

Abstract
Background & Aims
Foxp3+ T-regulatory cells (Tregs) maintain intestinal homeostasis under conditions of continuous challenge with inflammatory microbes. However, plasticity of the Treg population under certain conditions has been reported; Foxp3+ Tregs can be converted to Foxp3− CD4+ T cells.
Methods
We used mice with a T cell-induced colitis model to study the regulatory role of type I interferons (IFNs) in adaptive immunity. We transferred CD4+CD45RBhi (RBhi) T cells, with or without CD4+CD45RBlo CD25+ T cells, from wild-type or IFN-αβR−/− mice into Rag1−/− recipients. We analyzed induction of colitis by flow cytometry, confocal microscopy, and enzyme-linked immunosorbent assay and reverse-transcription polymerase chain reaction analyses. IFN-αβR−/−Rag−/− mice were given injections of recombinant IFN-a following transfer of IFN-αβR−/− RBhi T cells and CD4+Foxp3+ cells from Foxp3-eGFP mice.
Results
Signaling by type I IFNs was required for maintenance of Foxp3 expression and the suppressive activity of Tregs in mice. Transfer of CD4+CD45 RBloCD25+ Tregs from IFN-αβR−/− mice did not prevent T-cell induction of colitis in mice. Foxp3 expression by Tregs transferred from IFN-αβR−/− mice was significantly lower than that of Tregs from wild-type mice. Administration of recombinant IFN-α reduced T cell-mediated colitis by increasing the number of Foxp3+ Tregs and their suppressive functions.
Conclusions
Type I IFNs regulate intestinal homeostasis by maintaining Foxp3 expression on Tregs in colons of mice under inflammatory conditions.
Keywords: Adoptive Transfer, Inflammatory Bowel Disease, Mouse Model, Immune Regulation
The gastrointestinal (GI) tract harbors vast numbers of diverse bacteria and is exposed to numerous food-derived antigens.1 Interestingly, these combined immunologic stimuli do not provoke inflammatory reactions in the intestinal mucosa under steady-state conditions. One possible explanation for this phenomenon is that immune cells in the GI tract execute unique regulatory tasks that are uncommon in other organs.1 Hence, communication between different immune effector cells and T-regulatory cells (Tregs) is essential for the maintenance of GI homeostasis.2 Indeed, immune dysregulation in the GI mucosa leads to chronic inflammatory conditions such as celiac disease and inflammatory bowel disease.3
The transfer of naïve CD4+ T cells (CD4+CD45RBhi) into immune-deficient mice (eg, Rag−/− mice) is an established model of experimental colitis.1,4 The transferred naïve CD4+ T cells undergo activation and proliferation in response to the large array of microbial products in the intestinal lumen and consequently provoke colitis. The cotransfer of a CD4+CD45RBlo population inhibits this type of intestinal inflammation.5 Subsequent studies have shown that Tregs expressing the lineage-specific Foxp3 transcription factor mediate the inhibition of colitis in this model.6,7 Foxp3 is essential for the development and function of Tregs.7,8 The critical role of Foxp3 in the regulation of immune responses in mice and humans has been independently validated in the Foxp3-mutant scurfy mice and in patients with immunodysregulation polyendocrinopathy enteropathy X-linked syndrome.9,10 Continuous Foxp3 expression is essential for the suppressive/regulatory functions of Tregs.7,11 Therefore, it has been assumed that for Foxp3 to play an effective regulatory role, its expression must be stable, and that differentiation into a Foxp3-expressing cell must be unidirectional. However, recent reports have suggested that there is plasticity among Tregs in response to environmental cues, such as severe inflammation.8,12,13 The environmental stimuli that restrain the conversion of Tregs to effector CD4 T-helper cells remain undefined.
Type I interferons (IFNs) include the multiple gene products of IFN-α and a single gene product of IFN-β.14 They are induced by viral RNA and DNA and certain bacterial products (eg, lipopolysaccharide), which activate pattern recognition receptors expressed by different cell types and induce signaling via the heterodimeric receptor, IFN-αβR.14 Although type I IFNs were originally described to be antiviral agents, recent evidence has uncovered that they have immunomodulatory functions as well.14 In previous studies, we reported that TLR9-induced type I IFN signaling inhibits intestinal inflammation and protects the intestinal tissue from dextran sulfate sodium-induced colitis.15,16 Recently, it has been proposed that type I IFN signaling in dendritic cells and macrophages inhibits development of Th17 and that this role of type I IFN is critical for the alleviation of experimental autoimmune encephalomyelitis provoked by encephalitogenic Th17 cells.17,18
In this study, we explored the regulatory role of type I IFN in adaptive immunity using a murine T cell-mediated colitis model. We found that type I IFN is necessary for maintaining Foxp3 expression and Treg function in vivo. Consequently, CD4+CD45RBloCD25+ Tregs isolated from IFN-αβR−/− mice could not suppress CD4+ CD45RBhi T cell-mediated colitis in an adoptive transfer model. The administration of recombinant IFN-α ameliorated the severity of colitis by increasing the number of Foxp3+ cells and improving Treg function. Our results reveal a novel and unexpected role for type I IFN signaling in the regulation of Treg plasticity under inflammatory conditions and highlight its role in intestinal homeostasis.
Materials and Methods
Mice
Six-week-old wild-type (WT), Foxp3-eGFP, CD45.1, Rag1−/−, and IL-10−/− mice, all on the C57BL6/J (B6) background, were purchased from the Jackson Laboratory (Bar Harbor, ME) and bred in our animal facility. IFN-αβR−/− (KO) mice (B6) were described previously.15IL-10−/− mice were crossed to IFN-αβR−/− mice to generate IL-10−/−IFN-αβR−/− double knockout (DKO) mice. Rag1−/− mice were crossed to IFN-αβR−/− mice to generate Rag1−/−IFN-αβR−/− mice. All of the crossed mice were genotyped. All of the experimental animal procedures were approved by the University of California San Diego Animal Care Program or by Chonnam National University and were conducted in accordance with institutional guidelines for animal care and use.
