

Abstract
Elastin, the main component of elastic fibers, is synthesized only in early life and provides the blood vessels with their elastic properties. With aging, elastin is progressively degraded, leading to arterial enlargement, stiffening, and dysfunction. Also, elastin is a key regulator of vascular smooth muscle cell proliferation and migration during development since heterozygous mutations in its gene (Eln) are responsible for a severe obstructive vascular disease, supravalvular aortic stenosis, isolated or associated to Williams syndrome. Here, we have studied whether early elastin synthesis could also influence the aging processes, by comparing the structure and function of ascending aorta from 6- and 24-month-old Eln+/− and Eln+/+ mice. Eln+/− animals have high blood pressure and arteries with smaller diameters and more rigid walls containing additional although thinner elastic lamellas. Nevertheless, longevity of these animals is unaffected. In young adult Eln+/− mice, some features resemble vascular aging of wild-type animals: cardiac hypertrophy, loss of elasticity of the arterial wall through enhanced fragmentation of the elastic fibers, and extracellular matrix accumulation in the aortic wall, in particular in the intima. In Eln+/− animals, we also observed an age-dependent alteration of endothelial vasorelaxant function. On the contrary, Eln+/− mice were protected from several classical consequences of aging visible in aged Eln+/+ mice, such as arterial wall thickening and alteration of α1-adrenoceptor-mediated vasoconstriction. Our results suggest that early elastin expression and organization modify arterial aging through their impact on both vascular cell physiology and structure and mechanics of blood vessels.
INTRODUCTION
Elastic fibers, made of 90% elastin and 10% microfibrils, represent more than 50% of the ascending aorta dry weight.1 They are organized into circumferentially oriented lamellar units, that is, concentric elastic lamellae alternating with vascular smooth muscle cell layers (VSMC).2 By circumferential stretching, the elastic fibers of large arteries store part of the energy delivered by the ejected blood volume during systole, and they release this energy during the diastole by returning to their initial dimension. This is crucial for smoothing blood flow and pressure.3 Nevertheless, during the course of life, elastic fibers are progressively fragmented and lose their initial mechanical properties. This process, leading to a series of cellular and extracellular alterations and vascular dysfunctions, is probably the major contributor to the structural and functional alterations defining normal vascular aging4 (i.e., symptoms not related to specific diseases such as atherosclerosis or aneurysms). First, enzymatic cleavage of elastic fibers occurs progressively in normal vascular aging. The elastic fiber/lamella fragmentation leads to increases in arterial diameter and stiffness, as well as production of vasoactive elastin peptides.5–8 Lipid and calcium deposition on arterial elastic fibers also enhances the wall stiffening caused by fragmentation and may contribute to the progression of cardiovascular dysfunction.5,9 Another major feature of aging is the increased arterial wall thickness, which compensates for the effects of the elevated stress in the wall due to altered elastic fiber structure and hemodynamics, including elevated blood pressure.9 Finally, vascular cell function is also altered in normal aging, leading to a reduced response of vascular smooth muscle cells to adrenergic vasoconstrictors10 and a reduced production of the vasodilator NO by endothelial cells.11
To understand if elastin could regulate some of the structural and functional age-related/late-onset arterial changes, we have investigated whether an initial/genetic difference in vascular elastin quantity and organization could modify the course of arterial aging. To support this hypothesis, it was already known that elastin haploinsufficiency causes early-onset changes in the structure/function of the cardiovascular system. Supravalvular aortic stenosis (SVAS), isolated or in the setting of Williams syndrome, is an obstructive vascular disorder associated with heterozygous mutations/deletions in the elastin gene, leaving only a single functional allele. This pathology features, in humans, ascending aorta lumen narrowing due to deregulated hyperproliferation of VSMCs in the subendothelium, additional thinner elastic lamellae, abnormal mechanical properties of arteries, often increased blood pressure, and heart hypertrophy and failure, if not corrected.12–14 Also, elastin-null mice (Eln−/−) develop smaller blood vessels, almost occluded by proliferating VSMCs, and die within 72 h after birth.15 Eln+/− mice survive until adulthood although, similar to SVAS patients, they are hypertensive and have an aortic wall with an increased number of elastic lamellae that are two-fold thinner than in the Eln+/ + animals, both modifications leading to increased arterial stiffness.14 Hypertension, correlated with high circulating renin levels, could be a physiological adaptation allowing Eln+/− mice to maintain normal blood flow, despite a stiffer arterial wall.16
These observations showed that qualitative and quantitative defects in elastin expression contribute to structural alterations and dys-functions during arterial morphogenesis and maturation. To study whether elastin quantity can also induce late-onset arterial changes, we selected Eln+/− mice, which, although stable and nonpathologic, present elastin haploinsufficiency and a different cardiovascular system already in young animals.14,16 In these animals, we have studied the structure and mechanical properties of a large elastic artery, the ascending aorta, and the cardiac function in aging. Our results suggest that early elastin deficiency in Eln+/− mice induces long-term changes in arterial physiology, resulting in modifications of some of the major features of vascular aging.
MATERIALS AND METHODS
Animals
Eighty Eln+/+ male mice (C57B1/6J) or mice bearing a heterozygous deletion of exon 1 in the elastin gene (Eln+/−), which leads to a nonfunctional allele, were backcrossed for more than five generations into the C57B1/6J background and used in these experiments.14,15 Studies were performed in two age groups: adults, 5–7 months old (6 months = adults); and aged, 23–27 months (24 months = aged). Littermates from Eln+/− crosses were used whenever possible. All housing and surgical procedures were in accordance with institutional guidelines.
Blood pressure measurements and heart weight
Mice were anesthetized by intraperitoneal (i.p.) injection of 60 mg/kg ketamine (Merial, Lyon, France) and 6 mg/kg xylazine (Bayer, Puteaux, France) and placed on a heating table to maintain body temperature at 37°C. Mean arterial pressure was measured by inserting in the carotid artery a small catheter (Portex, Smiths Medical International Limited, Watford, United Kingdom) filled with a heparinized saline solution and connected to a pressure transducer (P23ID, Gould Instrument Co., Cleveland, OH).
In other animals, anesthetized with pentobarbital (60 mg/kg, i.p.), hearts and left ventricles were dissected, washed, and weighed (wet weight).
