

Abstract
Physiological homeostasis is essential for organism survival. Highly responsive neuronal networks are involved but constituent neurons are just beginning to be resolved. To query brain serotonergic neurons in homeostasis, we used a synthetic GPCR (Di)-based neuronal silencing tool, mouse RC∷FPDi, designed for cell type-specific, ligand (clozapine-N-oxide, CNO)-inducible and reversible suppression of action potential firing. In mice harboring Di-expressing serotonergic neurons, CNO administration by systemic injection attenuated the chemoreflex that normally increases respiration in response to tissue CO2 elevation and acidosis. At the cellular level, CNO suppressed firing rate increases evoked by CO2/acidosis. Body thermoregulation at room temperature was also disrupted following CNO triggering of Di; core temperatures plummeted, then recovered. This work establishes that serotonergic neurons regulate life-sustaining respiratory and thermoregulatory networks, and demonstrates a noninvasive tool for mapping neuron function.
Normal mammalian cell and organism function requires relative constancy around optimal internal physiological conditions. Maintaining this dynamic equilibrium involves neuronal networks affecting numerous brain and body systems. One such homeostatic reflex, the respiratory chemoreflex, controls ventilation in response to deviations in arterial and brainstem pH/PCO2 (partial pressure of CO2) (1-6). Various classes of brainstem cells (3, 7-11) have been implicated, with integration and redundancy among components likely essential. Serotonergic neurons of the lower brainstem have been proposed as one critical constituent (3, 12-16). They also have been implicated in other homeostatic circuitry such as the thermoregulatory network for body temperature maintenance (17-19). Fatal or life-threatening clinical disorders of homeostatic dysfunction also point to serotonergic involvement, such as in the sudden infant death syndrome (SIDS) (20, 21) and serotonin syndrome (22). Direct evidence demonstrating a requirement for serotonergic neurons in homeostasis, however, is only now emerging (15, 18) as tools with sufficient resolving power become available.
To test if serotonergic neuron activity is critical to homeostatic control, we engineered a genetic tool, allele RC∷FPDi (Fig. 1A), for suppressing neuron excitability in conscious mice inducibly, reversibly, with cell-subtype precision, and with minimal invasiveness. Conditional intersectional genetics (23-25) were used to switch on expression of the synthetic receptor Di (DREADD, hM4D) (26), a Gi/o-protein coupled receptor (GPCR) with engineered selectivity for the biologically inert synthetic ligand clozapine-N-oxide (CNO) while being refractive to endogenous ligands. Like endogenous Gi/o-GPCRs, the binding of CNO by Di has been shown capable of triggering cell-autonomous hyperpolarization and diminished cell excitability (26-28). RC∷FPDi, a knock-in ROSA26 allele utilizing the CAG promoter (29) offers non-viral means for Di expression, exploiting the dual-recombinase methodology endowing Cre and Flpe transgenics with the potential to switch-on Di expression in combinatorially-defined neuron subtypes, as previously demonstrated for various intersectional alleles (23-25).
Fig. 1.
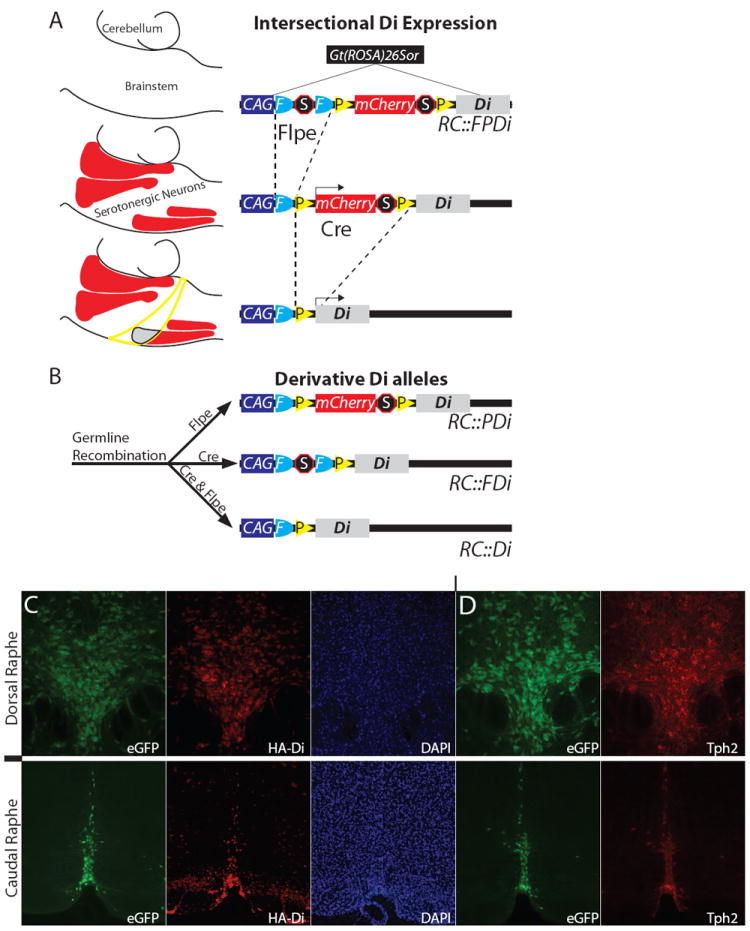
Cell-selective Di expression via RC∷FPDi. (A) This Gt (ROSA)26Sor knock-in allele consists of CAG regulatory elements, an FRT-flanked transcriptional Stop, a loxP-flanked mCherry-Stop, and HA-tagged-Di-encoding sequence. A hypothetical example (left) illustrates intersectional restriction of Di to a serotonergic neuron subset (gray area, lower schema) after Flpe- and Cre-recombination; mCherry marks Flpe-only cells (red areas represent serotonergic nuclei); neither Di nor mCherry are expressed in Cre-only cells (yellow circumscribed area). (B) Derivative Di alleles. (C, D) In RC∷PDi, RC∷rePe, Slc6a4-cre brainstems, we observed concurrent reproducible expression of HA-tagged-Di (far-red indirect HA-immunofluorescence), eGFP, and Tph2 (indirect immunofluorescence). DAPI, nuclear stain 4’,6-diamidino-2-phenylindole.
To meet needs for single recombinase-mediated Di expression (versus the dual-recombinase strategy), we generated derivatives of RC∷FPDi (Fig. 1B): RC∷PDi requires only Cre to switch-on Di expression because the FRT-cassette was excised in the ancestral germ line; reciprocally, RC∷FDi requires only Flpe to switch-on Di expression. By partnering RC∷PDi with Slc6a4-cre, we expressed Di in virtually all serotonergic neurons (Fig. 1C, D) and a small subset of thalamic neurons. We also incorporated a Cre-responsive GFP allele, RC∷rePe, to fluorescently visualize the Slc6a4-cre-lineage (Fig. 1C, D), thereby facilitating validation of lineage-specific Di expression (Fig. 1D) as well as electrophysiological study of Di-expressing neurons (Fig. 2).
Fig. 2.