Adoptive Transfer and Clinical Evaluation of Colitis
CD4+ T cells were enriched from a splenocyte cell suspension using a CD4-negative selection kit (Miltenyi Biotec, Auburn, CA) and were subsequently stained with PE-Cy5-anti-CD4 (L3T4; eBioscience, San Diego, CA), Alexa Fluor 488-conjugated anti-CD25 (PC61.5; eBioscience), and PE-anti-CD45RB (C363.16A; eBioscience). These cells were then sorted to obtain the CD4+CD45RBhi (naïve), CD4+CD45RBlo, and CD4+CD45RBlowCD25+ populations using a Mo-Flo cell sorter (DakoCytomation, Carpinteria, CA). A total of 3 × 105 CD4+CD4RBhigh cells were transferred to Rag1−/− mice via intraperitoneal injection. For the cotransfer, 3 × 105 CD4+ CD45RBlo cells were administered via intraperitoneal injection after the CD4+CD4RBhigh cell transfer. In certain experiments, 4 × 105 CD4+CD45RBhi cells, with or without 1 × 105 CD4+CD45RBloCD25+ cells, were transferred via intraperitoneal injection into the Rag1−/− mice. The combined score of weight loss and bleeding was calculated as the Disease Activity Index (DAI), as previously described.15,19,20
Histologic Scoring
Histologic scoring of H&E-stained colonic tissues was performed as described previously.15,20
Flow Cytometry
Isolated CD4+ cells were treated with Fc-Block (2.4G2; BD Biosciences) and then stained with the appropriate combinations of the following antibodies: APC-anti-CD4 (L3T4), FITC-anti-CD25 (7D4; BD Biosciences, San Jose, CA), PE-anti-CD45.1 (A20; eBioscience), FITC-anti-CD45.2 (104; eBioscience), and PerCp-Cy5.5-anti-TCRβ (H57-597; eBioscience). For intracellular staining of Foxp3, the surface-stained cells were fixed and permeabilized according to the manufacturer's instructions (eBioscience) and stained with PE anti-Foxp3 (FJK-16s; eBioscience). Flow cytometric data were acquired using a FACSCalibur cytometer with CellQuest software (BD Biosciences) and analyzed with FlowJo software (Tree Star, Ashland, OR).
Bromodeoxyuridine Staining
Rag1−/− mice were injected intraperitoneally with 4 × 105 CD4+CD45RBhi cells and 1 × 105 CD4+CD45RBloCD25+ Tregs from WT or IFN-αβR−/− mice. Two weeks after the transfer, the mice were injected intraperitoneally with bromodeoxyuridine (BrdU) solution (BD Biosciences) and killed 24 hours later. Incorporation of BrdU was detected in paraffin-embedded colon tissue and splenocytes using the BrdU In-Situ Kit (BD Biosciences) and the BrdU Flow Kit (BD Biosciences).
Culture of Colon Explants
Cytokine levels in the cultures of colon tissues (colon explants) were determined by enzyme-linked immunosorbent assay (ELISA), as described previously.20
ELISA
Cytokine concentrations in the various supernatants were measured by ELISA using commercially available kits (eBioscience).
Quantitative Reverse-Transcription Polymerase Chain Reaction
Gene expression levels from colon tissue and T-cell subtypes were analyzed by quantitative reverse-transcription polymerase chain reaction (qRT-PCR) analysis. Total RNA was isolated from distal colon tissues or from purified T cells using TRIzol (Invitrogen, Carlsbad, CA) and RNeasy kits (Qiagen, Valencia, CA), respectively. The total RNA was reverse transcribed using the Superscript III kit (Invitrogen) and an oligo-dT primer. Quantitative PCR was performed on an ABI 7300 (Applied Biosystems, Carlsbad, CA) using the Power SYBR Green PCR mixture (Applied Biosystems). Expression of hypoxanthineguanine phosphoribosyltransferase (HPRT) was used as internal reference in all PCR experiments. The relative messenger RNA (mRNA) quantities in the samples were calculated as described elsewhere.19,20
Preparation of Lamina Propria Lymphocyte Suspensions
The colon tissues were cut with scissors to produce smaller pieces of tissue and then were incubated with 0.5 mg/mL collagenase (type IV; Sigma, St Louis, MO) in RPMI 1640 media (Mediatech, Manassas, VA) containing 5% fetal calf serum (5% FCS RPMI 1640).21,22 Lamina propria lymphocyte (LPL) cells were recovered from the interface of a 40% and 70% discontinuous Percoll (GE Healthcare, Pittsburgh, PA) solution.
Immunostaining and Confocal Imaging
Whole colons from mice were embedded in OCT compound (Sakura Finetek, Tokyo, Japan) and cryosectioned for immunostaining and confocal imaging. Colon sections were fixed and permeabilized with Cytofix/Cytoperm (BD Biosciences) and stained as described previously.19 To stain immunocytes obtained from the colon, the sections were stained with the appropriate antibodies. The sections were counterstained with 4′,6-diamidino-2-phenylindole and then mounted with ProLong Gold antifade reagent (Life Technologies, Grand Island, NY). Confocal images were obtained with an Olympus FV1000 (Tokyo, Japan) confocal microscope at the UCSD Neuroscience Microscopy Shared Facility.
Treatment With PEG-IFN-α
Rag1−/−IFN-αβR−/− mice received 4 × 105 CD4+ CD45RBhigh cells from KO mice and CD4+Foxp3+ cells (0.4 × 105) from Foxp3-eGFP mice, as described earlier. The mice were injected intraperitoneally with 5 μg of pegylated recombinant human IFN-α-2a (PEG-IFN-α Pegaysys; Genentech, San Francisco, CA) or phosphate-buffered saline (PBS) on days 1, 7, 14, and 21. The mice were monitored and evaluated as described previously. The levels and percentages of Foxp3+-expressing cells from the spleens were determined by flow cytometry.
Statistics
All results are expressed as mean ± SEM unless otherwise stated. Statistical significance between 2 groups was analyzed using the unpaired Student t test unless otherwise stated.
Results
CD4+ CD45RBloCD25+ Cells From IFN-αβR−/− Mice Do Not Suppress CD4+ CD45RBhi-Mediated Colitis
To investigate the role of type I IFN signaling in CD4+ T cell-mediated colitis, we used the adoptive transfer model. We transferred naïve CD4+CD45RBhi (RBhi) T cells, with or without CD4+CD45RBlo (RBlo) cells, from WT (B6) or IFN-αβR−/− mice into Rag1−/− recipients. Body weight and clinical signs of colitis were monitored weekly. Although the cotransfer of WT RBhi with WT RBlo cells did not induce colitis, the transfer of these RBhi and RBlo cells from KO mice resulted in the development of severe intestinal inflammation (Supplementary Figure 1A–C). Histologic analyses (H&E staining) of the affected colons revealed the massive infiltration of inflammatory cells into the lamina propria, the elongation of crypts, epithelial hyperplasia, and the depletion of mucin-secret-ing cells in the colons of the KO RBhi and KO RBlo cotransfer group (Supplementary Figure 1D). Accordingly, colon explants from these recipients produced significantly higher levels of IFN-γ and tumor necrosis factor (TNF)-α compared with those obtained from WT cotransfer recipients (Supplementary Figure 1E). Moreover, the transfer of KO RBlo cells alone induced colitis 6 weeks after transfer (Supplementary Figure 2A and B).