Untrasound study
Ultrasound studies were performed as previously described,17,18 using a Toshiba Powervision 6000 device (SSA 370A, Toshiba, Medical France S.A., Puteaux, France) equipped with an 8- to 14-MHz linear transducer under isoflurane anesthesia (0.75–1.0% in oxygen) with spontaneous ventilation. In vivo ascending aorta systolic and diastolic diameters were measured from time-mode imaging as gated on R wave from simultaneous ECG tracing.
Arterial desmosine and hydroxyproline contents
Desmosine levels, considered as representative of the elastin content, were determined by radioimmunoassay, as described previously.19 Hydroxyproline levels, considered as representative of the collagen content, were determined by amino acid analysis using standard techniques.20 Results are expressed as amino acid mass per vessel segment length.
Cannulated aorta mechanics
Mechanical studies were performed on cannulated ascending aortas excised from mice anesthetized by intraperitoneal injection of pentobarbital (60 mg/kg). The inner and outer diameters were measured at different pressures upon inflation using a physiological solution. Below 125 mmHg, the inner diameter was calculated as described previously.16
Distensibility is the change in relative luminal volume (percentage) per mmHg.21 Here we calculated the distensibility per 25 mmHg increment (D25).16
Circumferential midwall strain (ε), circumferential wall stress (σ), and incremental elastic modulus (E) were calculated according to classic formulas22: ε, relative increase in diameter at a given pressure as compared to the diameter at no pressure; σ, forces that are circumferentially applied on a each small portion (surface) of the vessel wall; E, wall stiffness.
Cannulated aorta reactivity
Variations of the ascending aorta diameters in response to 5 μmol/L phenylephrine (PE), a VSMC-dependent vasoconstrictor mainly acting through the alpha1-adrenoceptors, then 5 μmol/L acetylcholine (Ach), an endothelial cell-dependent vasodilator, was assessed at 75 mmHg.16 Control diameters of Eln+/− mouse aortas before PE application were normalized to the mean control diameter of Eln+/ + mouse aortas prior to comparisons of the effect of treatment as a function of genotype.23
Histological examination
Vessels excised from anesthetized mice were fixed overnight in 4% paraformaldehyde (PFA) at 4°C and processed for paraffin embedding. Transverse sections (3μm) of the ascending aorta were stained with hematoxylin-eosin for cells staining, Weigert for elastic fibers, and picrosirius red for collagen.
Ultrastructure—transmission electron microscopy
Ascending aortas were routinely fixed by cardiac perfusion with 2.5% glutaraldehyde in 0.1 M sodium cacodylate buffer (pH 7.4), then embedded in Epon for ultrastructural analyses. Sections were stained according to a previously described method,24 or pretreated with tannic acid before uranyl acetate staining. Sections were examined on a JEM 1200 EX II (Jeol, Tokyo, Japan) electron microscope at an accelerating voltage of 80 kV.
Expression of tropoelastin, lysyl-oxidase, lysyl-oxidase-like-1, fibrillin-1, fibrillin-2, fibulin-5, and type I collagen in the mouse aorta
RNA isolation and reverse transcription from entire aortas was performed according to the classic method.25 The yield of total RNA from a single ascending aorta was typically 2.9–7.5 μg. Gene expressions were evaluated by real-time PCR using a Light-Cycler with the FastStart DNA Master SYBR Green Kit (Roche Diagnostics, Meylan, France) after mRNA reverse transcription of 400 ng total RNA in the presence of oligodT, then normalized to the housekeeping gene beta-actin. Measured transcript levels were reproducible at levels that were significantly above background. Oligo primers used were:
tropoelastin (TE): 5′-AAGCTGCTGCTAAG-GCTGC-3′ (antisense) and 5′-TGCAACTCC-TCCACCTGGGAA-3′ (sense),
lysyl-oxidase (LOX): 5′-AATTCAGCCACT-ATGACCTGCTTGA-3′ (antisense) and 5′-GTAGCGAATGTCACAGCGTACAACA-3′ (sense),
lysyl-oxidase-like-1 (LOXL1): 5′ CAGCTTCT-GCCTGGAGGACA-3′ (antisense) and 5′-CGTAGCGACCTGTGTAGTGGATG-3′ (sense),
fibrillin-1: 5′-TGCCAGCAGCGAGATGGAC-GA-3′ (antisense) and 5′-TGGCGAGGCT-CACGTTGGCTT-3′ (sense),
fibrillin-2: 5′-AGTAGCCGCAACCTCGTCAC-AAA-3′ (antisense) and 5′-AGCACTGGTA-ACTGCCCTTGGTGT-3′ (sense),
fibulin-5: 5′-ATATCAATGAATGTGAGCA-CC-3′ (antisense) and 5′-GTTGATGACAGT-GTTGACAGTGAT-3′ (sense),
type I collagen: 5′-GTTCGTGACCGTGAC-CTTGA-3′ (antisense) and 5′-GGTGAAGC-GACTGTTGCCT-3′ (sense),
endothelial nitric oxide synthase (eNOS): 5′-GCGTATGCGGCTTGTCACCT-3′ (antisense) and 5′-CGGCTGCCACCTGATCCTAA-3′ (sense).
mRNA expressions were normalized to the housekeeping gene beta-actin, amplified using the following primers: 5′-TGGAATCCTGTGG-CATCCATGAAAC-3′ (antisense) and 5′-TAA-AACGCAGCTCAGTAACAGTCCG-3′ (sense).
Statistics
Comparisons, as a function of age, of Eln+/− and Eln+/+ body weight, heart weight/body weight ratios, heart rate, mean blood pressure, ultrasonography parameters, mRNA dosages, desmosine and hydroxyproline contents and their ratios, electron microscopy image analysis, and vessel diameters (OD and ID) in response to vasoactive agonists were assessed using two-way ANOVA, followed when necessary by Fisher’s least significant difference test (LSD) or t-test for paired value comparisons. Vessel diameters were compared as a function of transmural pressure level (0–175 mmHg), KCN treatment (before or after), genotype (Eln+/− or Eln+/ +), and age (6- or 24-month-old) using a four-way ANOVA followed when necessary by LSD tests. Vessel midwall strain, stress, incremental elasticity modulus, distensibility, and local midwall strain comparisons were assessed using the nonparametric Mann-Whitney U test. Unless otherwise indicated, the results are presented as mean values ± standard error of the mean (SEM), and p values ≤ 0.05 were considered statistically significant.
RESULTS
General physiological parameters
No significant difference in life span was observed between Eln+/+ and Eln+/− mice (Chi2 test, P = 0.227) (Fig. 1A). Eln+/− mice were found substantially hypertensive throughout adulthood and aging, with a mean arterial pressure increased by 20–30% when compared to Eln+/+ animals, although no difference in heart rate could be detected. No age-dependent increase in blood pressure was observed (Table 1).
