-9 attenuates glomerulonephritis and lung injury in experimental lupus. J Am Soc Nephrol. 2007;18:1721–1731. doi: 10.1681/ASN.2006101162. [DOI] [PubMed] [Google Scholar]
- 12.Demaria O, Pagni PP, Traub S, de Gassart A, Branzk N, Murphy AJ, Valenzuela DM, Yancopoulos GD, Flavell RA, Alexopoulou L. TLR8 deficiency leads to autoimmunity in mice. J Clin Invest. 2010;120:3651–3662. doi: 10.1172/JCI42081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Santiago-Raber ML, Dunand-Sauthier I, Wu T, Li QZ, Uematsu S, Akira S, Reith W, Mohan C, Kotzin BL, Izui S. Critical role of TLR7 in the acceleration of systemic lupus erythematosus in TLR9-deficient mice. J Autoimmun. 2010;34:339–348. doi: 10.1016/j.jaut.2009.11.001. [DOI] [PubMed] [Google Scholar]
- 14.Christensen SR, Kashgarian M, Alexopoulou L, Flavell RA, Akira S, Shlomchik MJ. Toll-like receptor 9 controls anti-DNA autoantibody production in murine lupus. J Exp Med. 2005;202:321–331. doi: 10.1084/jem.20050338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Petri M. Update on anti-phospholipid antibodies in SLE: the Hopkins’ Lupus Cohort. Lupus. 2010;19:419–423. doi: 10.1177/0961203309360541. [DOI] [PubMed] [Google Scholar]
- 16.Sherer Y, Gorstein A, Fritzler MJ, Shoenfeld Y. Autoantibody explosion in systemic lupus erythematosus: more than 100 different antibodies found in SLE patients. Semin Arthritis Rheum. 2004;34:501–537. doi: 10.1016/j.semarthrit.2004.07.002. [DOI] [PubMed] [Google Scholar]
- 17.Meroni PL, Borghi MO, Raschi E, Tedesco F. Pathogenesis of antiphospholipid syndrome: understanding the antibodies. Nat Rev Rheumatol. 2011;7:330–339. doi: 10.1038/nrrheum.2011.52. [DOI] [PubMed] [Google Scholar]
- 18.Chen M, Kallenberg CG. ANCA-associated vasculitides—advances in pathogenesis and treatment. Nat Rev Rheumatol. 2010;6:653–664. doi: 10.1038/nrrheum.2010.158. [DOI] [PubMed] [Google Scholar]
- 19.Leddy JP, Wilkinson SL, Kissel GE, Passador ST, Falany JL, Rosenfeld SI. Erythrocyte membrane proteins reactive with IgG (warm-reacting) anti-red blood cell autoantibodies: II. Antibodies coprecipitating band 3 and glycophorin A. Blood. 1994;84:650–656. [PubMed] [Google Scholar]
- 20.Hashimoto Y, Kawamura M, Ichikawa K, Suzuki T, Sumida T, Yoshida S, Matsuura E, Ikehara S, Koike T. Anticardiolipin antibodies in NZW x BXSB F1 mice. A model of antiphospholipid syndrome. J Immunol. 1992;149:1063–1068. [PubMed] [Google Scholar]
- 21.Moll T, Martinez-Soria E, Santiago-Raber ML, Amano H, Pihlgren-Bosch M, Marinkovic D, Izui S. Differential activation of anti-erythrocyte and anti-DNA autoreactive B lymphocytes by the Yaa mutation. J Immunol. 2005;174:702–709. doi: 10.4049/jimmunol.174.2.702. [DOI] [PubMed] [Google Scholar]
- 22.Garcia-Romo GS, Caielli S, Vega B, Connolly J, Allantaz F, Xu Z, Punaro M, Baisch J, Guiducci C, Coffman RL, et al. Netting neutrophils are major inducers of type I IFN production in pediatric systemic lupus erythematosus. Sci Transl Med. 2011;3:73ra20. doi: 10.1126/scitranslmed.3001201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Leadbetter EA, I, Rifkin R, Hohlbaum AM, Beaudette BC, Shlomchik MJ, Marshak-Rothstein A. Chromatin-IgG complexes activate B cells by dual engagement of IgM and Toll-like receptors. Nature. 2002;416:603–607. doi: 10.1038/416603a. [DOI] [PubMed] [Google Scholar]