Because major differences between WT and KO cells were observed in the recipients transferred with the RBlo subpopulations, we reasoned that this effect might indicate that type I IFN affects the function of the Tregs within the RBlo population. The RBlo fraction is a heterogeneous population that contains Tregs and antigen-experienced cells. To elucidate whether the KO RBlo CD25+ Tregs within the transferred RBlo population are responsible for the lack of suppressive function, we transferred WT or KO CD4+CD45RBloCD25+ (Treg) cells into Rag1−/− recipients along with naïve WTRBhi cells. Rag1−/− mice that received WT RBhi cells cotransferred with KO, but not WT Tregs, developed severe colitis within 3 weeks after transfer (Figure 1A–C). The histologic analyses of these mice indicated massive infiltration of inflammatory cells into the lamina propria with crypt elongation and epithelial cell hyperplasia (Figure 1D). A significantly higher number of CD4+ cells was observed in the colons of mice that received the KO Tregs (Figure 1E). Consistent with these data, higher levels of IFN-γ and TNF-α were detected in the supernatants of colon explants obtained from mice cotransferred with KO Tregs (Figure 1F). To evaluate the proliferative responses of the adoptively transferred CD4+ T cells, we performed BrdU staining. Splenic CD4+ T cells isolated from recipients that were cotransferred with KO Tregs displayed higher BrdU incorporation than those in recipients cotransferred with WT Tregs (Figure 2A). Similar findings were observed in the colons of these 2 groups of mice (Figure 2B). BrdU was incorporated into the epithelial cells and the infiltrating cells in the colon lamina propria of recipients cotransferred with WT effector T cells (Teffs) and KO Tregs. In contrast, in the colons of recipients cotransferred with WT Tregs, BrdU+ cells were only detected in the epithelia (Figure 2B).
Figure 1.
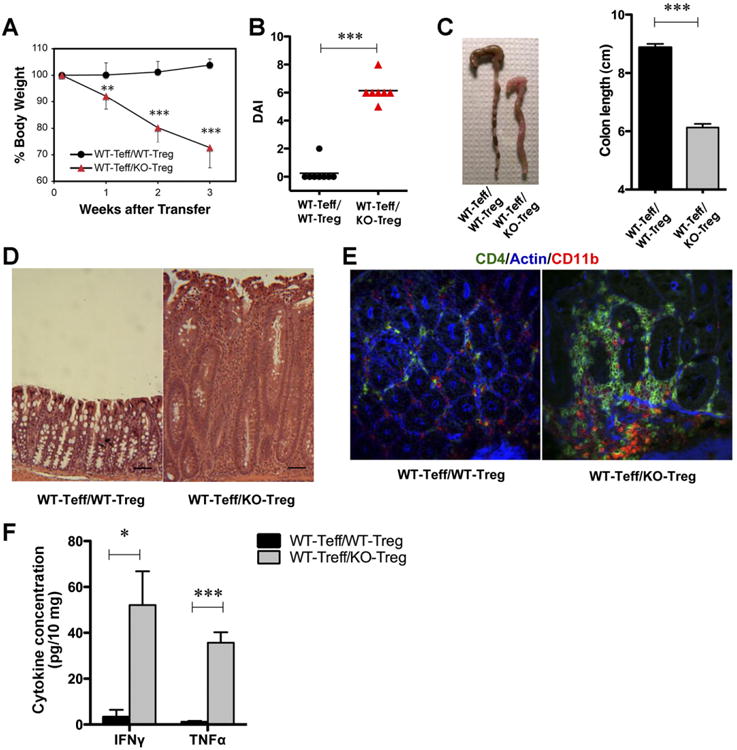
Type I IFN signaling is essential for Treg function. Rag1−/− mice were adoptively transferred with WT 4 × 105 CD4+CD45RBhi (Teff) cells and 1 × 105 CD4+CD45RBloCD25+ (Treg) cells from WT or KO mice. (A) Changes in body weight. The data represent the weights of 7–8 mice per group and are expressed as the means ± SDs. (B) DAI. (C) Photographs of representative colons from the 2 experimental groups, 3 weeks after transfer, and mean colon length (cm). (D) Representative H&E-stained micrographs from the 2 experimental groups (scale bar, 50 mm; original magnification 100×). (E) Confocal imaging of colon tissue from 2 representative experiments. (F) Colon explants were cultured for 16 hours. IFN-γ and TNF-α levels in the supernatants were determined by ELISA. Cytokine concentrations were normalized to the weight of the tissue. *P < .05, **P < .01, ***P < .001.
Figure 2.

KO Tregs are unable to inhibit proliferation of WT CD4+CD45RBhi cells in Rag1−/− recipients. Rag1−/− mice were adoptively transferred with 4 × 105 CD4+CD45RBhi (Teff) and 1 × 105 CD4+CD45RBloCD25+ (Treg) cells from WT or KO mice. (A) BrdU incorporation in CD4+ splenocytes was determined by flow cytometry 2 weeks after the transfer. (B) BrdU incorporation (brown color) was detected in epithelial cells (blue arrows) and cells infiltrating the colon lamina propria (red arrows) of recipients that were cotransferred with KO Tregs (scale bar, 20 mm; original magnification 200×).
We then tested whether the CD4+CD45RBloCD25+ Treg subpopulation alone could induce colitis without the cotransfer of RBhi cells. Rag1−/− mice were injected intraperitoneally with fluorescence-activated cell sorted (FACS) CD4+CD45RBloCD25+ cells (95% Foxp3+; Supplementary Figure 3A) from WT or KO mice. Neither recipient group developed clinical signs of colitis during the first 6 weeks posttransfer (Supplementary Figure 3B), and CD4+TCRβ+ cells were not recovered from the spleens of these mice (data not shown). The histologic analyses confirmed that neither WT nor KO CD4+CD45RBloCD25+ Tregs induced colitis (Supplementary Figure 3C). These data indicate that KO CD4+CD45RBloCD25+ Tregs alone cannot survive in vivo after they are transferred into Rag1−/− mice unless they are also cotransferred with effector cells. Moreover, a small fraction of KO CD4+CD45RBloCD25+Foxp3− (<5%; see Supplementary Figure 3A) cells do not outgrow to induce colitis. Taken together, these results suggest that the KO CD4+CD45RBloCD25+ Treg population is dysfunctional in this model of T cell-mediated colitis.
Development of Tregs in KO Mice
To compare development of Tregs in KO and WT mice, we assessed the percentage and number of Foxp3+ cells in the thymuses, spleens, and LPLs of these mice. No significant differences were observed in the percentage of CD4+Foxp3+ cells in these specimens between WT and KO mice (Supplementary Figure 4A–C). The transcription levels of Treg-related genes in CD4+CD45RBloCD25+ cells were determined by qRT-PCR (Supplementary Table 1) using FACS CD4+CD45RBloCD25+ cells from WT and KO mice. The expression levels of Foxp3, CD25, CTLA-4, GITR, CD39, CD73, LFA-1, L-selectin, CD103, CD2, and Ebi-323–28 were similar in WT and KO Tregs (Supplementary Figure 4D). These results, combined with those discussed previously (Figure 1), strongly suggest that KO Tregs undergo normal development in KO mice, while they acquire a dysfunctional immunosuppressive phenotype after their cotransfer with naïve CD4+ T cells into Rag1−/− recipients.