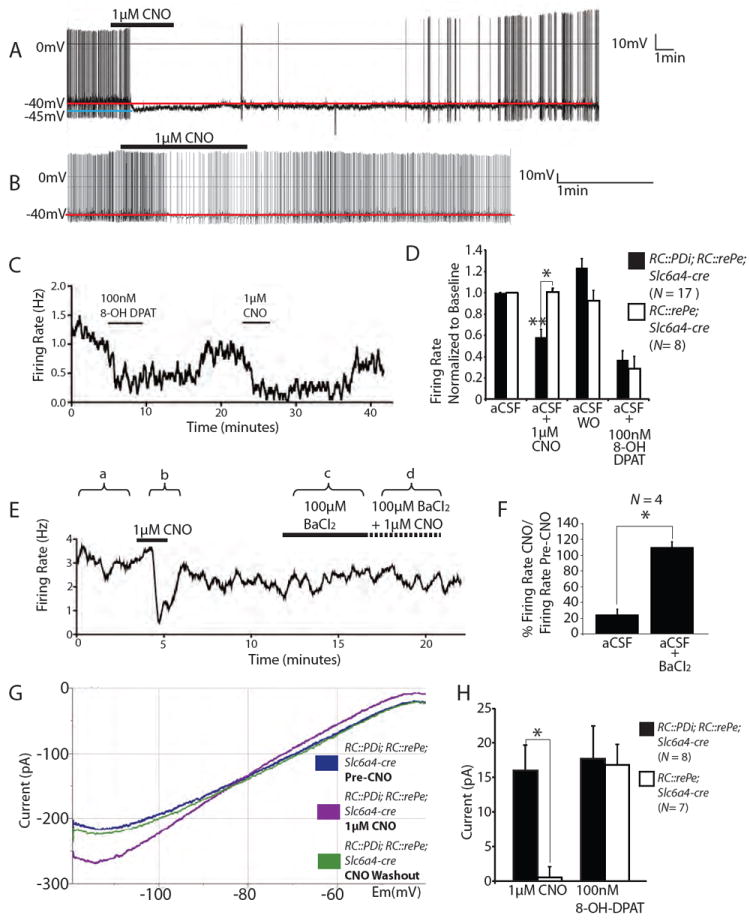
Inducible and reversible suppression of serotonergic neuron excitability using RC∷PDi. (A, B) Recordings of cultured medullary serotonergic neurons from RC∷PDi; RC∷rePe; Slc6a4-cre mice showing CNO-induced abolishment (A) or moderate suppression (B) of action potential firing, with recovery following return to artificial cerebrospinal fluid (aCSF) superfusate. (C) Firing rate during sequential applications of 8-OH-DPAT and CNO. (D) Average firing rates (normalized to baseline ± SEM) of Di/GFP-expressing versus control neurons. Inhibition by CNO was observed for Di-neurons compared to: pre-CNO, post-CNO aCSF superfusate (aCSF washout (WO) of CNO), and to CNO-exposed control neurons, **p < 0.0001 (Friedman test), *p < 0.005 (Mann-Whitney test). CNO-inhibition was comparable to that of 8-OH-DPAT (N = 9 RC∷PDi; RC∷rePe; Slc6a4-cre neurons; N = 8 control neurons). (E) Sample trace of BaCl2-mediated block of CNO-induced suppression. (F) Average ratio of firing rate for CNO versus pre-CNO, expressed as a percentage ± SEM. Neurons were assayed in aCSF (a and b in (E)) and upon subsequent application of BaCl2 (c and d) *p < 0.05 (Friedman test). (G) Voltage clamp recording showing the current-voltage relationship of a Di-expressing serotonergic neuron with or without CNO. (H) Average current elicited by CNO and 8-OH-DPAT at -60 mV, *p = 0.002 (Mann-Whitney test).
To test Di in modulating serotonergic neuron activity, as driven by RC∷PDi, we made current-clamp membrane potential recordings (Fig. 2) from lower brainstem serotonergic neurons cultured from RC∷PDi; Slc6a4-cre mice, again coupled with RC:rePe to fluorescently identify Cre- and thus Di-expressing neurons. CNO reduced the firing rate of Di-expressing neurons by ~40% on average (Fig. 2D) – a reduction similar to that successively observed in the same neurons following application of 8-OH-DPAT (Fig. 2C, D), an agonist for the endogenous inhibitory serotonin autoreceptor 5HT1A, which is a GPCR known to act through Gi/o and Kir3 channels to inhibit serotonergic neuron excitability (27, 30). The response onset to CNO occurred on average 1.3 ± 0.1 (SEM) min after switching the perfusion port from a CSF to CNO, with bath superfusate exchange taking ~1 minute, suggesting response onset within seconds. Peak responses to CNO occurred 2.8 ± 0.3 (SEM) min after port switching, thus within ~2 min of CNO exposure. The CNO-induced suppression of action potential firing reversed on return to control superfusate (Fig. 2A-D), reaching full recovery on average in 12.4 ± 2.9 (SEM) min (though with notable variation from 0.9-25.4 min) loosely correlating with duration of CNO exposure. The CNO-induced suppression could be blocked by the potassium channel inhibitor barium chloride (300μM) (Fig. 2E,F), consistent with a Kir3 mechanism of neuron inhibition. There was neuron-to-neuron variation in firing rate suppression not only in response to CNO (0.58 ± 0.07 (SEM) normalized to baseline values pre-CNO, Fig. 2D) but also to 8-OH-DPAT (0.37 ± 0.09 (SEM)), likely reflecting endogenous heterogeneity among 5-HT neuron subtypes – not all serotonergic neurons express 5HT1A autoreceptors (31), and they may also not all express Kir3-type channels.
The current-voltage relationships characterizing Di-expressing serotonergic neurons revealed activation, by CNO/Di, of an inwardly rectifying conductance reversing near the predicted reversal potential for potassium (predicted EK = -81 mV, measured EK = -78 ± 1.9 (SEM) mV) (Fig. 2G). Responsive neurons (8 of 17 in this assay) demonstrated an increase in slope conductance averaging 2.7 ± 1.1 (SEM) pS between -110 and -90 mV, and a hyperpolarizing current of 16 ± 3.6 (SEM) pA at -60 mV (Fig. 2H) ranging between 5-45 pA across potentials of -60 to -50 mV – a resting-potential range inclusive of most serotonergic neurons (16, 32). The response to 8-OH-DPAT in the same neurons was similar, with hyperpolarizing currents of 17.7 ± 4.7 (SEM) pA at -60 mV, ranging between 5-85 pA across -50 to -60 mV, and an increase in slope conductance of 4.7 ± 1.3 (SEM) pS between -110 and -90 mV. Control serotonergic neurons (Fig. 2D, H) exhibited a similarly robust response to 8-OH-DPAT but not to CNO, indicating that CNO in the absence of Di does not measurably affect membrane conductance or action potential firing rate. Collectively, these findings point to CNO/Di, like 8-OH-DPAT and the 5HT1A autoreceptor, evoking hyperpolarizing outward potassium currents via Kir3-type channels.