- 24.Isnardi I, Ng YS, Srdanovic I, Motaghedi R, Rudchenko S, von Bernuth H, Zhang SY, Puel A, Jouanguy E, Picard C, et al. IRAK-4- and MyD88-dependent pathways are essential for the removal of developing autoreactive B cells in humans. Immunity. 2008;29:746–757. doi: 10.1016/j.immuni.2008.09.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chen C, Li H, Tian Q, Beardall M, Xu Y, Casanova N, Weigert M. Selection of anti-double-stranded DNA B cells in autoimmune MRL-lpr/ lpr mice. J Immunol. 2006;176:5183–5190. doi: 10.4049/jimmunol.176.9.5183. [DOI] [PubMed] [Google Scholar]
- 26.Shlomchik MJ. Sites and stages of autoreactive B cell activation and regulation. Immunity. 2008;28:18–28. doi: 10.1016/j.immuni.2007.12.004. [DOI] [PubMed] [Google Scholar]
- 27.Nickerson KM, Christensen SR, Cullen JL, Meng W, Luning Prak ET, Shlomchik MJ. TLR9 promotes tolerance by restricting survival of anergic anti-DNA B cells, yet is also required for their activation. J Immunol. 2013;190:1447–1456. doi: 10.4049/jimmunol.1202115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Tabeta K, Hoebe K, Janssen EM, Du X, Georgel P, Crozat K, Mudd S, Mann N, Sovath S, Goode J, et al. The Unc93b1 mutation 3d disrupts exogenous antigen presentation and signaling via Toll-like receptors 3, 7 and 9. Nat Immunol. 2006;7:156–164. doi: 10.1038/ni1297. [DOI] [PubMed] [Google Scholar]
- 29.Kim YM, Brinkmann MM, Paquet ME, Ploegh HL. UNC93B1 delivers nucleotide-sensing toll-like receptors to endolysosomes. Nature. 2008;452:234–238. doi: 10.1038/nature06726. [DOI] [PubMed] [Google Scholar]
- 30.Santiago-Raber ML, Haraldsson MK, Theofilopoulos AN, Kono DH. Characterization of reciprocal Lmb1-4 interval MRL-Faslpr and C57BL/6-Faslpr congenic mice reveals significant effects from Lmb3. J Immunol. 2007;178:8195–8202. doi: 10.4049/jimmunol.178.12.8195. [DOI] [PubMed] [Google Scholar]
- 31.Haraldsson MK, dela Paz NG, Kuan JG, Gilkeson GS, Theofilopoulos AN, Kono DH. Autoimmune alterations induced by the New Zealand Black Lbw2 locus in BWF1 mice. J Immunol. 2005;174:5065–5073. doi: 10.4049/jimmunol.174.8.5065. [DOI] [PubMed] [Google Scholar]
- 32.Aït-Azzouzene D, Kono DH, Gonzalez-Quintial R, McHeyzer-Williams LJ, Lim M, Wickramarachchi D, Gerdes T, Gavin AL, Skog P, McHeyzer-Williams MG, et al. Deletion of IgG-switched autoreactive B cells and defects in Fas(lpr) lupus mice. J Immunol. 2010;185:1015–1027. doi: 10.4049/jimmunol.1000698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Scatizzi JC, Haraldsson MK, Pollard KM, Theofilopoulos AN, Kono DH. The Lbw2 locus promotes autoimmune hemolytic anemia. J Immunol. 2012;188:3307–3314. doi: 10.4049/jimmunol.1103561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Nishimura H, Strominger JL. Involvement of a tissue-specific autoantibody in skin disorders of murine systemic lupus erythematosus and autoinflammatory diseases. Proc Natl Acad Sci USA. 2006;103:3292–3297. doi: 10.1073/pnas.0510756103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Izui S, Berney T, Shibata T, Fulpius T. IgG3 cryoglobulins in autoimmune MRL-lpr/lpr mice: immunopathogenesis, therapeutic approaches and relevance to similar human diseases. Ann Rheum Dis. 1993;52(Suppl 1):S48–S54. doi: 10.1136/ard.52.suppl_1.s48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Herlands RA, Christensen SR, Sweet RA, Hershberg U, Shlomchik MJ. T cell-independent and toll-like receptor-dependent antigen-driven activation of autoreactive B cells. Immunity. 2008;29:249–260. doi: 10.1016/j.immuni.2008.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Gilbert D, Lopez B, Parain J, Koutouzov S, Tron F. Overlap of the anti-cardiolipin and anti-nucleosome responses of the (NZW X BXSB)F1 mouse strain: a new pattern of cross-reactivity for lupus-related autoantibodies. Eur J Immunol. 2000;30:3271–3280. doi: 10.1002/1521-4141(200011)30:11<3271::AID-IMMU3271>3.0.CO;2-6. [DOI] [PubMed] [Google Scholar]