In Vivo Survival and Function of KO Tregs
Because the data presented previously indicate that KO Tregs display a dysfunctional phenotype after they are transferred to Rag1−/− mice, we evaluated whether type I IFN signaling is necessary for survival and/or function of Tregs in vivo.
We used CD45.1 and CD45.2 congenic mice as donors of naïve CD4+RBhi and CD4+RBloCD25+ Tregs, respectively. Rag1−/− recipients were injected intraperitoneally with naïve CD45.1+RBhi and CD45.2+RBloCD25+ cells from WT or KO mice. Three weeks after transfer, we analyzed the number and percentage of CD45.1 (Teff) and CD45.2 (Treg) cells in the reconstituted spleens and lamina propria of the recipients. The number of recovered KO Tregs (CD45.2+) from the spleens was comparable to that observed for WT Tregs (Figure 3A). However, the ratio of CD45.1 (Teff) to CD45.2 (Treg) cells in the spleens was 7:3 in recipients transferred with WT Tregs vs 9:1 in recipients transferred with KO Tregs (Figure 3B). The percentage of CD45.1 cells cotransferred with WT CD45.2 Tregs (67.9% ± 4.9%) was significantly lower than that observed on transfer with KO CD45.2 Tregs (93.2% ± 2.6%; P < .05). Furthermore, the percentage of CD45.1 cells cotransferred with WT CD45.2 Tregs (80.9% ± 6.1%) was significantly lower than that observed on transfer with KO CD45.2 Tregs (96.4% ± 1.0%; P < .05) in the lamina propria (Figure 4A). These data, taken together with the data presented previously, indicate that (1) KO Tregs survive at rates similar to WT Tregs under the specified in vivo conditions and (2) KO Tregs fail to restrain WT Teff proliferation and fail to inhibit colitogenic properties (Figure 1).
Figure 3.
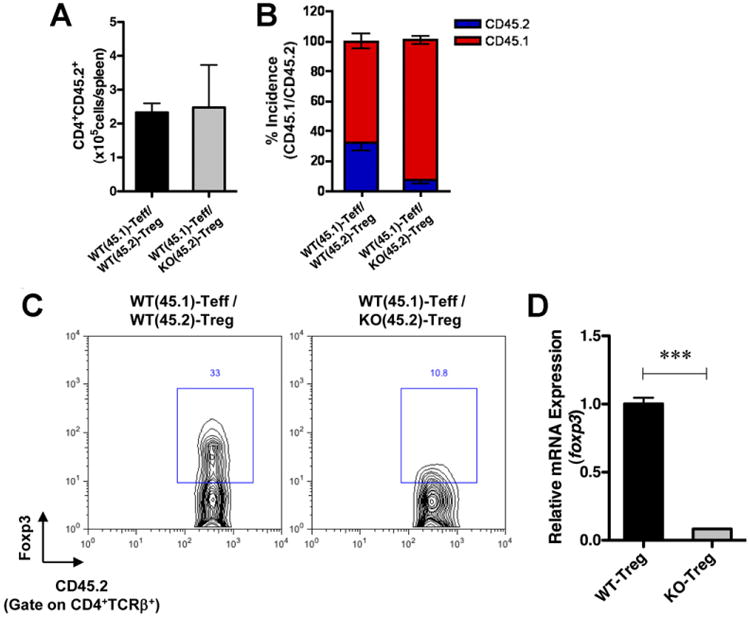
KO Tregs lose Foxp3 expression after adoptive transfer into Rag1−/− recipients. CD4+ CD45RBhi cells from congenic CD45.1 mice were transferred along with CD4+CD45RBloCD25+ cells from WT (CD45.2) or KO (CD45.2) into Rag1−/− mice. (A) The number of Tregs (CD4+ CD45.2+) in the spleens of the recipient mice (n = 3 for each group) was determined by flow cytometry at 3 weeks after transfer. (B) The percentage of CD45.1 vs CD45.2 cells gated on CD4+ cells in the spleens was determined by flow cytometry. (C) Foxp3 expression in CD4+TCR/3+CD45.2+ cells was determined by flow cytometry. The data are representative of 3 independent experiments. (D) Foxp3 mRNA expression levels were determined in recovered Tregs. Congenic Tregs were recovered from the transferred mice 4 weeks after transfer, and the expression levels of Foxp3 were analyzed by real-time RT-PCR as described in Materials and Methods. ***P < .001.
Figure 4.

KO Tregs lose Foxp3 after adoptive transfer into Rag1−/− recipients in the LPL. CD4+CD45RBhi cells from congenic CD45.1 mice were transferred with CD4+CD45RBloCD25+ cells from WT (CD45.2) or KO (CD45.2) into Rag1−/− mice. (A) The percentages of CD45.1 vs CD45.2 cells gated on CD4+ cells in the LPLs were determined by flow cytometry. (B) Foxp3 expression in CD4+ TCRβ+CD45.2+ cells was determined by flow cytometry. The data are representative of 3 independent experiments.
Type I IFN Signaling Is Essential for the Maintenance of Foxp3 Expression in Tregs in the Colitis Environment
Because Foxp3 is the master regulator of Treg development and function, we analyzed its expression levels in dysfunctional Tregs in vivo. Rag1−/− recipients were coinjected intraperitoneally with naïve CD45.1+RBhi and CD45.2+RBloCD25+ cells from WT or KO mice. Three weeks after the transfer, we analyzed the Foxp3 expression levels in CD45.2+ cells after gating the CD4+TCRβ+ population. Surprisingly, 33% of WT CD4+TCR|3+CD45.2+ cells expressed Foxp3, whereas only 10.8% of KO CD4+TCRβ+CD45.2+ cells expressed Foxp3 in the spleens (Figure 3C). To analyze the transcript levels of Foxp3 in Tregs under these conditions, CD45.2+TCRβ+CD45RBloCD25+ splenocytes underwent FACS from the recipients and the mRNA levels were determined by qRT-PCR. The Foxp3 mRNA levels were reduced in the KO Tregs compared with WT Tregs (Figure 3D). The same observation was also detected in isolated LPLs, in which 24.7% of the WT CD4+TCRβ+CD45.2+ cells expressed Foxp3 compared with only 5.6% of the KO CD4+TCRβ+CD45.2+ cells (Figure 4B). These data strongly suggest that continuous type I IFN signaling is essential for the maintenance of Foxp3 expression in the inflammatory environment.