FIG. 1.

Effect of aging and genotype on the survival rate (A), length of the ascending aorta of 6- and 24-month-old Eln+/+ and Eln+/− mice (B), and histological examination of paraffin-embedded cross-sections of the ascending aorta of 6- and 24-month-old Eln+/+ and Eln+/− mice (C). (a–d) eosin/hematoxylin; (e–h) Weigert; and (I–l) picrosirius red stainings (For color version, see <www.liebertonline.com/rej>.). Bar sizes: 200 μm (a–d) and 50 μm (e–l). In each group, n = 39–50 (A) and n = 4–5 (B). lm, lumen; d, distal; p, proximal.
Table 1.
Physiological Parameters and Cardiovascular Ultrasound Study of 6- and 24-Month-Old Eln+/+ and Eln+/− Mice. In Each Group, n = 12–13 for Body and Heart Weights and n = 4 for Mean Arterial Pressure
| 6 month-old mice | 24 month-old mice | |||
|---|---|---|---|---|
| Eln+/+ | Eln+/− | Eln+/+ | Eln+/− | |
| BW(g) | 36.0 ± 1.1 | 35.3 ± 1.3 | 35.3 ± 0.8 | 33.0 ±1.0 |
| Heart weight/BW (%) | 0.43 ± 0.01 | 0.50 ± 0.02G | 0.53 ± 0.03A | 0.62 ± 0.02G |
| Left ventricle/BW (%) | 0.35 ± 0.01 | 0.40 ± 0.02G | 0.43 ± 0.02A | 0.49 ± 0.02G |
| Mean arterial pressure (mmHg) | 103 ± 11 | 123 ± 2G | 107 ± 4 | 138 ± 8G |
| Ultrasound study: | ||||
| Number of animals | 5 | 4 | 10 | 9 |
| Systolic aorta diameter (mm) | 1.95 ± 0.08 | 1.68 ± 0.10G | 2.24 ± 0.15 | 1.85 ± 0.13G |
| Diastolic aorta diameter (mm) | 1.8 ± 0.07 | 1.54 ± 0.10G | 2.12 ± 0.15 | 1.75 ± 0.13G |
| Heart rate (beats per minute) | 504 ± 11 | 499 ± 3 | 508 ± 16 | 496 ± 17 |
| LVEDD/BW (mm) | 0.11 ± 0.002 | 0.12 ± 0.01 | 0.12 ± 0.01 | 0.13 ± 0.01 |
| Left atrium dimension-La (mm) | 3.14 ± 0.05 | 3.17 ± 0.14 | 3.26 ± 0.20 | 3.00 + 0.14 |
| Shortening fraction-FS (%) | 41 ± 3 | 41 ± 2 | 40 ± 5 | 39 + 3 |
| Vcfc (circ/s) | 2.80 ± 0.20 | 2.70 ± 0.20 | 3.01 ± 0.29 | 3.00 ± 0.16 |
| Sa (cm/s) | 3.5 ± 0.3 | 3.1 ± 0.2 | 2.8 ± 0.4 | 2.7 ± 0.3 |
| Spw (cm/s) | 3.80 ± 0.10 | 3.60 ± 0.30 | 3.60 ± 0.20 | 2.97 ± 0.40 |
| Isovolumic relaxation time (ms) | 15.0 ± 0.5 | 17.0 ± 3.0 | 19.5 ± 0.7A | 17.5 ± 1.5 |
| Ea (cm/s) | 4.3 ± 0.3 | 5.3 ± 0.5 | 4.1 ± 0.3 | 5.3 ± 0.4 |
| Epw (cm/s) | 4.1 ± 0.3 | 4.6 ± 0.2 | 5.0 ± 0.6 | 4.1 ± 0.4 |
| E/Ea | 6.0 | 20.0 ± 3.0 | 25.0 ± 2.3 | 19.0 ± 2.1 |
Significant difference with adult animals of the same genotype.
Significant difference with Eln+/+ animals of matching age.
BW, body weight; LVEDD, left ventricle end diastolic diameter; Vcfc, mean shortening velocity of circumferential fibers, corrected for time; Sa, Spw: maximal systolic velocity of the mitral annulus and posterior wall, respectively. Ea, Epw: maximal diastolic velocity of the mitral annulus and posterior wall, respectively. E/Ea: maximal velocity of LV mitral inflow to Ea.
Body weights were found similar in all animal groups (33–36 g), yet heart weight increased with age in both genotypes (+25%). Cardiac hypertrophy was observed in adult and aged Eln+/− mice, with left ventricles ~15% heavier. The heart weight of adult Eln+/− mice was the same as those of aged Eln+/+ animals (Table 1).
Histology and ultrastructure of the ascending aorta wall
The ascending aorta length was significantly increased by aging (+23–30% in both genotypes) and elastin haploinsufficiency (+20–26% at both ages): 2.57 ± 0.15 vs. 3.24 ± 0.14 mm in adults, and 3.34 ± 0.23 vs. 4.00 ± 0.17 mm in aged Eln+/+ and Eln+/− mice, respectively (two-way ANOVA, p ≤ 0.05) (Fig. 1B). In Eln+/− mice, the arterial diameter was decreased (Table 1, pFig. 1C). Two to three additional lamellar units were present compared to wild-type animals while, within each genotype, aging had no effect on the number of elastic lamellae (Fig. 1C): 8.8 ± 0.4 vs. 11 ± 0.1 lamellae in adults, and 8.6 ± 0.3 vs. 10.7 ± 0.0 in aged Eln+/+ and Eln+/− mice, respectively (two-way ANOVA, ≤ 0.05).
Ascending aorta semi-thin sections (data not shown) and electron microscopy images showed that in Eln+/− mice at both ages: (1) elastic lamellae were thinner, (2) the distance between successive lamellae was shorter, and (3) elastic lamellae were more fragmented or branched, with irregular contours, compared to Eln+/+ elastic lamellae (fragmentation was similar in adult Eln+/− and aged Eln+/+ mice) (Figs. 2A[c,d], 2B[c,d], 3A(C). Moreover, the distance between elastic lamellae significantly increased with age in Eln+/+ mice only, but not in Eln+/− animals (Fig. 3B). Also, thickness of the vascular smooth muscle cell layers was increased more significantly with age in Eln+/+ (+55%) than in Eln+/− mice (+19%) (Fig. 3D).