- 38.Hao Y, O’Neill PJ, Naradikian MS, Scholz JL, Cancro MP. A B-cell subset uniquely responsive to innate stimuli accumulates in aged mice. Blood. 2011;118:1294–1304. doi: 10.1182/blood-2011-01-330530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Rubtsov AV, Rubtsova K, Fischer A, Meehan RT, Gillis JZ, Kappler JW, Marrack P. Toll-like receptor 7 (TLR7)-driven accumulation of a novel CD11c+ B-cell population is important for the development of autoimmunity. Blood. 2011;118:1305–1315. doi: 10.1182/blood-2011-01-331462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Asselin-Paturel C, Brizard G, Chemin K, Boonstra A, O’Garra A, Vicari A, Trinchieri G. Type I interferon dependence of plasmacytoid dendritic cell activation and migration. J Exp Med. 2005;201:1157–1167. doi: 10.1084/jem.20041930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Dörner T. SLE in 2011: Deciphering the role of NETs and networks in SLE. Nat Rev Rheumatol. 2012;8:68–70. doi: 10.1038/nrrheum.2011.200. [DOI] [PubMed] [Google Scholar]
- 42.Sorice M, Circella A, Misasi R, Pittoni V, Garofalo T, Cirelli A, Pavan A, Pontieri GM, Valesini G. Cardiolipin on the surface of apoptotic cells as a possible trigger for antiphospholipids antibodies. Clin Exp Immunol. 2000;122:277–284. doi: 10.1046/j.1365-2249.2000.01353.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Price BE, Rauch J, Shia MA, Walsh MT, Lieberthal W, Gilligan HM, O’Laughlin T, Koh JS, Levine JS. Anti-phospholipid autoantibodies bind to apoptotic, but not viable, thymocytes in a beta 2-glycoprotein I-dependent manner. J Immunol. 1996;157:2201–2208. [PubMed] [Google Scholar]
- 44.Amagai M, Stanley JR. Desmoglein as a target in skin disease and beyond. J Invest Dermatol. 2012;132:776–784. doi: 10.1038/jid.2011.390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Cheng J, Torkamani A, Peng Y, Jones TM, Lerner RA. Plasma membrane associated transcription of cytoplasmic DNA. Proc Natl Acad Sci USA. 2012;109:10827–10831. doi: 10.1073/pnas.1208716109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Yoshinobu K, Baudino L, Santiago-Raber ML, Morito N, Dunand-Sauthier I, Morley BJ, Evans LH, Izui S. Selective up-regulation of intact, but not defective env RNAs of endogenous modified polytropic retrovirus by the Sgp3 locus of lupus-prone mice. J Immunol. 2009;182:8094–8103. doi: 10.4049/jimmunol.0900263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Allam R, Anders HJ. The role of innate immunity in autoimmune tissue injury. Curr Opin Rheumatol. 2008;20:538–544. doi: 10.1097/BOR.0b013e3283025ed4. [DOI] [PubMed] [Google Scholar]
- 48.Baccala R, Gonzalez-Quintial R, Lawson BR, Stern ME, Kono DH, Beutler B, Theofilopoulos AN. Sensors of the innate immune system: their mode of action. Nat Rev Rheumatol. 2009;5:448–456. doi: 10.1038/nrrheum.2009.136. [DOI] [PubMed] [Google Scholar]