Administration of Type I IFN Improves Treg Function In Vivo
To investigate whether administration of type I IFN can modulate Treg function in vivo, we administered PEG-IFN-α to the recipient mice. To rule out the possibility that IFN-α induces any immunomodulatory effects on various non-Treg cells, Rag1−/−IFN-αβR−/− and IFN-αβR−/− mice were used as the recipients and donors, respectively. We cotransferred FACS WT Foxp3+ reporter Tregs (>99% purity) with KO CD4+RBhi into Rag1−/−IFN-αβR−/− recipients (Teff: Treg = 10:1). The recipient mice were treated with PEG-IFN-α or PBS intraperitoneally once per week. Whereas the PBS-treated mice developed colitis, PEG-IFN-α–treated mice maintained their original body weight and displayed lower DAIs and histologic scores compared with the PBS-treated mice (Figure 5A–C). Consistent with the severity of colitis, IFN-γ and TNF-α levels were significantly lower in the colon explants from PEG-IFN-α–treated recipients (Figure 5D). To evaluate the effects of PEG-IFN-α administration on Foxp3+ cells, we isolated, permeabilized, and stained splenocytes from the transferred mice using an anti-Foxp3 antibody. The percentage of CD4+Foxp3+ cells was significantly increased in the PEG-IFN-α–treated mice compared with the PBS-treated group (Figure 5E).
Figure 5.
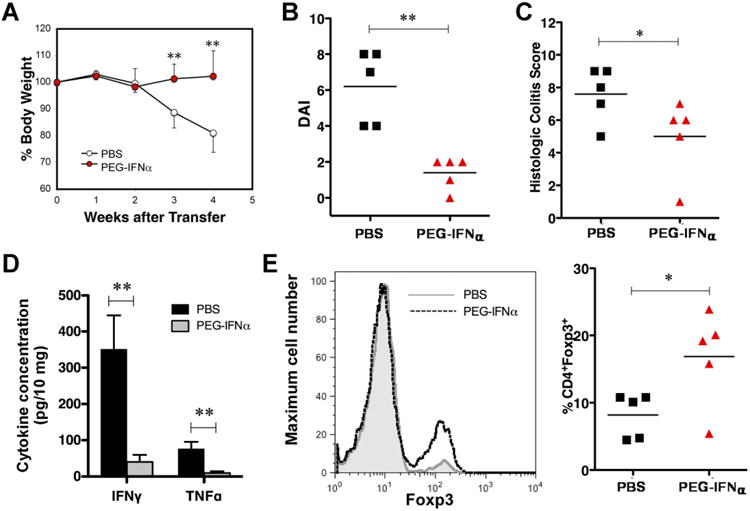
Treatment with PEG-IFN-α enhances Treg function in the transfer model. Rag1−/−IFN-αβR−/− mice were injected with CD4+CD45RBhigh cells (4 × 105) from KO mice and Foxp3+ cells (0.4 × 105) from Foxp3-eGFP mice. The mice were injected intraperitoneally weekly with PEG-IFN-α. (A) Changes in body weight. The data represent 5 mice per group and are expressed as the means ± SDs. (B) DAI. (C) Histologic scores of the colons from PEG-IFN-α–treated and untreated recipients. P values were calculated using the Mann–Whitney test (n = 5 for each group). (D) Colon explants were cultured for 16 hours, and the levels of IFN-γ and TNF-α were determined by ELISA. Cytokine concentrations were normalized to the weights of the tissues. (E) A representative histogram of 5 independent experiments. The number of CD4+Foxp3+ splenocytes in recipient mice (n = 5 for each group) was determined by flow cytometry at 4 weeks after transfer. *P < .05; **P < .01.
PEG-IFN-α is widely used for the treatment of various cancers and viral infections. Because it has been reported that polyethylene glycol (PEG) enhances colonic barrier function and ameliorates trinitrobenzene sulfonic acid-induced experimental colitis in rats,29 we tested whether PEG itself could affect intestinal barrier function (Supplementary Materials and Methods). The intraperitoneal administration of PEG (40 kilodaltons; 13.3 mg/mouse), under the conditions presented in Figure 6, did not alter intestinal permeability, as determined by fluorescein isothiocyanate-dextran measurement,30 in a dextran sulfate sodium–induced colitis model.15 Intraperitoneally administered PEG also did not affect DAI (Supplementary Figure 5A) and the intestinal permeability function (Supplementary Figure 5B). An equal amount of orally administered PEG also did not influence the intestinal barrier function (Supplementary Figure 5A and B).
Figure 6.
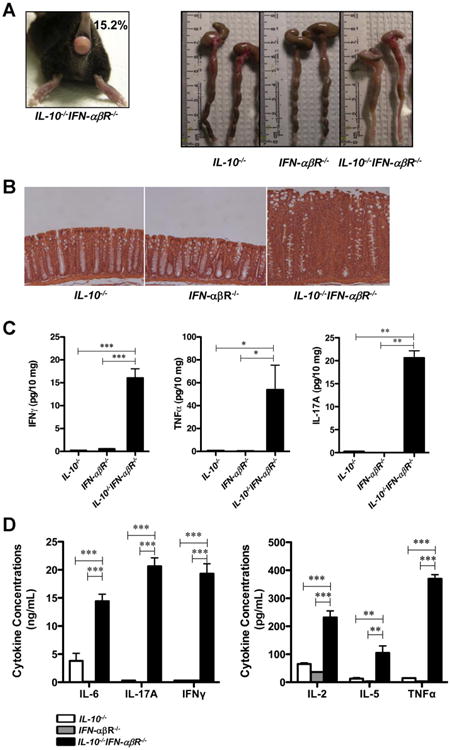
Type I IFN and IL-10 have a nonredundant protective role in experimental colitis. (A) IL-10−/−IFN-αβR−/− DKO mouse with rectal prolapse, and photographs of colons from 17-weekold mice. (B) Micrographs of paraffin-embedded colon samples from IL-10−/−, IFN-αβR−/−, and IL-10−/−IFN-−/−αβR−/− mice were sectioned and stained with H&E (original magnification 100×). (C) Colon explants from the mice were cultured for 16 hours, and IFN-γ, TNF-α, and IL-17A were detected in the supernatants by ELISA (n = 4 for each group). Cytokine concentrations were normalized to the weights of the tissues. (D) Colon LPLs isolated from IL-10−/−, IFN-αβR−/−, or IL-10−/−IFNαβR−/− mice (n = 4 each) were activated with anti-CD3/28 antibodies for 48 hours, and the cytokine levels in the supernatants were measured by ELISA. ND, not detectable; *P < .05; **P < .01; ***P < .001.