FIG. 2.

Transmission electron microscopy images of the ascending aorta in mice aged of 6 (A) and 24 months (B). (a,c) Eln+/+; (b,d) Eln+/−; (a,b) intima; (c,d) media. Arrows, disruptions of the elastic lamella; asterisks, elastin deposit in margin of elastic lamellas; squares, subendothelial accumulation of extracellular matrix; endo, endothelium; el, elastic lamella; lm: lumen; smc, smooth muscle cell. Bar size: 1 μm.
FIG. 3.

Morphometric analysis of the transmission electron microscopy images of the ascending aorta in Eln+/+ and Eln+/− mice aged of 6 (adult) and 24 months (old). Thickness of the elastic laminae (A), distance between successive elastic laminae (B), fragmentation of elastic laminae (C), thickness of VSMC layer (D), distance between the internal elastic lamina (IEL) and endothelial cells (EC) (E) or smooth muscle cells (SMC) (F). Number of measurements performed on different sections from several animals in each group: 89 (A), 102 (B), 20 (C), 57 (D), 64 (E), 75 (F). #significant difference compared to adult animals of matching genotype. Two-way ANOVA: *general effect of genotype; °general effect of age; n.s., not significant.
In Eln+/− animals, picrosirius red staining suggested a more dispersed distribution of collagen in the aorta wall (Fig.1C), and unusual elastin deposits were observed in the margin of elastic lamellae (Fig. 2A and 2B[b,d]). Also, in animals from both genotypes, aging was correlated with an increase in cytoplasmic extrusions due to cell stretching and accumulation of extracellular matrix (ECM), which tends to make more discontinuous the contacts between elastic lamellae and vascular cells. ECM accumulation, however, was limited to the media in wild-type mice, whereas it was present in both the media and the intima in the aorta of Eln+/− animals. As a consequence, the endothelial cells, which lied on a basement membrane directly connected to the internal elastic lamina (IEL) in Eln+/+ arteries, were clearly separated from the IEL by a subendothelial accumulation of ECM and had a basal side which turned to crenelated in Eln+/− mice. A similar phenomenon increased the distance between the IEL and the first medial layer of smooth muscle cells in Eln+/− animals (Figs. 2A[a,b], 2B[a,b], 3E–F).
Desmosine and hydroxyproline contents in the ascending aorta
The contents of desmosine and hydroxyproline remained constant with age in both genotypes. However, it was observed at both ages that, while hydroxyproline content was similar in mice of both genotypes, desmosine content and desmosine/hydroxyproline ratios were about 40–50% lower in Eln+/− than in Eln+/+ ascending aortas (Fig. 4A–C). However, the desmosine/hydroxyproline ratios were unchanged by aging within each genotype (Fig. 4C).
FIG. 4.
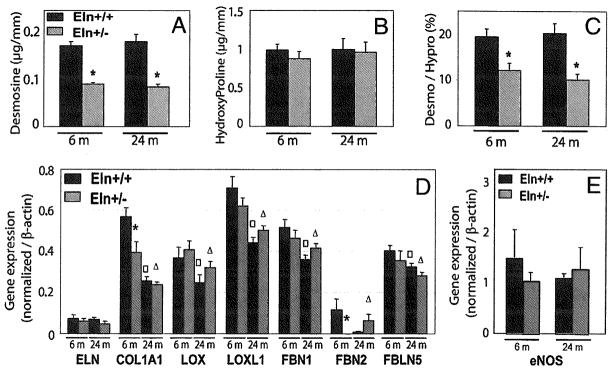
Desmosine and hydroxyproline contents and ratios (A–C), and gene expressions quantified by real-time PCR (D and E) in the ascending aorta of 6- and 24-month-old Eln+/+ and Eln+/− mice. ELN, tropoelastin; COL1A1, α1 chain of collagen-I; LOX, lysyl-oxidase; LOXL1, lysyl-oxidase-like-1; FBN1, fibrillin-1; FBN2, fibrillin-2; FBLN5, fibulin-5; eNOS, endothelial nitric oxide synthase.*, □, △Significant difference between Eln+/+ and Eln+/− mice of matching age, between 6- and 24-month-old Eln+/+ mice, and between 6- and 24-month-old Eln+/− mice, respectively. n = 7–11 in each group.
Extracellular matrix gene expression
The expression of the following genes involved in elastic fiber assembly were investigated: tropoelastin (the elastin precursor), lysyl-oxidase, lysyl-oxidase-like-1, fibrillin-1, fibrillin-2, and fibulin-5. The expression levels of all these genes were generally low in adult and aged animals (i.e., several fold lower than in growing animals [data not shown]). Tropoelastin and fibrillin-2 showed particularly low expression levels at both ages. The expression levels of all the genes studied, with the exception of tropoelastin, decreased with age independently of the genotype. Only two genotype-related differences were noticed: significantly less collagen-I and fibrillin-2 were expressed in ascending aortas of 6-month-old Eln+/− mice compared to that of wild-type animals of matching age, while no genotype-related difference could be detected in 24-month-old animals. The expression of collagen-I and fibrillin-2 in adult Eln+/− mice was close to the levels observed in aged Eln+/+ animals (Fig. 4D). In addition, no significant difference in the expression of endothelial nitric oxide synthase (eNOS) could be detected between all groups of mice (Fig. 4E).
In vivo cardiovascular function
Functional characterization by echocardiography in anesthetized mice showed left ventricle and left atrium enlargement in Eln+/− mice. No significant differences were found for left ventricle contractility and diastolic function (Table 1), except for isovolumic relaxation time, which normally increased with age26 in Eln+/+ mice (+30%), but not in Eln+/− animals. In vivo ascending aorta diameters were assessed in anesthetized mice at the time of echocardiography. Systolic and diastolic diameters of the ascending aorta were smaller (~−15%) in Eln+/− as compared to Eln+/+ animals, at both ages (Table 1).
Dimensions and mechanics of the cannulated ascending aorta
Study of cannulated vessels confirmed that in adult and aged animals the Eln+/− ascending aorta has a smaller diameter than does the Eln+/+ at any given pressure (Fig. 5A). However, when compared at their respective in vivo mean arterial pressures (~100 and 125–135 mmHg for Eln+/+ and Eln+/−, respectively), the diameters were similar between Eln+/+ and Eln+/− animals at both ages. It was also observed that aging in both genotypes leads to a general increase in inner diameter, except at the highest pressures where the maximum diameters of mice of a given genotype tend to be similar at both ages, or perhaps slightly decreased in wild-type animals (Fig. 5A).