Hypercapnic acidosis, a decrease in tissue pH caused by PCO2 elevation from ventilatory dysfunction or cellular metabolism, is a powerful respiratory stimulus. To assess a requirement for serotonergic neuron activity in the CO2-driven respiratory chemoreflex, we measured the ventilatory response of adult RC∷PDi; Slc6a4-cre mice and sibling controls to an increase in inspired CO2 from 0% (room air) to 5% (a modest rise) before and after CNO injection (Fig. 3A-C). The typical increase in ventilation, for restoring normal arterial pH/PCO2 levels, was reduced by ~50% in RC∷PDi; Slc6a4-cre mice within minutes following CNO administration (Fig. 3B). Prior to CNO, CO2 exposure evoked a ventilatory response comparable to that seen in control siblings, indicating that Di expression alone does not affect the chemoreflex. Nor is CNO alone inhibitory, given the normal ventilatory response of CNO-treated control siblings (Fig. 3C). Similar results were obtained when the Slc6a4-cre driver was replaced with Pet1∷Flpe (24), an alternative serotonergic neuron driver, and partnered with RC∷FDi (Fig. S1), there by independently confirming attribution of function to serotonergic neurons.
Fig. 3.
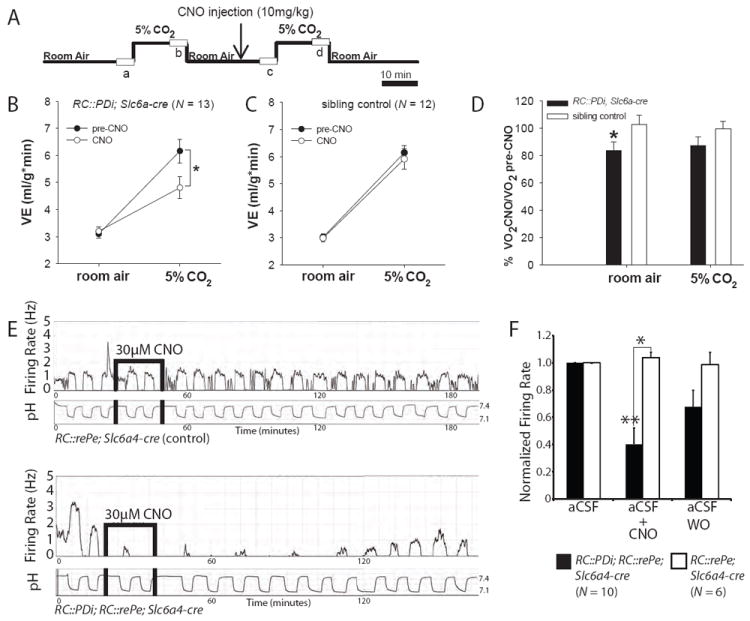
CNO/Di-perturbation of serotonergic neurons disrupts the central respiratory CO2 chemoreflex. (A) Protocol for plethysmographic assessment at 34°C of respiratory responses to inspired CO2 in the awake animal before and after CNO administration; boxes (a, b, c, d) represent points analyzed from continuous recordings. (B) RC∷PDi; Slc6a4-cre mice upon CNO administration showed reduced minute ventilation responses to inspired CO2 as compared to pre-CNO baselines, *p = 0.002 (RM ANOVA followed by Tukey post-hoc). (C) Pre-CNO and CNO responses to CO2 were indistinguishable for control mice and from the pre-CNO response of RC∷PDi;Slc6a4-cre mice. (D) RC∷PDi;Slc6a4-cre mice exhibited reduced VO2 upon CNO administration as compared to controls, *p < 0.05 (paired t-test). (E) Firing rate and simultaneous bath pH recordings from cultured serotonergic neurons from controls (top) compared to RC∷PDi; RC∷rePe; Slc6a4-cre mice (bottom). Changing CO2 from 5% to 9% shifted pH from ~7.4 to ~7.16 and induced an increase in firing rate. In Di-neurons (bottom), this response was inhibited by CNO and reversed on return to aCSF. (F) Peak firing rates (normalized to baseline) in 9% CO2-saturated aCSF were suppressed with CNO application in RC∷PDi; RC∷rePe;Slc6a4-cre neurons, *p < 0.05 (Mann-Whiteney test).
Room air ventilation was unaffected following CNO/Di manipulation of Slc6a4-cre-expressing neurons (Fig. 3A-D). This is reminiscent of phenotypes reported for adult mice in which embryonic deletion of the gene lmx1b in Pet1-expressing cells results in developmental loss of most if not all serotonergic neurons (18). Notably, following CNO injection, oxygen consumption in room air dropped from within to below normal range (0.053 ± 0.003(SEM) ml/g/min to 0.043 ± 0.003(SEM) ml/g/min) in RC∷PDi; Slc6a4-cre mice but not controls (Fig. 3D). Thus, Di-mediated perturbation of Slc6a4-cre-expressing neurons led to a decrease in metabolic rate without proportionately matching effects on ventilation. An additional role for serotonergic neurons may thus be supported – in influencing metabolic rate and the ability of ventilation to properly track with metabolic state.
Triggering the chemoreflex requires sensing and transducing milieu changes in pH/PCO2 into cellular activity changes such as in action potential firing rates capable of affecting respiratory circuit output and breathing. We found that cultured serotonergic neurons from the lower brainstem reproducibly increased their firing rate 2-3 fold in response to acidosis (0.2-0.3 pH units, a magnitude of physiological relevance) caused by increasing the CO2 bubbled in the superfusate from 5 to 9% (Fig. 3E and Fig. S2), consistent with previous findings in serotonergic neurons from rats (13, 16). Approximately 70% of medullary serotonergic neurons exhibit chemosensitivity, in line with the view that not all medullary serotonergic neurons project to brainstem respiratory centers and that this property likely distinguishes a specific functional subset of serotonergic neurons. Because recordings were performed under conditions of relative synaptic isolation (blockade of GABAA, NMDA, and AMPA/kainate receptors by picrotoxin, AP5, and CNQX, respectively), the observed chemosensitivity may be an intrinsic property of these serotonergic neurons. CNO administration inhibited this chemoresponsiveness (1μ M (Fig. 2S) or 30 μM (Fig. 3E, F), concentrations less than or comparable to that delivered in vivo), which could be restored on return to control superfusate; control, non-Di-expressing serotonergic neurons showed no response to CNO (Fig 3E, upper panel). CNO was continuously applied over 2-3 pH cycles for on average 20 min (versus the ~5 min exposure during firing rate assays (Fig. 2A-D)); recovery times were longer (76.3 ± 60.7 (SEM) min) following this extended CNO exposure.
These data support the parsimonious model whereby a serotonergic neuron subset in the lower brainstem act as central respiratory chemoreceptors capable of regulating the downstream respiratory network, and thus lung ventilation, in an attempt to restore normal arterial pH/PCO2 (3, 12, 14, 18, 33). PCO2 stabilization is also served by Phox2b-expressing glutamatergic neurons of the brainstem retrotrapezoid nucleus (34, 35). It will be important to determine how these and likely other (3, 7-9, 35) neural systems integrate to control breathing and under what developmental stages, arousal states, and conditions they may be differentially employed. Astrocytes along the brainstem surface have also been implicated as respiratory chemosensors, capable of influencing retrotrapezoid neuron activity (11, 36); it will be important to assess whether astrocytes also influence serotonergic neurons and whether their effects on chemoreception persist in conscious animals.