IL-10 and Type IIFN Signaling Have Nonredundant Protective Roles in Experimental Colitis
The IL-10−/−IFN-αβR−/− DKO mice developed severe colitis at 4 months of age, whereas IL-10−/− or IFN-αβR−/− single KO mice did not develop intestinal inflammation under the same housing conditions. Among the more than 100 mice that we observed, 15.2% of the DKO mice developed rectal prolapse within 17 weeks. Moreover, macroscopic signs of intestinal inflammation such as thickening of the intestinal wall and diarrhea were evident only in the colon of DKO mice (Figure 6A). Histologic evaluation revealed the infiltration of inflammatory cells to the lamina propria and crypt elongation in the colons of the DKO mice (Figure 6B). Colon explants from the DKO mice secreted higher levels of inflammatory cytokines such as IFN-γ, TNF-α and interleukin (IL)-17A (Figure 6C). Similar results were obtained after anti-CD3/28 antibody stimulation of colon LPLs (Figure 6D). To further explore the expression of the inflammatory mediators in colon tissues, we performed qPCR analysis. The mRNA levels of IL-1, IL-17A, GM-CSF, KC, MIP1−/−αβ × –γ, MIP1β, CXCL10, and CCL22 were elevated in the colon tissues obtained from DKO mice compared with those obtained from IL-10−/− or IFN-αβR−/− mice (Table 1). Taken together, these results indicate that IL-10 and type I IFN signaling have nonredundant protective roles in experimental colitis.
Table 1. Relative mRNA Expression Level in Colon Tissues.
| Genes | IL-10−/− | IFN-αβR−/− | IL-10−/−IFN-αβR−/− | |
|---|---|---|---|---|
| Cytokines | IL1β | 1.0 ± 0.1 | 1.8 ± 0.6 | 33.8 ± 8.7a |
| IL-17 | 1.0 ± 0.2 | 1.1 ± 0.2 | 3.3 ± 1.3 | |
| GM-CSF | 1.0 ± 0.4 | 1.2 ± 0.5 | 5.4 ± 2.0 | |
| IFNγ | 1.0 ± 0.2 | 0.6 ± 0.2 | 23.6 ± 0.1b | |
| TNFα | 1.0 ± 0.2 | 0.7 ± 0.2 | 9.0 ± 1.3b | |
| Cell markers | CD3d | 1.0 ± 0.2 | 1.2 ± 0.3 | 11.6 ± 2.1b |
| F4/80 | 1.0 ± 0.2 | 0.6 ± 0.1 | 2.9 ± 0.7 | |
| Chemokines | KC | 1.0 ± 0.2 | 1.8 ± 0.9 | 67.3 ± 12.5b |
| MIP1α | 1.0 ± 0.1 | 0.4 ± 0.1b | 3.7 ± 0.9a | |
| MIP1β | 1.0 ± 0.2 | 2.4 ± 0.3a | 25.8 ± 8.7a | |
| CXCL10 | 1.0± 0.2 | 2.6 ± 0.6 | 33.1 ± 17.9 | |
| CCL22 | 1.0 ± 0.1 | 0.4 ± 0.2a | 8.4 ± 2.9 |
NOTE. Total RNA was isolated from the distal colon tissue, and mRNA expression levels were measured by qRT-PCR for the indicated genes in 17-week-old IL-10−/−, IFN-αβR−/−, and IL-10−/−IFN-αβR−/− mice (n = 4 each) was measured by qPCR. The data represent 3–4 mice per group. Significant differences were calculated in comparison with the IL-10−/− mice.
P < .05.
P < .01.
Discussion
In this study, we showed that type I IFN signaling is important for maintaining Foxp3 expression in the inflamed mucosa in a model of T cell-mediated colitis and that the administration of recombinant IFN-α ameliorates the severity of colitis by increasing the number of Foxp3+ cells and by improving their function.
The CD4+ T-cell transfer model of experimental colitis has been instrumental in elucidating the physiologic roles of Tregs.1,4 We observed that the CD4+RBlo subset isolated from IFN-abR−/− (KO) mice was unable to suppress colitis induced by the cotransferred CD4+RBhi subset (Supplementary Figure 1). Moreover, the CD4+RBlo subset alone (ie, isolated from KO mice) could induce colitis in Rag1−/− recipients even without the cotransfer of the CD4+RBhi subset (Supplementary Figure 2). Because we repeatedly failed to identify any defect of the KO Tregs on WT CD4+ T-cell function (eg, proliferative response) under in vitro conditions (data not shown), we applied an in vivo analysis. Using a cotransfer of congenic CD4+RBhi and CD4+RBloCD25+ subsets, we identified that the number of recovered KO Tregs is similar to that observed for WT Tregs in recipient mice (Figure 3A). Surprisingly, the expression levels of the Foxp3 protein were significantly reduced in KO Tregs compared with WT Tregs in the spleen and lamina propria (Figures 3C and 4B). A similar profile was observed for Foxp3 mRNA levels (Figure 3D). These results clearly suggest a novel role for type I IFN signaling in the immune regulation of intestinal homeostasis through the maintenance of Foxp3 expression. One of the central features of Tregs is their low production of IL-2. Therefore, Tregs are highly dependent on an exogenous supply of IL-27,11,31,32 for their function and/or proliferation in vivo. Because CD4+TCRβ+ cells were not recovered from Rag1−/− mice that were injected with CD4+CD45RBloCD25+ Tregs alone (data not shown), the cotransferred Teffs most likely support their proliferation and function of Tregs by supplying IL-2. Although type I IFN signaling is essential for the suppressive function of Tregs in the transfer model, KO mice do not develop spontaneous colitis or autoimmune disease unless they are challenged with various agents.15,17,18 Indeed, no significant differences were observed in the percentages of CD4+Foxp3+ cells or in the mRNA levels of Treg-related genes in the KO vs WT mice (Supplementary Figure 4). Collectively, these data indicate that the dysfunctional phenotype of the KO Tregs was acquired only after their transfer into the Rag1−/− recipients.
To determine whether type I IFN signaling is therapeutically efficacious and whether it mediates its beneficial effects via Foxp3 stabilization, we cotransferred Teffs from KO mice and Tregs from WT mice into IFN-αβR−/−Rag1−/− mice. PEG-IFN-α treatment ameliorated T cell-mediated experimental colitis and maintained Foxp3+ expression in CD4+ cells (Figure 5E). Type I IFN treatment has been documented to improve the frequency and suppressive function of Tregs in patients with relapsing-remitting multiple sclerosis,33 and there was also increased Foxp3 mRNA expression in the peripheral blood mononuclear cells from these patients. Our observations can therefore explain the beneficial effects of type I IFN treatment on this disease, and they suggest the potential therapeutic application of type I IFNs in the modulation of T cell–mediated intestinal inflammation.