FIG. 5.

Diameter pressure curves and derived mechanical parameters of the ascending aorta of 6- and 24-month-old Eln+/+ and Eln+/− mice. *, +Significant difference between 6-month-old Eln+/+ and Eln+/− mice or between 24 month-old Eln+/+ and Eln+/− mice, respectively. □, △Significant difference between 6 month-old and 24 month-old values for Eln+/+ and Eln+/− mice, respectively. §Significant difference between Eln+/+ and Eln+/− of matching age at their respective mean arterial pressure. n = 5–10 in each group.
Distensibility (D25) generally decreased with pressure in all animals. In adult Eln+/− animals, distensibility was increased at low intraluminal pressure and decreased at the highest pressures compared to that of Eln+/+ mice, whereas aorta distensibility was almost identical in aged animals of both genotypes over the entire pressure range. Compared to adults, aged animals of both genotypes presented elevated aortic distensibility at low pressure and faster distensibility drop with increasing pressure. At the highest pressures, including physiological/working blood pressures, aged Eln+/− aortas presented a distensibility that was only slightly changed compared to that of adult Eln+/− vessels, whereas the distensibility of aged Eln+/+ aortas was substantially decreased compared to that of adult Eln+/+ arteries (Fig. 5B).
Despite an increased number of elastic lamellae, the aorta wall thickness of Eln+/− mice was not increased (Fig. 5C) because of the decreased lamina thickness and interlamellar distance (Fig. 3A and B). The evolution with age of ascending aorta wall thickness was surprisingly different in Eln+/+ and Eln+/− mice. In adults, the wall thickness of Eln+/− mice was not increased and even slightly lower than that of Eln+/+ animals. In the older mice, the wild-type animals exhibited a classic and substantial age-dependent increase in wall thickness, while no wall thickening was observed with aging in Eln+/− animals.
Elastin haploinsufficency and aging also had an influence on aorta mechanical parameters (Fig. 5D–G). At the respective in vivo blood pressures of Eln+/+ and Eln+/− mice: (1) circumferential midwall strain (ε) was higher in Eln+/− than in Eln+/+ adult mice and was similar in both genotypes in aged animals (Fig. 5D); (2) circumferential wall stress (σ) was elevated in Eln+/− mice at both ages (Fig. 5E), (3) with age, aortic stress was decreased in Eln+/+ and increased in Eln+/− mice when ε> 0.8 (Fig. 5F); and (4) incremental elastic modulus (E) was virtually unchanged by aging in Eln+/+ animals, while a strong age-dependent increase in E was observed in Eln+/− mice above 100 mm Hg (Fig. 5G).
Ascending aorta reactivity
Response to the vasoconstrictor phenylephrine (PE) was similar in adult animals of both genotypes. However, a substantial age-dependent decrease in PE-induced constriction (~−50%) was observed in wild-type mice while no age-related change could be observed in Eln+/− mice (Fig. 6A and B). When acetylcholine (Ach) was applied to the ascending aorta, no difference in vasodilation was observed between adult Eln+/+ and Eln+/− mice. In aged mice, Ach-induced relaxation of the aorta was found to be similar to that of adults in wild-type animals, whereas it was significantly reduced by approximately 25% compared to adult animals in Eln+/− mice (Fig. 6A and C).
FIG. 6.

Reactivity to vasoactive agents of the ascending aorta of 6- and 24-month-old Eln+/+ and Eln+/− mice. (A) 5 μmol/L phenylephrine (PE) or 5 μmol/L phenylephrine + 5 μmol/L acetylcholine (PE + Ach). PE-induced vasoconstriction is also represented as the decrease in inner diameter, in percent of the control diameter (B), and Ach-induced vasodilation is represented as the reversal of the PE-induced vasoconstriction, in percent (C). #Significant difference between control and post-treatment values at same age and genotype. +, □, △,Significant difference for the same treatment between Eln+/+ and Eln+/− of matching ages, between 6- and 24-month-old Eln+/+, and between 6- and 24-month-old Eln+/− mice, respectively. n = 5–9 in each animal group.
DISCUSSION
Elastin is an essential determinant of arterial morphogenesis.15 VSMCs hyperproliferate when elastin is present in below normal levels, leading to abnormal structure and function of arteries. This has been documented in the genetic diseases supravalvular aortic stenosis or Williams syndrome.12,13,27 We previously showed that Eln+/− mice survive until adulthood, although they exhibit some features of human disease such as hypertension, cardiac hypertrophy, and, in arteries, an increased number of elastic lamellae, increased stiffness, and smaller diameters.16 In the present study, we have investigated whether early modifications of the Eln+/− mouse arterial structure and function, resulting from elastin hemizygosity, could have other consequences latter in life and alter the normal cardiovascular aging processes.
Our results indicated that some parameters are similar in adult and aged Eln+/+ and Eln+/− animals: no difference in life span, body weight, heart rate, and most cardiac functional parameters. Elastic fiber-related gene expression was generally similar in the two genotypes and decreased similarly with age in Eln+/+ and Eln+/− mice, except for tropoelastin whose expression level did not significantly vary between 6- and 24-months of age in all animals.
We also found that some features are specifically modified in the ascending aorta of adult and aged Eln+/− mice compared to Eln+/ + animals of matching ages. This includes decreased elastin content and elastin/hydroxyproline-containing collagen ratio, hypertension, and ascending aorta lengthening, consistent with the previously described carotid artery and abdominal aorta lengthening in Eln+/− young mice.28 Vessels were smaller in diameter at a given pressure and there was a substantial subendothelial accumulation of extracellular matrix (ECM) separating the endothelial cells from the internal elastic lamina. Further investigation may determine whether this subendothelial ECM accumulation corresponds to the increase in fibronectin, proteoglycans, and collagen contents (although no structured collagen was observed here) occurring in the age-dependent intimal thickening observed in humans and rats.29 Additionally, in Eln+/− mice, the elastic lamellae were thinner, increased in number, and had irregular borders. Elastin droplet-like structures, separate from the elastic lamellae, were also observed and could correspond to either abnormal longitudinally oriented elastic lamellae or isolated non-structurally organized and non-mechanically functional elastin deposited by deregulated VSMCs. Moreover, collagen-I and fibrillin-2 expressions were lower in adult Eln+/− than in Eln+/+ mice, and a surprizing reinduction of fibrillin-2 expression was observed in aged Eln+/− animals only.