During the whole-body plethysmography (Fig. 3), chamber temperature was held at ~34°C to reduce effects of body temperature fluctuations on respiratory parameters and oxygen consumption; under these conditions, core body temperature remained within ± 0.65 (SEM) °C of baseline for controls and Di-mice. Absent this thermoneutral environment, double transgenic RC∷PDi; Slc6a4-cre mice dropped body temperature within minutes of CNO administration. While housed individually at room temperature (~23°C), RC∷PDi; Slc6a4-cre mice dropped body temperature from 36.9 ± 0.2(SEM)°C to 30.33 ± 0.2 (SEM)°C within 30 min of CNO injection, dipping to 27.1 ± 0.9(SEM)°C by ~2.5 h (Fig. 4A), after which recovery ensued with restoration to within normal range by 11.8 ± 1.3(SEM) h. Sibling controls receiving CNO showed normal thermoregulation (37.2 ± 0.03 (SEM)°C). These findings establish that Slc6a4-cre-expressing neuron activity is required for adult thermal homeostasis, with most thermoregulatory capacity lost given the near equilibration of body temperature to ambient room temperature. The kinetics of body temperature change (Fig. 4A) reflect, to a large degree, body thermal inertia, contrasting the more rapid response observed for individual neurons (Fig. 2). Similar body temperature dysregulation was observed on replacing Slc6a4-cre; RC∷PDi with Pet1∷Flpe (24); RC∷FDi (Fig. S3), thereby independently confirming attribution of function to serotonergic neurons (albeit the extent of temperature dysregulation is less severe, likely because the Pet1∷Flpe driver shows mosaicism in recombinase expression in medullary serotonergic neurons). Delineating the effectors of body temperature homeostasis acting downstream of serotonergic circuitry will be an important next step.
Fig. 4.
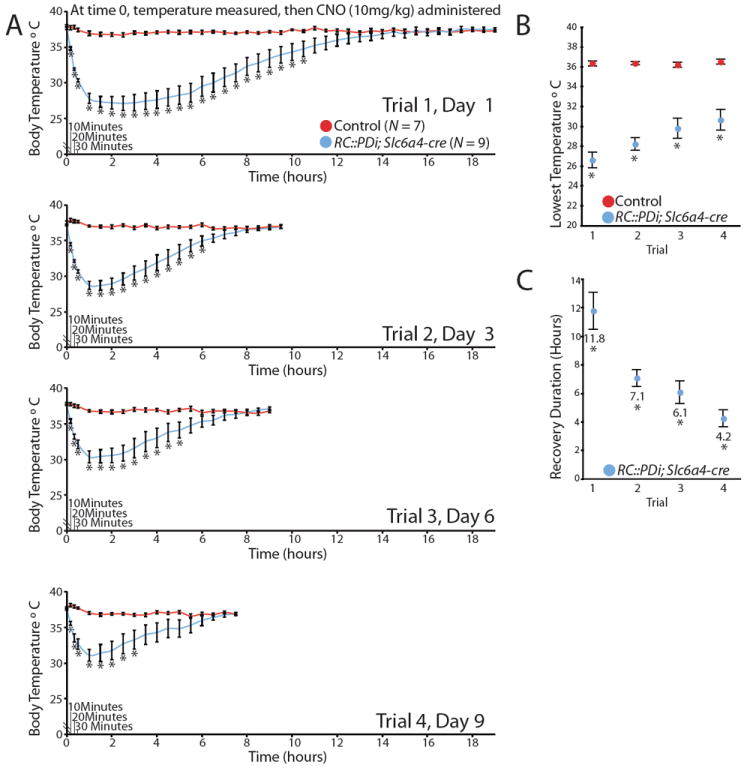
CNO/Di-inhibition of serotonergic neurons induced severe yet reversible and repeatable hypothermia. Trials consisted of body temperature assessments taken at room temperature just before a single CNO administration and then every 10 min for the first half hour, followed by every 30 min until recovery. Animals underwent 4 sequential trials. (A) Body temperature averages of RC∷PDi; Slc6a4-cre mice versus controls before and after CNO injection, *p < 0.05 (unpaired t-test). (B) Average lowest temperatures achieved per trial, *p < 0.05 (a one-way repeated measures ANOVA). (C) Average duration to return to 36°Cfollowing CNO injection, *p < 0.01 (a one-way repeated measures ANOVA).
By contrast to the response evoked upon acute serotonergic neuron inhibition, adult mice devoid of serotonin-producing neurons from midgestation onward are able to maintain a near wild-type body temperature (~36-38°C) while housed at room temperature (~24°C) (14, 18). This phenotypic difference indicates that compensatory circuitry arises in the face of developmental loss of serotonergic neurons and highlights how acute neuron perturbation avoids such confounds.
Repeated neuron perturbation in the same animal is possible. With each round of CNO administration, body temperature plummeted within minutes (Fig. 4A); the severity of the induced hypothermia, though, showed modest adaptation with each subsequent trial (Fig. 4B). Time required for return to baseline body temperature also showed adaptation (Fig. 4C). We observed no overt behavioral deficits (short- or long-term) following these bouts of CNO/Di-triggered hypothermia, perhaps not surprising given rodents’ capacity for daily torpor.
Using this RC∷FPDi strategy, we have revealed impaired respiratory and body temperature control upon acute perturbation of serotonergic neuron activity, providing direct evidence that serotonergic neurons play key roles in the central chemoreflex and thermoregulation. These findings offer potential mechanistic explanation for fatal or life-threatening disorders of homeostasis that associate with serotonin abnormalities, such as in SIDS and the serotonin syndrome. In SIDS, the leading cause of death in children between 1 month and 1 year of age (21), multiple abnormalities in the brainstem serotonergic system have been identified including serotonin insufficiency (20, 21). Our findings suggest that such insufficiency might compromise an infant’s respiratory response to hypercapnic acidosis that may occur upon rebreathing exhaled air as a result of sleeping prone in the face-down position (SIDS infants are often found prone), contributing to respiratory failure and death. By contrast, serotonin syndrome is a disorder of serotonin excess and extreme hyperthermia, shivering, seizures, coma and in some cases death that can result acutely from serotonin drug interactions (22). This association between serotonin excess and hyperthermia is consistent with our reciprocal findings of serotonergic inhibition inducing hypothermia. We presume that in vivo CNO/Di signaling suppresses action potential firing, resulting in net inhibition of serotonergic neuron activity. Other scenarios are possible, such as net excitation, but seem improbable given: the whole animal phenotypes observed upon CNO/Di signaling in serotonergic neurons; the molecular mechanism by which Di appears to act; and the paucity of evidence that net excitation could occur. For example, such net excitation has not been observed in cultured serotonergic neurons nor in extensive studies on 5HT1A receptor agonists in vivo or ex vivo, which act mechanistically like CNO/Di (37, 38). Assessing the precise in vivo electrophysiological consequences of CNO/Di signaling on serotonergic neuron activity including chemoresponsiveness awaits means to record from individual medullary serotonergic neurons over the course of hours (like Fig. 3E) in unanesthetized, conscious mice. By contrast, recordings under either anesthesia (15, 39-41) or decerebration (42, 43) suffer from the confound of perturbing serotonergic neuron activity and chemoreflex properties – the very processes under examination.
The present studies, as well as our analyses of broad constitutive Di expression and activity (Fig. S4), establish RC∷FPDi as a neuronal perturbation tool featuring in vivo: ligand-inducibility within seconds-to-minutes, inhibition for minutes-to-hours, reversibility within hours, intra-animal repeatability, and high cell-subtype selectivity (including anatomically dispersed populations) and thus resolution for functional mapping. This tool can be applied in the awake, freely behaving animal without confounding interference from anesthesia, surgical procedures like cannulation, or compensatory changes in circuitry that can develop in response to constitutive genetic alterations. RC∷FPDi can be applied to map other behaviors in which dysregulation of specific populations has been implicated.