It has been reported that IL-10−/− mice develop spontaneous chronic enterocolitis,35 which is largely attributed to dysfunctional Tregs.36 These reports led us to investigate the effects of type I IFN and IL-10 on Treg function. The IL-10−/− mice were crossed to generate IL-10−/−IFN-αβR−/− DKO mice. We found that the IFN-αβR−/− and IL-10−/− KO mice did not develop spontaneous colitis in our animal facility20 because only DKO mice developed overt signs of colitis. This result shows the nonredundant roles that the type I IFNs and IL-10 have with regard to Treg function and colitis protection.
The molecular mechanisms by which type I IFNs exert their immunomodulatory functions and the efficacy of these mechanisms in various immunopathological conditions are still not completely understood. Given their beneficial effects on intestinal homeostasis in animal models,14 type I IFNs have been tested as therapeutic agents in human inflammatory bowel disease with controversial results.37–40 Because IFN-αβR, a common receptor for type I IFNs, is expressed by nearly all cell types and its activation can result in different biological properties,14 it is possible that these diverse effects might blunt the Foxp3 stabilizing effects observed in this study. We propose to consider the application of type I IFNs in immune-mediated diseases associated with decreased Treg function.
Supplementary Material
Acknowledgments
The authors thank Kyoung-Oh Cho (Chonnam National University, Gwangju, Korea) for critical support during the histologic evaluations.
Funding: E.R. was supported by National Institutes of Health grants AI068685, AI095623, AI077989, and DK35108. E.R. and J.M.G.-N. were supported by a grant from Crohn's & Colitis Foundation of America. S.E.L. was supported by grant 2011-0026156 (MEST) and grant RTI05-01-01 (MKE) from the Republic of Korea.
Abbreviations used in this paper
- BrdU
bromodeoxyuridine
- DAI
Disease Activity Index
- DKO
double knockout
- ELISA
enzyme-linked immunosorbent assay
- FACS
fluorescence-activated cell sorted
- HPRT
hypoxanthine-guanine phosphoribosyltransferase
- IFN
interferon
- IL
interleukin
- KO
knockout
- LPL
lamina propria lymphocyte
- qRT-PCR
quantitative reverse-transcription polymerase chain reaction
- Teff
effector T cell
- TNF
tumor necrosis factor
- Treg
regulatory T cell
- WT
wild type
Footnotes
Conflicts of interest: The authors disclose no conflicts.
Supplementary Material: Note: To access the supplementary material accompanying this article, visit the online version of the journal at www.gastrojournal.org, and at http://dx.doi.org/10.1053/j.gastro.2012.03.042.
References
- 1.Izcue A, Coombes JL, Powrie F. Regulatory lymphocytes and intestinal inflammation. Annu Rev Immunol. 2009;27:313–338. doi: 10.1146/annurev.immunol.021908.132657. [DOI] [PubMed] [Google Scholar]
- 2.Xavier RJ, Podolsky DK. Unravelling the pathogenesis of inflammatory bowel disease. Nature. 2007;448:427–434. doi: 10.1038/nature06005. [DOI] [PubMed] [Google Scholar]
- 3.Zenewicz LA, Antov A, Flavell RA. CD4 T-cell differentiation and inflammatory bowel disease. Trends Mol Med. 2009;15:199–207. doi: 10.1016/j.molmed.2009.03.002. [DOI] [PubMed] [Google Scholar]
- 4.Izcue A, Coombes JL, Powrie F. Regulatory T cells suppress systemic and mucosal immune activation to control intestinal inflammation. Immunol Rev. 2006;212:256–271. doi: 10.1111/j.0105-2896.2006.00423.x. [DOI] [PubMed] [Google Scholar]
- 5.Powrie F, Leach MW, Mauze S, et al. Phenotypically distinct subsets of CD4+ T cells induce or protect from chronic intestinal inflammation in C. B-17 scid mice. Int Immunol. 1993;5:1461–1471. doi: 10.1093/intimm/5.11.1461. [DOI] [PubMed] [Google Scholar]
- 6.Belkaid Y, Tarbell K. Regulatory T cells in the control of hostmicroorganism interactions (*) Annu Rev Immunol. 2009;27:551–589. doi: 10.1146/annurev.immunol.021908.132723. [DOI] [PubMed] [Google Scholar]
- 7.Sakaguchi S, Yamaguchi T, Nomura T, et al. Regulatory T cells and immune tolerance. Cell. 2008;133:775–787. doi: 10.1016/j.cell.2008.05.009. [DOI] [PubMed] [Google Scholar]
- 8.Hori S. Developmental plasticity of Foxp3+ regulatory T cells. Curr Opin Immunol. 2010;22:575–582. doi: 10.1016/j.coi.2010.08.004. [DOI] [PubMed] [Google Scholar]
- 9.Brunkow ME, Jeffery EW, Hjerrild KA, et al. Disruption of a new forkhead/winged-helix protein, scurfin, results in the fatal lymphoproliferative disorder of the scurfy mouse. Nat Genet. 2001;27:68–73. doi: 10.1038/83784. [DOI] [PubMed] [Google Scholar]
- 10.Bennett CL, Christie J, Ramsdell F, et al. The immune dysregulation, polyendocrinopathy, enteropathy, X-linked syndrome (IPEX) is caused by mutations of FOXP3. Nat Genet. 2001;27:20–21. doi: 10.1038/83713. [DOI] [PubMed] [Google Scholar]
- 11.Josefowicz SZ, Rudensky A. Control of regulatory T cell lineage commitment and maintenance. Immunity. 2009;30:616–625. doi: 10.1016/j.immuni.2009.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Oldenhove G, Bouladoux N, Wohlfert EA, et al. Decrease of Foxp3+ Treg cell number and acquisition of effector cell phenotype during lethal infection. Immunity. 2009;31:772–786. doi: 10.1016/j.immuni.2009.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhou X, Bailey-Bucktrout S, Jeker LT, et al. Plasticity of CD4(+) FoxP3(+) T cells. Curr Opin Immunol. 2009;21:281–285. doi: 10.1016/j.coi.2009.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gonzalez-Navajas JM, Lee J, David M, et al. Immunomodulatory functions of type I interferons. Nat Rev Immunol. 2012;12:125–135. doi: 10.1038/nri3133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Katakura K, Lee J, Rachmilewitz D, et al. Toll-like receptor 9-induced type I IFN protects mice from experimental colitis. J Clin Invest. 2005;115:695–702. doi: 10.1172/JCI22996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gonzalez-Navajas JM, Law J, Nguyen KP, et al. Interleukin 1 receptor signaling regulates DUBA expression and facilitates Tolllike receptor 9-driven antiinflammatory cytokine production. J Exp Med. 2010;207:2799–2807. doi: 10.1084/jem.20101326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Guo B, Chang EY, Cheng G. The type I IFN induction pathway constrains Th17-mediated autoimmune inflammation in mice. J Clin Invest. 2008;118:1680–1690. doi: 10.1172/JCI33342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Shinohara ML, Kim JH, Garcia VA, et al. Engagement of the type I interferon receptor on dendritic cells inhibits T helper 17 cell development: role of intracellular osteopontin. Immunity. 2008;29:68–78. doi: 10.1016/j.immuni.2008.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lee J, Mo JH, Katakura K, et al. Maintenance of colonic homeostasis by distinctive apical TLR9 signalling in intestinal epithelial cells. Nat Cell Biol. 2006;8:1327–1336. doi: 10.1038/ncb1500. [DOI] [PubMed] [Google Scholar]