Some cardiovascular alterations that appear with age in wild-type animals were already present in adult Eln+/− animals. This included cardiac hypertrophy, higher fragmentation of the elastic lamellae than in Eln+/+ mice, and decreased aortic distensibility. In aging, these alterations were maintained or enhanced in Eln+/− mice while they were just appearing in Eln+/+ animals. Hence, these specific features of adult Eln+/− mice resemble premature aging compared to Eln+/+ mouse aging. Conversely, other classic characteristics of normal aging in wild-type animals did not appear in aged Eln+/− mice. In many species, including mice and humans, normal arterial aging features a dilation of the large systemic and pulmonary arteries together with a fragmentation of the elastic lamellae, and a substantial wall thickening.9,30 This is consistent with the age-dependent wall thickening (40%) that we observed in Eln+/+ mice. In Eln+/− mice, aging-induced arterial dilation and elastic lamellae fragmentation were also observed yet no arterial wall thickening could be detected. This is surprising, since age-related wall thickening, due to ECM accumulation and VSMC hypertrophy, is an adaptive compensatory mechanism for maintaining close to normal circumferential forces, that is, stress (stress = pressure X radius/thickness), directed against the arterial wall. When blood pressure and/or arterial radius increase with age, stress would increase in the absence of wall thickening. In the absence of wall thickening, the age-dependent increase in blood pressure and/or arterial diameter would lead to increased wall stress. In aging Eln+/+ mice, although no change in mean arterial pressure (MAP) could be detected, the arterial diameter substantially increased in the physiological blood pressure range (~100 mmHg) and the wall thickened. These phenomena were probably due to VSMC hypertrophy (+55%) and production of ECM and probably non-collagenic since there were no age-related increases in hydroxyproline content and collagen-I expression in response to circumferential mechanical forces. In Eln+/− mice, however, there was a smaller age-dependent increase in arterial diameter in their working range of pressure (MAP, 125–135 mmHg) and stress did not, or only slightly, increased with age. As a result, there is no need for an increase in wall thickness to maintain the wall stress at the level measured in adults. Our results suggest that the limited increase in aortic diameter in Eln+/− mice aging is caused by a lower age-related VSMC hypertrophy (+19%) and premature fragmentation of the elastic lamellae. This degradation could decrease the role of elastic fibers (elasticity/extensibility) and increase the role of the collagen fibers (stiffness) in Eln+/− arteries. This would lead to stiffer Eln+/− vessels in adults, working at high pressure close to their extensibility limit, resembling the observations in aged Eln+/+ animals. Finally, VSMC-dependent aorta constriction in response to alpha1-adrenoceptor stimulation is classically decreased in normal aging,10,31 potentially in relation with the decreased number of adrenergic receptors evidenced in several tissues: alpha1b-subtype in the aorta,32 alpha-type in ventricular cells,33 and beta-type in blood cells.34 This is consistent with our results in phenylephrine-treated Eln+/+ aortas, whereas no such age-dependent loss of response to this agonist was seen in Eln+/− mice. On the contrary, an age-related decrease in response to the endothelium-dependent vasodilator acetylcholine was observed in the aorta of Eln+/− animals, but not in Eln+/+ mice, suggesting a stronger age-dependent dysfunction of the endothelium in Eln+/− mice.
Taken together, our results show that the Eln+/− mice do not present any major pathology of the heart and aorta, and have a life span similar to wild-type animals. This suggests that genetic elastin deficiency can be efficiently compensated for in adult and aged mice by different mechanisms, including VSMC proliferation and modified elastic fiber deposition, leading to production of additional elastic lamellae. These processes result in a different state of the cardiovascular physiology of Eln+/− mice, in which the combination of elevated blood pressure and modified arterial mechanics and reactivity does not affect life span. Our studies expand to aging the concept that a decreased deposition of insoluble elastin/elastic fibers stimulates VSMC proliferative activity and vascular changes, such as in SVAS or WS patients27 or fibulin-5 deficient mice.35 However, in Eln+/− mice, despite the enhanced VSMC proliferation, the distance between successive elastic lamellae was shorter than in Eln+/+ mice. This is explained by the fact that, in Eln+/− mice, VSMC proliferation does not lead to accumulation of cells into the same cell layer. The additional cells migrate to form separate/additional concentric cell layers and corresponding additional elastic lamellae.14 As a consequence of the different morphologies of the elastic lamellae and VSMCs, the interlamellar distance and lamellar unit thickness are even lower in Eln+/− than in Eln+/+ mice, especially in aged animals.
In addition, the specific cardiovascular aging features observed in Eln+/− mice with high blood pressure are not caused by early hypertension-induced remodeling. Investigations in humans or murine models show that hypertension results in increased large artery diameter and wall thickness, as well as mortality,9,30,36 which were not observed in Eln+/− mice. It is likely then that particular maturation and aging processes develop from the initial and sustained elastin deficiency of Eln+/− animals. However, high blood pressure observed in Eln+/− mice is correlated to increased circulating renin levels,16 and activation of the renin-angiotensin system is known, in humans and animals, to be a major effector of secondary hypertension originating in renal artery atherosclerosis or other diameter narrowing, and subsequent altered renal blood flow.37 Moreover, because atherosclerosis generally progresses in the elderly, the frequency of renovascular hypertension increases with age, reaching at least 7% of the human population above age 65.38 It would therefore be interesting to investigate the histology and function of renal arteries in aging Eln+/− mice to search for the presence of processes leading to elevated blood pressure. The fact that patients with Williams syndrome often present hypertension and renal artery stenosis39 supports this hypothesis. Also, hypertension and endothelial dysfunction, present in Eln+/− mice, are known to favor cerebrovascular diseases such as stroke and aneurysms in humans or animal models.40–42 However, these pathologies are probably not enhanced in Eln+/− mice since the life span of these animals was not decreased. Further investigation of the cerebral arteries or microvasculature of Eln+/− mice could determine whether structural or functional changes are present in these vessels.
Finally, our study suggests that there is not a unique process for vascular aging, allowing adaptive mechanisms leading to normal life span (i.e., there is certain plasticity of aging). As elastin is no longer synthesized in adulthood, the initial conditions established during embryogenesis and early life (i.e., cardiovascular structure and function) determine the way that arteries will evolve with age. In particular, initial elastin quantity, quality, and organization might have an impact on the VSMC functions, including response to agonists, synthesis activity, proliferation, and migration, and could be a determinant of the events that will occur later in life, dramatically changing the consequences of aging on the arterial structure and function.