MATERIALS AND METHODS
VECTOR CONSTRUCTION AND MOUSE DERIVATION
The RC∷FPDi targeting vector was derived from our pRC∷FPmodular plasmid which contains CAG promoter/enhancer sequence (45), an FRT- flanked cassette consisting of PKG-neo sequence (for positive selection of homologous recombinants) and the lox2 transcriptional stop cassette derived from pBS302 (46), a loxP-flanked cassette containing mCherry-encoding sequence followed by a concatemer of SV40pA stop sequences (47), and followed by sequence encoding HA-tagged Di (Di template plasmid (48) provided by Dr. Bryan Roth). Once validated in cell culture for recombination efficacy, this entire transgene was inserted into the p15a_Kan_R26 plasmid containing R26 sequence as targeting homology. The completed targeting vector, pRC∷FPDi, was linearized and electroporated into ES (Tc-1) cells and resulting gancyclovir resistant colonies screened by PCR for homologous recombination at the R26 locus;, 3’ recombination detection: primer RR712 – GGGCGTACTTGGCATATGAT and primer RR754 - CGCCTAAAGAAGAGGCTGTG amplifying a 1472 bp product; 5’ recombination detection: primer RR751 – CCAGATTGTGACGAAGCAGA and primer RR763 – TCTCCCCTCAGAGAAATGGA, amplifying a 4844 bp product. Using standard methods, ES cells from a single recombinant ES clone were used to derive RC∷FPDi chimeric mice, and germline derivation achieved by crossing to C57BL/6J mice. RC∷PDi, RC∷FDi, and RC∷Di were derived by crossing RC∷FPDi mice to either129S4/SvJaeSor-Gt (ROSA)26Sortm1(FLP1)Dym mice (49) to remove the FRT-flanked stop cassette, or to Tg (ACTB-cre)2Mrt (50) to remove the loxP-flanked stop cassette.
IMMUNOHISTOCHEMISTRY
Anesthetized RC∷PDi; RC∷rePe; Slc6a-cre mice (N = 3) were perfused with phosphate buffered saline (PBS) followed by 4% paraformaldehyde in PBS. Brains were extracted, soak-fixed in 4% paraformaldehyde at 4°C for 2 h, cryoprotected in 30% sucrose/PBS and then cryosectioned (40 μm) and processed as floating sections. Sections were bleached in 1% H2O2/PBS for 30 min, blocked with 5% donkey serum/0.1% TritonX-100/PBS and detected with an anti-HA antibody for 72 h at 4°C. Biotin-conjugated donkey anti-rabbit secondary antibody was reacted with avidinbiotin complexing reagents and developed with tyramide conjugated Alexafluor 647 (far red channel) for visualization. Similarly, GFP was detected using a chick anti-GFP antibody and visualized with a FITC-conjugated donkey anti-chick antibody (green channel). Tph2 (rate limiting enzyme in serotonin synthesis in the CNS, thus a key serotonergic neuron identity marker) was detected using a rabbit polyclonal antibody and visualized by donkey anti-rabbit antibody conjugated to Cy5 (far red channel). Cell nuclei were visualized with DAPI, 4’,6-diamidino-2-phenylindole.
PLETHYSMOGRAPY AND EKG
Awake, freely moving mice were assayed for respiratory parameters and heart rate using continuously recorded whole body plethysmography and EKG. For double transgenic RC∷PDi; Slc6a4-cre mice and sibling controls, mice were placed in a room-air plethysmograph chamber held at 34°C, acclimatized for 20 min of which recordings from the last 5 min (window “a” in Fig. 3A) were used later for data analyses. Chamber gases were then shifted to 5%CO2 (in air) for 15 min, of which the last 5 min of recordings (window “b” in Fig. 3A) were used for data analyses. Chamber gas was then switched back to room air for 15 min, mice were removed briefly for an intraperitoneal injection of CNO dissolved in saline (1 mg/ml) to an effective concentration of 10mg/kg CNO and then returned immediately to the room-air chamber for an additional 10 min of recordings, of which the last 5 min (window “c” in Fig. 3A) were used for data analyses. Mice were then exposed to a 5%CO2 gas mixture for 15 min, of which the last 5 min of recordings (window “d” in Fig. 3A) were used for data analyses.
In these plethysmography experiments, respiratory airflow was assayed in a 140 ml water-jacketed temperature controlled glass chamber attached to a differential pressure transducer and reference chamber. Water temperature was warmed to 35.1°C, resulting in a chamber temperature of 34°C (thermoneutral zone for mouse) as required to maintain constancy in respiratory and metabolic parameters from trial to trial as well as to maintain linearity of ventilatory pressure changes due to humidification, rarefaction, and thermal expansion and contraction during inspiration and expiration. Gas flow through the chamber was regulated by inline pre- and post-gas flow meters. Flow rate was ~ 325 ml/min. Volume calibrations were performed by repeated injections of a known volume from a 1 ml syringe. Oxygen consumption was determined by measuring the difference between oxygen concentration of gas into the chamber and gas exiting the chamber with an oxygen sensor and oxygen analyzer. Humidified gas flow into the chamber was either room air or a 5%CO2 mix, balanced with air (medical grade) as needed for each experiment. For EKG, the ventral thoracic region was prepped with a depilatory cream. A vest made from stretchable bandage material was used to hold the EKG electrodes to the chest with EKG conductive paste. EKG was measured by a telemetry amplifier and transmitted to a base station for recording. Mice were assayed for core body temperature both before and after recording in the chamber. Readings from the pressure transducer, oxygen sensor and EKG were acquired at 1kHz and analyzed for peak amplitude, peak frequency, and average voltage off-line. Respiratory volume was determined by the following formula
Where:
A=peak of breath signal in volts
B=peak of signal for injection volume
C=volume injection (in ml)
D=mouse body temperature (in °C)
E=chamber temperature (in °C)
F=barometric pressure (in mmHg)
G=pressure of water vapor of mouse = 1.142+(0.8017*D)-(0.012*D2)+ (0.0006468*D3)
H=pressure of water vapor of chamber = 1.142+(0.8017*E)-(0.012*E2)+(0.0006468*E3)
Results were compared using two-way RM ANOVA followed by Tukey post-hoc analysis for RC∷PDi; Slc6a4-cre ventilation experiments, and paired t-test was used for comparison of oxygen consumption. RC∷FDi; Pet1∷Flpe minute ventilation experiments were analyzed in the same way as RC∷PDi; Slc6a4-cre ventilation experiments. Standard error of the mean (SEM) is shown for all plethysmographic data.
For RC∷Di genotypes and sibling controls, mice were placed in a room-air plethysmograph chamber held at 34°C, again as required to maintain constancy in respiratory and metabolic parameters from trial to trial as well as to maintain linearity of ventilatory pressure changes due to humidification, rarefaction, and thermal expansion and contraction during inspiration and expiration. Mice were acclimatized for 15 min, and then assayed for respiratory and heart rate values for 5 min. Mice were then briefly removed from the chamber for intraperitoneal injection of CNO dissolved in saline (1mg/ml) to an effective concentration of 10mg/kg. Mice were immediately returned to the chamber and respiratory and heart rate assessments resumed.