- 49.Guiducci C, Tripodo C, Gong M, Sangaletti S, Colombo MP, Coffman RL, Barrat FJ. Autoimmune skin inflammation is dependent on plasmacytoid dendritic cell activation by nucleic acids via TLR7 and TLR9. J Exp Med. 2010;207:2931–2942. doi: 10.1084/jem.20101048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Baccala R, Gonzalez-Quintial R, Blasius AL, Rimann I, Ozato K, Kono DH, Beutler B, Theofilopoulos AN. Dependence of lupus development on IRF8 and Slc15a4 implicates plasmacytoid dendritic cells in disease pathogenesis. Proc Natl Acad Sci. 2013;110:2940–2945. doi: 10.1073/pnas.1222798110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Boulé MW, Broughton C, Mackay F, Akira S, Marshak-Rothstein A, Rifkin IR. Toll-like receptor 9-dependent and -independent dendritic cell activation by chromatin-immunoglobulin G complexes. J Exp Med. 2004;199:1631–1640. doi: 10.1084/jem.20031942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Means TK, Latz E, Hayashi F, Murali MR, Golenbock DT, Luster AD. Human lupus autoantibody-DNA complexes activate DCs through cooperation of CD32 and TLR9. J Clin Invest. 2005;115:407–417. doi: 10.1172/JCI23025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Urbonaviciute V, Fürnrohr BG, Meister S, Munoz L, Heyder P, De Marchis F, Bianchi ME, Kirschning C, Wagner H, Manfredi AA, et al. Induction of inflammatory and immune responses by HMGB1-nucleosome complexes: implications for the pathogenesis of SLE. J Exp Med. 2008;205:3007–3018. doi: 10.1084/jem.20081165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Teichmann LL, Ols ML, Kashgarian M, Reizis B, Kaplan DH, Shlomchik MJ. Dendritic cells in lupus are not required for activation of T and B cells but promote their expansion, resulting in tissue damage. Immunity. 2010;33:967–978. doi: 10.1016/j.immuni.2010.11.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Koehn J, Huesken D, Jaritz M, Rot A, Zurini M, Dwertmann A, Beutler B, Korthaüer U. Assessing the function of human UNC-93B in Toll-like receptor signaling and major histocompatibility complex II response. Hum Immunol. 2007;68:871–878. doi: 10.1016/j.humimm.2007.07.007. [DOI] [PubMed] [Google Scholar]
- 56.Crane MJ, Gaddi PJ, Salazar-Mather TP. UNC93B1 mediates innate inflammation and antiviral defense in the liver during acute murine cytomegalovirus infection. PLoS ONE. 2012;7:e39161. doi: 10.1371/journal.pone.0039161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Caetano BC, Carmo BB, Melo MB, Cerny A, dos Santos SL, Bartholomeu DC, Golenbock DT, Gazzinelli RT. Requirement of UNC93B1 reveals a critical role for TLR7 in host resistance to primary infection with Trypanosoma cruzi. J Immunol. 2011;187:1903–1911. doi: 10.4049/jimmunol.1003911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Cervantes JL, Weinerman B, Basole C, Salazar JC. TLR8: the forgotten relative revindicated. Cell Mol Immunol. 2012;9:434–438. doi: 10.1038/cmi.2012.38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Pifer R, Benson A, Sturge CR, Yarovinsky F. UNC93B1 is essential for TLR11 activation and IL-12-dependent host resistance to Toxoplasma gondii. J Biol Chem. 2011;286:3307–3314. doi: 10.1074/jbc.M110.171025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Koblansky AA, Jankovic D, Oh H, Hieny S, Sungnak W, Mathur R, Hayden MS, Akira S, Sher A, Ghosh S. Recognition of profilin by Toll-like receptor 12 is critical for host resistance to Toxoplasma gondii. Immunity. 2013;38:119–130. doi: 10.1016/j.immuni.2012.09.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Hidmark A, von Saint Paul A, Dalpke AH. Cutting edge: TLR13 is a receptor for bacterial RNA. J Immunol. 2012;189:2717–2721. doi: 10.4049/jimmunol.1200898. [DOI] [PubMed] [Google Scholar]
- 62.Oldenburg M, Krüger A, Ferstl R, Kaufmann A, Nees G, Sigmund A, Bathke B, Lauterbach H, Suter M, Dreher S, et al. TLR13 recognizes bacterial 23S rRNA devoid of erythromycin resistance-forming modification. Science. 2012;337:1111–1115. doi: 10.1126/science.1220363. [DOI] [PubMed] [Google Scholar]