Acknowledgments
The authors wish to thank S. Bouillot, B. Mariko, M. Coquand-Gandit, and F. Aïnoun for their help. The authors acknowledge the following institutions for funding: European Commission (contracts TELASTAR, 5th PCRD, no. QLK6-CT-2001-00332; and ELAST-AGE, 6th PCRD, no. LSHM-CT-2005-018960), Fondation pour la Recherche Médicale fellowship (MP), and Association Francaise contre les Myopathies.
References
- 1.Rosenbloom J, Abrams WR, Mecham R. Extracellular matrix 4: the elastic fiber. Faseb J. 1993;7:1208–1218. [PubMed] [Google Scholar]
- 2.Wolinsky H, Glagov S. A lamellar unit of aortic medial structure and function in mammals. Circ Res. 1967;20:99–111. doi: 10.1161/01.res.20.1.99. [DOI] [PubMed] [Google Scholar]
- 3.Belz GG. Elastic properties and Windkessel function of the human aorta. Cardiovasc Drugs Ther. 1995;9:73–83. doi: 10.1007/BF00877747. [DOI] [PubMed] [Google Scholar]
- 4.Izzo JL, Jr, Mitchell GF. Aging and arterial structure-function relations. Adv Cardiol. 2007;44:19–34. doi: 10.1159/000096701. [DOI] [PubMed] [Google Scholar]
- 5.Robert L. Aging of the vascular-wall and atherosclerosis. Exp Gerontol. 1999;34:491–501. doi: 10.1016/s0531-5565(99)00030-3. [DOI] [PubMed] [Google Scholar]
- 6.Jacob MP. Extracellular matrix remodeling and matrix metalloproteinases in the vascular wall during aging and in pathological conditions. Biomed Pharmacother. 2003;57:195–202. doi: 10.1016/s0753-3322(03)00065-9. [DOI] [PubMed] [Google Scholar]
- 7.Faury G, Ristori MT, Verdetti J, Jacob MP, Robert L. Effect of elastin peptides on vascular tone. J Vasc Res. 1995;32:112–119. doi: 10.1159/000159084. [DOI] [PubMed] [Google Scholar]
- 8.Lograno MD, Bisaccia F, Ostuni A, Daniele E, Tamburro AM. Identification of elastin peptides with vasorelaxant activity on rat thoracic aorta. Int J Biochem Cell Biol. 1998;30:497–503. doi: 10.1016/s1357-2725(98)00008-9. [DOI] [PubMed] [Google Scholar]
- 9.Laurent S, Lacolley P, Girerd X, Boutouyrie P, Bezie Y, Safar M. Arterial stiffening: opposing effects of age-and hypertension-associated structural changes. Can J Physiol Pharmacol. 1996;74:842–849. [PubMed] [Google Scholar]
- 10.Docherty JR. The effects of ageing on vascular alpha-adrenoceptors in pithed rat and rat aorta. Eur J Pharmacol. 1988;146:1–5. doi: 10.1016/0014-2999(88)90480-3. [DOI] [PubMed] [Google Scholar]
- 11.Taddei S, Virdis A, Ghiadoni L, Versari D, Salvetti A. Endothelium, aging, and hypertension. Curr Hyper-tens Rep. 2006;8:84–89. doi: 10.1007/s11906-006-0045-4. [DOI] [PubMed] [Google Scholar]
- 12.Wessel A, Motz R, Pankau R, Bursch JH. Arterial hypertension and blood pressure profile in patients with Williams-Beuren syndrome. Z Kardiol. 1997;86:251–257. doi: 10.1007/s003920050056. [DOI] [PubMed] [Google Scholar]
- 13.Chowdhury T, Reardon W. Elastin mutation and cardiac disease. Pediatr Cardiol. 1999;20:103–107. doi: 10.1007/s002469900415. [DOI] [PubMed] [Google Scholar]
- 14.Li DY, Faury G, Taylor DG, Davis EC, Boyle WA, Mecham RP, Stenzel P, Boak B, Keating MT. Novel arterial pathology in mice and humans hemizygous for elastin. J Clin Invest. 1998;102:1783–1787. doi: 10.1172/JCI4487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Li DY, Brooke B, Davis EC, Mecham RP, Sorensen LK, Boak BB, Eichwald E, Keating MT. Elastin is an essential determinant of arterial morphogenesis. Nature. 1998;393:276–280. doi: 10.1038/30522. [DOI] [PubMed] [Google Scholar]
- 16.Faury G, Pezet M, Knutsen RH, Boyle WA, Heximer SP, McLean SE, Minkes RK, Blumer KJ, Kovacs A, Kelly DP, Li DY, Starcher B, Mecham RP. Developmental adaptation of the mouse cardiovascular system to elastin haploinsufficiency. J Clin Invest. 2003;112:1419–1428. doi: 10.1172/JCI19028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Parlakian A, Charvet C, Escoubet B, Mericskay M, Molkentin JD, Gary-Bobo G, De Windt LJ, Ludosky MA, Paulin D, Daegelen D, Tuil D, Li Z. Temporally controlled onset of dilated cardiomyopathy through disruption of the SRF gene in adult heart. Circulation. 2005;112:2930–2939. doi: 10.1161/CIRCULATIONAHA.105.533778. [DOI] [PubMed] [Google Scholar]
- 18.Royer A, van Veen TA, Le Bouter S, Marionneau C, Griol-Charhbili V, Leoni AL, Steenman M, van Rijen HV, Demolombe S, Goddard CA, Richer C, Escoubet B, Jarry-Guichard T, Colledge WH, Gros D, de Bakker JM, Grace AA, Escande D, Charpentier F. Mouse model of SCN5A-linked hereditary Lenegre’s disease: age-related conduction slowing and myocardial fibrosis. Circulation. 2005;111:1738–1746. doi: 10.1161/01.CIR.0000160853.19867.61. [DOI] [PubMed] [Google Scholar]
- 19.Starcher B, Conrad M. A role for neutrophil elastase in the progression of solar elastosis. Connect Tissue Res. 1995;31:133–140. doi: 10.3109/03008209509028401. [DOI] [PubMed] [Google Scholar]
- 20.Brown-Augsburger P, Tisdale C, Broekelmann T, Sloan C, Mecham RP. Identification of an elastin cross-linking domain that joins three peptide chains. Possible role in nucleated assembly. J Biol Chem. 1995;270:17778–17783. doi: 10.1074/jbc.270.30.17778. [DOI] [PubMed] [Google Scholar]