TEMPERATURE ASSAYS
For RC∷PDi; Slc6a4-cre experiments, temperature was taken rectally with a lubricated thermocouple probe. Mice were weighed and temperature was taken before 10mg/kg CNO administration. After CNO intraperitoneal injection, temperature was taken every 10 min for the first half hour and every half hour thereafter until all mice recovered to a temperature of at least 36°C or higher. Because of the extremely long duration of the temperature assays to examine recovery, mice were held in standard cages at room temperature 21-23°C with water and food ad libitum and no 4°C cold challenge was used. For initial RC∷FDi; Pet1∷Flpe; experiments, mice were weighed and temperature taken before CNO administration (10mg/kg). After CNO administration, using a protocol modeled after Hodges et al. (51) mice were kept at room temperature for 30 min, their temperature taken, and they were then transferred to pre-chilled cages in a 4°C cold room. Body temperature was then assessed every 10 min for the first hour and every half hour for the second hour. The experiment was concluded after two hours of 4°C exposure.
The results between RC∷PDi; Slc6a4-cre and control siblings were compared using an unpaired t-test for each temperature assessment, similarly for RC∷FDi; Pet1∷Flpe and control mice. For comparing recovery times and lowest achieved temperature across trials in RC∷PDi; Slc6a4-cre experiments, a one-way repeated measures ANOVA analysis was used. SEM is shown on the graphs.
PRIMARY NEURONAL CULTURES
For primary mouse neuronal cultures, a ventral wedge shaped block of tissue (approximately 1.5-2mm per side with the apex oriented dorsally and the base spanning the ventral medullary surface) running the length of the medulla and caudal pons was dissected from postnatal (P0-P3) mice into ice-cold HEPES buffer (in mM: NaCl 130, KCl 4, MgCl2 1, CaCl2 1.5, HEPES 10, Dextrose 10, NaOH 3), digested for 30 minutes at 37°C in HEPES buffer with 1.3% papain, washed with complete MEM (MEM with 10% FBS, 1% Pen/Strep) with 0.15% trypsin inhibitor and 0.15% bovine serum albumin, triturated with a fire polished Pasteur pipette, and plated on 12mm round, poly L-ornithine pretreated coverslips in 12-well plates. After allowing cells to settle onto the coverslips for 45-60 min, complete MEM conditioned by mouse glial cultures was added. After 24 h, half of the medium for each well was exchanged with Neurobasal medium with B27 (1:50 Neurobasal), FGF-5 (10ng/ml) and BFGF (1ng/ml). After a week of culturing, supportive glial beds had formed and Ara-C (0.5-1μM) was added to attenuate glial growth. Cells were maintained at 37° C and 5% CO2 with weekly half-medium changes with Neurobasal/B27 medium for 4-12 weeks.
ELECTROPHYSIOLOGY
Neuron cultures were placed in a recording chamber mounted on a fixed-stage upright microscope equipped with 5x and 40x water immersion objectives, Nomarski optics, epi-fluorescence illumination and filter sets for detection of GFP. The recording chamber was continuously superfused (2.5-3ml/min) at room temperature with one of the solutions described below. Epi-fluorescence microscopy was used to identify GFP+ neurons. Patch electrodes were fabricated from capillary glass on a horizontal pipette puller to a DC resistance of 5-10MΩ after filling with intracellular solutions and connected to the headstage of an amplifier.
A CNO dose of 30μM was initially calculated as comparable to the 10mg/kg dose given to the mice during in vivo assays assuming ~70% of body weight is soluble to CNO and CNO is evenly distributed. As in vitro assays progressed, we found that 1μM CNO elicited similar responses. Except where otherwise noted, all electrophysiological assays used 1μM CNO concentrations.
For experiments measuring the current-voltage relationship, patch pipettes filled with intracellular solution (in mM: KOH 135, methanesulfonic acid 135, KCl 10, Hepes 5, EGTA 1, ATP 3, GTP 0.5, pH7.2) were used to form a gigaseal. The membrane was then ruptured to allow whole-cell access. Experiments were carried out in voltage clamp mode using pCLAMP 10 software. A stopcock system was used to switch between baseline Ringer’s (aCSF) solution (in mM: NaCl 121, KCl 6, MgCl2 2, CaCl2 2, NaH2PO4 1.3, NaHCO3 26, dextrose 10; pH 7.4 at 5%CO2/95%O2) and identical solutions with the addition of either 1 μM CNO or 100 nM 8-OH-DPAT with an approximate lag time of 1 min. Holding potential was -60mV. A slow (3 s) voltage ramp protocol from -120 to -40mV was repeated every 3 s. Sweeps under each condition were averaged. Current differences were analyzed at -60mV. A Shapiro-Wilk normality test suggested a non-normal data set, therefore a nonparametric Mann-Whitney test was used to compare RC∷PDi; RC∷rePe; Slc6a4-cre and control neurons.
For firing rate experiments, neurons were continuously superfused with Ringer’s solution (aCSF) containing synaptic blockers (in mM: NaCl 124, KCl 3, MgCl2 2, CaCl2 2, NaH2PO4 1.3, NaHCO3 26, dextrose 10, picrotoxin 0.1, AP-5 0.05, and CNQX 0.01; pH 7.4 at 5%CO2/95% O2) and current clamp membrane potential recordings were made using the perforated patch recording technique (48μM gramicidin included in intracellular solution). If neurons were not spontaneously active, depolarizing current was injected such that baseline firing rate was 1-2 Hz. A subset of neurons was tested with both CNO and 8-OH DPAT in random order. A Shapiro-Wilk normality test suggested a non-normal data set, thus results were compared using a Friedman test (a nonparametric repeated measures test comparing multiple groups), and a nonparametric Mann-Whitney test was used for comparison of RC∷FPDi; RC∷rePe; Slc6a4-cre and RC∷rePe; Slc6a4-cre firing rates upon CNO application, normalized to baseline.
To test sensitivity of the response to BaCl2 during current clamp membrane potential recordings, neurons were first superfused with 1μM CNO to establish their responsiveness. After washout of CNO, neurons were superfused with standard Ringer’s (aCSF) solution with synaptic blockers supplemented with 100μM BaCl2. Once a steady baseline was established with BaCl2 present, a solution containing both 100μM BaCl2 and 1μM CNO was applied. The firing rate during the first CNO application was normalized to baseline (standard Ringer’s aCSF, no BaCl2) and the firing rate during the second CNO application (with BaCl2) were normalized to the firing rate in standard Ringer’s supplemented with BaCl2. These ratios were represented as percentages and graphed with standard error of the mean. Data was compared with a Friedman test.
To test CO2/pH sensitivity, changing the bath solution to 9% CO2 / 91% O2 resulted in a change in extracellular pH from 7.4 to approximately 7.16 while bath pH was continuously measured with a pH electrode at the inflow to the recording chamber. Recordings were made from cultures 20-35 days in vitro, a range when serotonergic neurons demonstrate the maximum chemosensitivity index (52). Peak firing rates during 9% CO2 were determined for 1) challenges pre-CNO (baseline aCSF), 2) challenges during CNO application plus 3 challenges after CNO application (aCSF + CNO) and 3) the last 2 challenges of the recording (aCSF WO). For each neuron, the averages for condition 2 (aCSF + CNO) and 3 (aCSF WO) were normalized to condition 1 (baseline aCSF). The first half of the data was collected using 30μM CNO, however, as stated above, we found that 1μM CNO elicited similar responses in this assay. No differences were seen, thus data using 30μM and 1μM CNO was grouped for this assay. Data was compared using a Friedman test. A Mann-Whitney test was used for comparison of RC∷FPDi; RC∷rePe; Slc6a4-cre and RC∷rePe; Slc6a4-cre responses to CNO.