- 20.Gonzalez-Navajas JM, Fine S, Law J, et al. TLR4 signaling in effector CD4+ T cells regulates TCR activation and experimental colitis in mice. J Clin Invest. 2010;120:570–581. doi: 10.1172/JCI40055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mucida D, Park Y, Kim G, et al. Reciprocal TH17 and regulatory T cell differentiation mediated by retinoic acid. Science. 2007;317:256–260. doi: 10.1126/science.1145697. [DOI] [PubMed] [Google Scholar]
- 22.Weigmann B, Tubbe I, Seidel D, et al. Isolation and subsequent analysis of murine lamina propria mononuclear cells from colonic tissue. Nat Protoc. 2007;2:2307–2311. doi: 10.1038/nprot.2007.315. [DOI] [PubMed] [Google Scholar]
- 23.Collison LW, Workman CJ, Kuo TT, et al. The inhibitory cytokine IL-35 contributes to regulatory T-cell function. Nature. 2007;450:566–569. doi: 10.1038/nature06306. [DOI] [PubMed] [Google Scholar]
- 24.Deaglio S, Dwyer KM, Gao W, et al. Adenosine generation catalyzed by CD39 and CD73 expressed on regulatory T cells mediates immune suppression. J Exp Med. 2007;204:1257–1265. doi: 10.1084/jem.20062512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kaser A, Blumberg RS. Adaptive immunity in inflammatory bowel disease: state of the art. Curr Opin Gastroenterol. 2008;24:455–461. doi: 10.1097/MOG.0b013e328304d60d. [DOI] [PubMed] [Google Scholar]
- 26.Sakaguchi S, Sakaguchi N, Asano M, et al. Immunologic self-tolerance maintained by activated T cells expressing IL-2 receptor alpha-chains (CD25). Breakdown of a single mechanism of self-tolerance causes various autoimmune diseases. J Immunol. 1995;155:1151–1164. [PubMed] [Google Scholar]
- 27.Wing K, Onishi Y, Prieto-Martin P, et al. CTLA-4 control over Foxp3+ regulatory T cell function. Science. 2008;322:271–275. doi: 10.1126/science.1160062. [DOI] [PubMed] [Google Scholar]
- 28.Zheng Y, Josefowicz SZ, Kas A, et al. Genome-wide analysis of Foxp3 target genes in developing and mature regulatory T cells. Nature. 2007;445:936–940. doi: 10.1038/nature05563. [DOI] [PubMed] [Google Scholar]
- 29.Videla S, Lugea A, Vilaseca J, et al. Polyethylene glycol enhances colonic barrier function and ameliorates experimental colitis in rats. Int J Colorectal Dis. 2007;22:571–580. doi: 10.1007/s00384-006-0232-4. [DOI] [PubMed] [Google Scholar]
- 30.Bergstrom KS, Kissoon-Singh V, Gibson DL, et al. Muc2 protects against lethal infectious colitis by disassociating pathogenic and commensal bacteria from the colonic mucosa. PLoS Pathog. 2010;6:e1000902. doi: 10.1371/journal.ppat.1000902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yamaguchi T, Wing JB, Sakaguchi S. Two modes of immune suppression by Foxp3(+) regulatory T cells under inflammatory or non-inflammatory conditions. Semin Immunol. 2011;23:424–430. doi: 10.1016/j.smim.2011.10.002. [DOI] [PubMed] [Google Scholar]
- 32.Koreth J, Matsuoka K, Kim HT, et al. Interleukin-2 and regulatory T cells in graft-vs-host disease. N Engl J Med. 2011;365:2055–2066. doi: 10.1056/NEJMoa1108188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Qiu H, Fan Y, Joyee AG, et al. Type I IFNs enhance susceptibility to Chlamydia muridarum lung infection by enhancing apoptosis of local macrophages. J Immunol. 2008;181:2092–2102. doi: 10.4049/jimmunol.181.3.2092. [DOI] [PubMed] [Google Scholar]
- 34.Vandenbark AA, Huan J, Agotsch M, et al. Interferon-beta-1a treatment increases CD56bright natural killer cells and CD4+CD25+ Foxp3 expression in subjects with multiple sclerosis. J Neuroimmunol. 2009;215:125–128. doi: 10.1016/j.jneuroim.2009.08.007. [DOI] [PubMed] [Google Scholar]
- 35.Kuhn R, Lohler J, Rennick D, et al. Interleukin-10-deficient mice develop chronic enterocolitis. Cell. 1993;75:263–274. doi: 10.1016/0092-8674(93)80068-p. [DOI] [PubMed] [Google Scholar]
- 36.Kim SC, Tonkonogy SL, Albright CA, et al. Variable phenotypes of enterocolitis in interleukin 10-deficient mice monoassociated with two different commensal bacteria. Gastroenterology. 2005;128:891–906. doi: 10.1053/j.gastro.2005.02.009. [DOI] [PubMed] [Google Scholar]
- 37.Madsen SM, Schlichting P, Davidsen B, et al. An open-labeled, randomized study comparing systemic interferon-alpha-2A and prednisolone enemas in the treatment of left-sided ulcerative colitis. Am J Gastroenterol. 2001;96:1807–1815. doi: 10.1111/j.1572-0241.2001.03875.x. [DOI] [PubMed] [Google Scholar]
- 38.Nikolaus S, Rutgeerts P, Fedorak R, et al. Interferon beta-1a in ulcerative colitis: a placebo controlled, randomised, dose escalating study. Gut. 2003;52:1286–1290. doi: 10.1136/gut.52.9.1286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Tilg H, Vogelsang H, Ludwiczek O, et al. A randomised placebo controlled trial of pegylated interferon alpha in active ulcerative colitis. Gut. 2003;52:1728–1733. doi: 10.1136/gut.52.12.1728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Mannon PJ, Hornung RL, Yang Z, et al. Suppression of inflammation in ulcerative colitis by interferon-beta-1a is accompanied by inhibition of IL-13 production. Gut. 2011;60:449–455. doi: 10.1136/gut.2010.226860. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.