- 21.Smith JJ, Kampine JP. Circulatory Physiology—The Essentials. 3. Williams & Wilkins; Baltimore: 1990. [Google Scholar]
- 22.Gibbons CA, Shadwick RE. Functional similarities in the mechanical design of the aorta in lower vertebrates and mammals. Experientia. 1989;45:1083–1088. doi: 10.1007/BF01950164. [DOI] [PubMed] [Google Scholar]
- 23.Faury G, Chabaud A, Ristori MT, Robert L, Verdetti J. Effect of age on the vasodilatory action of elastin peptides. Mech Ageing Dev. 1997;95:31–42. doi: 10.1016/s0047-6374(96)01842-8. [DOI] [PubMed] [Google Scholar]
- 24.Franc S, Garrone R, Bosch A, Franc JM. A routine method for contrasting elastin at the ultrastructural level. J Histochem Cytochem. 1984;32:251–258. doi: 10.1177/32.2.6198356. [DOI] [PubMed] [Google Scholar]
- 25.Chomczynski P, Sacchi N. Single-step method of RNA isolation by acid guanidinium thiocyanate-phenol-chloroform extraction. Anal Biochem. 1987;162:156–159. doi: 10.1006/abio.1987.9999. [DOI] [PubMed] [Google Scholar]
- 26.Lim CC, Liao R, Varma N, Apstein CS. Impaired lusitropy-frequency in the aging mouse: role of Ca(2+)-handling proteins and effects of isoproterenol. Am J Physiol. 1999;277:H2083–2090. doi: 10.1152/ajpheart.1999.277.5.H2083. [DOI] [PubMed] [Google Scholar]
- 27.Urban Z, Riazi S, Seidl TL, Katahira J, Smoot LB, Chitayat D, Boyd CD, Hinek A. Connection between elastin haploinsufficiency and increased cell proliferation in patients with supravalvular aortic stenosis and Williams-Beuren syndrome. Am J Hum Genet. 2002;71:30–44. doi: 10.1086/341035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wagenseil JE, Nerurkar NL, Knutsen RH, Okamoto RJ, Li DY, Mecham RP. Effects of elastin haploinsufficiency on the mechanical behavior of mouse arteries. Am J Physiol Heart Circ Physiol. 2005;289:H1209–1217. doi: 10.1152/ajpheart.00046.2005. [DOI] [PubMed] [Google Scholar]
- 29.Lakatta EG. Arterial and cardiac aging: major shareholders in cardiovascular disease enterprises. Part III: cellular and molecular clues to heart and arterial aging. Circulation. 2003;107:490–497. doi: 10.1161/01.cir.0000048894.99865.02. [DOI] [PubMed] [Google Scholar]
- 30.Zhu BH, Ueno M, Matsushita T, Fujisawa H, Seriu N, Nishikawa T, Nishimura Y, Hosokawa M. Effects of aging and blood pressure on the structure of the thoracic aorta in SAM mice: a model of age-associated degenerative vascular changes. Exp Gerontol. 2001;36:111–124. doi: 10.1016/s0531-5565(00)00179-0. [DOI] [PubMed] [Google Scholar]
- 31.Johnson MD, Wray A. Alpha 1 adrenergic receptor function in senescent Fischer 344 rat aorta. Life Sci. 1990;46:359–366. doi: 10.1016/0024-3205(90)90015-j. [DOI] [PubMed] [Google Scholar]
- 32.Gurdal H, Tilakaratne N, Brown RD, Fonseca M, Friedman E, Johnson MD. The expression of alphal adrenoceptor subtypes changes with age in the rat aorta. J Pharmacol Exp Ther. 1995;275:1656–1662. [PubMed] [Google Scholar]
- 33.Partilla JS, Hoopes MT, Ito H, Dax EM, Roth GS. Loss of rat ventricular alpha 1-adrenergic receptors during aging. Life Sci. 1982;31:2507–2512. doi: 10.1016/0024-3205(82)90757-3. [DOI] [PubMed] [Google Scholar]
- 34.Schocken DD, Roth GS. Reduced beta-adrenergic receptor concentrations in ageing man. Nature. 1977;267:856–858. doi: 10.1038/267856a0. [DOI] [PubMed] [Google Scholar]
- 35.Spencer JA, Hacker SL, Davis EC, Mecham RP, Knutsen RH, Li DY, Gerard RD, Richardson JA, Olson EN, Yanagisawa H. Altered vascular remodeling in fibulin-5-deficient mice reveals a role of fibulin-5 in smooth muscle cell proliferation and migration. Proc Natl Acad Sci USA. 2005;102:2946–2951. doi: 10.1073/pnas.0500058102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Remsberg KE, Siervogel RM. A life span approach to cardiovascular disease risk and aging: the Fels Longitudinal Study. Mech Ageing Dev. 2003;124:249–257. doi: 10.1016/s0047-6374(02)00192-6. [DOI] [PubMed] [Google Scholar]
- 37.Ram CV, Clagett GP, Radford LR. Renovascular hypertension. Semin Nephrol. 1995;15:152–174. [PubMed] [Google Scholar]
- 38.Textor SC. Managing renal arterial disease and hypertension. Curr Opin Cardiol. 2003;18:260–267. doi: 10.1097/00001573-200307000-00004. [DOI] [PubMed] [Google Scholar]
- 39.Rose C, Wessel A, Pankau R, Partsch CJ, Bürsch J. Anomalies of the abdominal aorta in Williams-Beuren syndrome—another cause of arterial hypertension. Eur J Pediatr. 2001;160:655–658. doi: 10.1007/s004310100835. [DOI] [PubMed] [Google Scholar]
- 40.Sekhar LN, Heros RC. Origin, growth, and rupture of saccular aneurysms: a review. Neurosurgery. 1981;8:248–260. doi: 10.1227/00006123-198102000-00020. [DOI] [PubMed] [Google Scholar]
- 41.Ramirez-Lassepas M. Stroke and the aging of the brain and the arteries. Geriatrics. 1998;53 (Suppl 1):44–48. [PubMed] [Google Scholar]
- 42.Weinberger J. Diagnosis and prevention of atherosclerotic cerebral infarction. CNS Spectr. 2005;10:553–564. doi: 10.1017/s1092852900010208. [DOI] [PubMed] [Google Scholar]