In all cases, data are shown as mean ± SEM
Supplementary Material
Acknowledgments
This work was supported by the following National Institutes of Health Grants, F32HD063257-01A1 (RR); 5R21DA023643-02 (RB, SD); 5R21MH083613-02 (RR, JCK, SD); 5P01HD036379-13 (RR, AC, RB, JCK, EN, GR, SD); 5R01HL028066-30 (AC, EN); 5R01HD052772 (GR). Clozapine-N-Oxide used in some preliminary experiments was provided through NIH Rapid Access to Intervention Development Program. The authors wish to thank Bryan Roth for providing the Di-encoding cDNA, and Drs Bryan Roth, Wade Regehr, Court Hull, and Bernardo Sabatini and members of the Dymecki lab for helpful discussions as well as Yuanming Wu and Jia Jia Mai for technical assistance.
Footnotes
Supporting Online Material
Materials and Methods
References (45-53)
The authors declare no conflict of interests
References
- 1.Leusen IR. Chemosensitivity of the respiratory center; influence of CO2 in the cerebral ventricles on respiration. Am J Physiol. 1954 Jan;176:39. doi: 10.1152/ajplegacy.1953.176.1.39. [DOI] [PubMed] [Google Scholar]
- 2.Kety SS, Forster RE, Comroe Julius H., Jr March 13, 1911-July 31, 1984. Biogr Mem Natl Acad Sci. 2001;79:66. [PubMed] [Google Scholar]
- 3.Nattie E, Comroe Julius H., Jr distinguished lecture: central chemoreception: then … and now. J Appl Physiol. 2011 Jan;110:1. doi: 10.1152/japplphysiol.01061.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Guyenet PG, Stornetta RL, Bayliss DA. Central respiratory chemoreception. J Comp Neurol. 2010 Oct 1;518:3883. doi: 10.1002/cne.22435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Smith CA, Rodman JR, Chenuel BJ, Henderson KS, Dempsey JA. Response time and sensitivity of the ventilatory response to CO2 in unanesthetized intact dogs: central vs. peripheral chemoreceptors. J Appl Physiol. 2006 Jan;100:13. doi: 10.1152/japplphysiol.00926.2005. [DOI] [PubMed] [Google Scholar]
- 6.Haldane JS, Priestley JG. The regulation of the lung-ventilation. J Physiol. 1905 May 9;32:225. doi: 10.1113/jphysiol.1905.sp001081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mitchell RA, Loeschcke HH, Massion WH, Severinghaus JW. Respiratory responses mediated through superficial chemosensitive areas on the medulla. Journal of Applied Physiology. 1963 May 1;18:523. doi: 10.1152/jappl.1963.18.3.523. [DOI] [PubMed] [Google Scholar]
- 8.Elam M, Yao T, Thoren P, Svensson TH. Hypercapnia and hypoxia: chemoreceptor-mediated control of locus coeruleus neurons and splanchnic, sympathetic nerves. Brain Res. 1981 Oct 19;222:373. doi: 10.1016/0006-8993(81)91040-4. [DOI] [PubMed] [Google Scholar]
- 9.Williams RH, Jensen LT, Verkhratsky A, Fugger L, Burdakov D. Control of hypothalamic orexin neurons by acid and CO2. Proc Natl Acad Sci U S A. 2007 Jun 19;104:10685. doi: 10.1073/pnas.0702676104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Li A, Emond L, Nattie E. Brainstem catecholaminergic neurons modulate both respiratory and cardiovascular function. Adv Exp Med Biol. 2008;605:371. doi: 10.1007/978-0-387-73693-8_65. [DOI] [PubMed] [Google Scholar]
- 11.Gourine AV, et al. Astrocytes control breathing through pH-dependent release of ATP. Science. 2010 Jul 30;329:571. doi: 10.1126/science.1190721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Corcoran AE, et al. Medullary serotonin neurons and central CO2 chemoreception. Respir Physiol Neurobiol. 2009 Aug 31;168:49. doi: 10.1016/j.resp.2009.04.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Richerson GB. Response to CO2 of neurons in the rostral ventral medulla in vitro. J Neurophysiol. 1995 Mar;73:933. doi: 10.1152/jn.1995.73.3.933. [DOI] [PubMed] [Google Scholar]
- 14.Hodges MR, Richerson GB. The role of medullary serotonin (5-HT) neurons in respiratory control: contributions to eupneic ventilation, CO2 chemoreception, and thermoregulation. J Appl Physiol. 2010 May;108:1425. doi: 10.1152/japplphysiol.01270.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Depuy SD, Kanbar R, Coates MB, Stornetta RL, Guyenet PG. Control of breathing by raphe obscurus serotonergic neurons in mice. J Neurosci. 2011 Feb 9;31:1981. doi: 10.1523/JNEUROSCI.4639-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wang W, Richerson GB. Development of chemosensitivity of rat medullary raphe neurons. Neuroscience. 1999 Mar;90:1001. doi: 10.1016/s0306-4522(98)00505-3. [DOI] [PubMed] [Google Scholar]
- 17.Morrison SF, Nakamura K, Madden CJ. Central control of thermogenesis in mammals. Exp Physiol. 2008 Jul;93:773. doi: 10.1113/expphysiol.2007.041848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hodges MR, et al. Defects in breathing and thermoregulation in mice with near-complete absence of central serotonin neurons. J Neurosci. 2008 Mar 5;28:2495. doi: 10.1523/JNEUROSCI.4729-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cano G, et al. Anatomical substrates for the central control of sympathetic outflow to interscapular adipose tissue during cold exposure. J Comp Neurol. 2003 Jun 2;460:303. doi: 10.1002/cne.10643. [DOI] [PubMed] [Google Scholar]
- 20.Duncan JR, et al. Brainstem serotonergic deficiency in sudden infant death syndrome. JAMA. 2010 Feb 3;303:430. doi: 10.1001/jama.2010.45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kinney HC, Richerson GB, Dymecki SM, Darnall RA, Nattie EE. The brainstem and serotonin in the sudden infant death syndrome. Annu Rev Pathol. 2009;4:517. doi: 10.1146/annurev.pathol.4.110807.092322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sternbach H. The serotonin syndrome. Am J Psychiatry. 1991 Jun;148:705. doi: 10.1176/ajp.148.6.705. [DOI] [PubMed] [Google Scholar]
- 23.Awatramani R, Soriano P, Rodriguez C, Mai JJ, Dymecki SM. Cryptic boundaries in roof plate and choroid plexus identified by intersectional gene activation. Nat Genet. 2003 Sep;35:70. doi: 10.1038/ng1228. [DOI] [PubMed] [Google Scholar]
- 24.Jensen P, et al. Redefining the serotonergic system by genetic lineage. Nat Neurosci. 2008 Apr;11:417. doi: 10.1038/nn2050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kim JC, et al. Linking genetically defined neurons to behavior through a broadly applicable silencing allele. Neuron. 2009 Aug 13;63:305. doi: 10.1016/j.neuron.2009.07.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Armbruster BN, Li X, Pausch MH, Herlitze S, Roth BL. Evolving the lock to fit the key to create a family of G protein-coupled receptors potently activated by an inert ligand. Proc Natl Acad Sci U S A. 2007 Mar 20;104:5163. doi: 10.1073/pnas.0700293104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mark MD, Herlitze S. G-protein mediated gating of inward-rectifier K+ channels. Eur J Biochem. 2000 Oct;267:5830. doi: 10.1046/j.1432-1327.2000.01670.x. [DOI] [PubMed] [Google Scholar]
- 28.Ferguson SM, et al. Transient neuronal inhibition reveals opposing roles of indirect and direct pathways in sensitization. Nat Neurosci. 2011 Jan;14:22. doi: 10.1038/nn.2703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Dymecki SM, Kim JC. Molecular neuroanatomy’s “Three Gs”: a primer. Neuron. 2007 Apr 5;54:17. doi: 10.1016/j.neuron.2007.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Penington NJ, Kelly JS, Fox AP. Whole-cell recordings of inwardly rectifying K+ currents activated by 5-HT1A receptors on dorsal raphe neurones of the adult rat. J Physiol. 1993 Sep;469:387. doi: 10.1113/jphysiol.1993.sp019819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bonnavion P, Bernard JF, Hamon M, Adrien J, Fabre V. Heterogeneous distribution of the serotonin 5-HT (1A) receptor mRNA in chemically identified neurons of the mouse rostral brainstem: Implications for the role of serotonin in the regulation of wakefulness and REM sleep. J Comp Neurol. 2010 Jul 15;518:2744. doi: 10.1002/cne.22331. [DOI] [PubMed] [Google Scholar]
- 32.Wang W, Pizzonia JH, Richerson GB. Chemosensitivity of rat medullary raphe neurones in primary tissue culture. J Physiol. 1998 Sep 1;511(Pt 2):433. doi: 10.1111/j.1469-7793.1998.433bh.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Dias MB, Li A, Nattie E. Focal CO2 dialysis in raphe obscurus does not stimulate ventilation but enhances the response to focal CO2 dialysis in the retrotrapezoid nucleus. J Appl Physiol. 2008 Jul;105:83. doi: 10.1152/japplphysiol.00120.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Dubreuil V, et al. Defective respiratory rhythmogenesis and loss of central chemosensitivity in Phox2b mutants targeting retrotrapezoid nucleus neurons. J Neurosci. 2009 Nov 25;29:14836. doi: 10.1523/JNEUROSCI.2623-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Marina N, et al. Essential role of Phox2b-expressing ventrolateral brainstem neurons in the chemosensory control of inspiration and expiration. J Neurosci. 2010 Sep 15;30:12466. doi: 10.1523/JNEUROSCI.3141-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wenker IC, Kreneisz O, Nishiyama A, Mulkey DK. Astrocytes in the retrotrapezoid nucleus sense H+ by inhibition of a Kir4.1-Kir5.1-like current and may contribute to chemoreception by a purinergic mechanism. J Neurophysiol. 2010 Dec;104:3042. doi: 10.1152/jn.00544.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Pineyro G, Blier P. Autoregulation of serotonin neurons: role in antidepressant drug action. Pharmacol Rev. 1999 Sep;51:533. [PubMed] [Google Scholar]
- 38.Innis RB, Aghajanian GK. Pertussis toxin blocks 5-HT1A and GABAB receptor-mediated inhibition of serotonergic neurons. Eur J Pharmacol. 1987 Nov 10;143:195. doi: 10.1016/0014-2999(87)90533-4. [DOI] [PubMed] [Google Scholar]
- 39.Gobbi G, et al. Neurokinin 1 receptor antagonism requires norepinephrine to increase serotonin function. Eur Neuropsychopharmacol. 2007 Apr;17:328. doi: 10.1016/j.euroneuro.2006.07.004. [DOI] [PubMed] [Google Scholar]
- 40.Patel AJ, et al. Inhalational anesthetics activate two-pore-domain background K+ channels. Nat Neurosci. 1999 May;2:422. doi: 10.1038/8084. [DOI] [PubMed] [Google Scholar]
- 41.Washburn CP, Sirois JE, Talley EM, Guyenet PG, Bayliss DA. Serotonergic raphe neurons express TASK channel transcripts and a TASK-like pH- and halothane-sensitive K+ conductance. J Neurosci. 2002 Feb 15;22:1256. doi: 10.1523/JNEUROSCI.22-04-01256.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hayashi F, Sinclair JD. Respiratory patterns in anesthetised rats before and after anemic decerebration. Respir Physiol. 1991 Apr;84:61. doi: 10.1016/0034-5687(91)90019-f. [DOI] [PubMed] [Google Scholar]
- 43.St-John WM, Paton JF. Characterizations of eupnea, apneusis and gasping in a perfused rat preparation. Respir Physiol. 2000 Nov;123:201. doi: 10.1016/s0034-5687(00)00177-8. [DOI] [PubMed] [Google Scholar]
- 45.Niwa H, Yamamura K, Miyazaki J. Efficient selection for high-expression transfectants with a novel eukaryotic vector. Gene. 1991 Dec 15;108:193. doi: 10.1016/0378-1119(91)90434-d. [DOI] [PubMed] [Google Scholar]
- 46.Sauer B. Manipulation of transgenes by site-specific recombination: use of Cre recombinase. Methods Enzymol. 1993;225:890. doi: 10.1016/0076-6879(93)25056-8. [DOI] [PubMed] [Google Scholar]
- 47.Dymecki SM, Kim JC. Molecular neuroanatomy’s “Three Gs”: a primer. Neuron. 2007 Apr 5;54:17. doi: 10.1016/j.neuron.2007.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Armbruster BN, Li X, Pausch MH, Herlitze S, Roth BL. Evolving the lock to fit the key to create a family of G protein-coupled receptors potently activated by an inert ligand. Proc Natl Acad Sci U S A. 2007 Mar 20;104:5163. doi: 10.1073/pnas.0700293104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Farley FW, Soriano P, Steffen LS, Dymecki SM. Widespread recombinase expression using FLPeR (flipper) mice. Genesis. 2000 Nov-Dec;28:106. [PubMed] [Google Scholar]
- 50.Lewandoski M, Meyers EN, Martin GR. Analysis of Fgf8 gene function in vertebrate development. Cold Spring Harb Symp Quant Biol. 1997;62:159. [PubMed] [Google Scholar]
- 51.Hodges MR, et al. Defects in breathing and thermoregulation in mice with near-complete absence of central serotonin neurons. J Neurosci. 2008 Mar 5;28:2495. doi: 10.1523/JNEUROSCI.4729-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wang W, Richerson GB. Development of chemosensitivity of rat medullary raphe neurons. Neuroscience. 1999 Mar;90:1001. doi: 10.1016/s0306-4522(98)00505-3. [DOI] [PubMed] [Google Scholar]
- 53.Cunliffe-Beamer TL, Les EP. In: The UFAW Handbook on the Care & Management of Laboratory Animals. 6. Poole TB, editor. 1987. pp. 275–308. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.