

Abstract
Specific short peptides derived from motifs found in full-length proteins, in our case HIV-1 Nef, not only retain their biological function, but can also competitively inhibit the function of the full-length protein. A set of 20 Nef scanning peptides, 20 amino acids in length with each overlapping 10 amino acids of its neighbor, were used to identify motifs in Nef responsible for its induction of apoptosis. Peptides containing these apoptotic motifs induced apoptosis at levels comparable to the full-length Nef protein. A second peptide, derived from the Secretion Modification Region (SMR) of Nef, retained the ability to interact with cellular proteins involved in Nef's secretion in exosomes (exNef). This SMRwt peptide was used as the "bait" protein in co-immunoprecipitation experiments to isolate cellular proteins that bind specifically to Nef's SMR motif. Protein transfection and antibody inhibition was used to physically disrupt the interaction between Nef and mortalin, one of the isolated SMR-binding proteins, and the effect was measured with a fluorescent-based exNef secretion assay. The SMRwt peptide's ability to outcompete full-length Nef for cellular proteins that bind the SMR motif, make it the first inhibitor of exNef secretion. Thus, by employing the techniques described here, which utilize the unique properties of specific short peptides derived from motifs found in full-length proteins, one may accelerate the identification of functional motifs in proteins and the development of peptide-based inhibitors of pathogenic functions.
Keywords: Virology, Issue 76, Biochemistry, Immunology, Infection, Infectious Diseases, Molecular Biology, Medicine, Genetics, Microbiology, Genomics, Proteins, Exosomes, HIV, Peptides, Exocytosis, protein trafficking, secretion, HIV-1, Nef, Secretion Modification Region, SMR, peptide, AIDS, assay
Introduction
With the advent of anti-retroviral therapy, the AIDS epidemic in the western world has been slowed, but not curtailed, and the spread of HIV continues to be a major health burden throughout the world. With the exception of the marginally effective RV144 Thai trial, HIV vaccines have so far shown failure to protect from infection. Thus, research into additional, potential therapeutic targets is still warranted.
Along with CD4 T-cell depletion, persistent generalized immune activation is a hallmark of HIV infection. This Chronic Immune Activation (CIA) leads to increases in cell turnover, activated and differentiated lymphocytic subpopulations, cellular exhaustion and senescence, and the killing of T cells and B cells via Activation-Induced Cell Death (AICD)1,2,3, and it is well established as one of the strongest predictors of disease progression4,5,6,7,8,9,10,11,12. However, the mechanisms underlying CIA and CD4 T-cell depletion in HIV infection remain to be fully elucidated.
Evidence from our lab and others led us to a model for disease progression (Figure 1) wherein the HIV protein Nef (Negative Regulatory Factor) induces its secretion in exosomes from HIV-1 infected cells13,14. These Nef-containing exosomes (exNef) induce apoptosis in a number of cell lineages including uninfected CD4 T-cells15,16. Alternatively, in monocytes/macrophages exNef alters gene expression patterns, e.g. cytokine expression, and appears to induce a state of unscheduled immune activation. This body of evidence suggests an important role for exNef in CIA and CD4 T-cell depletion.
Understanding the mechanisms underlying Nef's ability to manipulate the exosomal trafficking pathway will be useful in engineering novel inhibitors of exNef secretion. Inhibition of exNef secretion should diminish the CD4 T-cell depletion and CIA that drive HIV/AIDS pathogenesis.
To gather the evidence that led to our model for HIV/AIDS disease progression and the subsequent data that built onto this model, we developed a number of novel reagents and methodologies that allowed us to analyze the genetics of exNef secretion, and begin to determine the cellular proteins involved. In the initial work, we found that Nef protein induces apoptosis in bystander cells and is released extracellularly from Nef-transfected and HIV-infected cells15. Peptides derived from SDF-1α (Stromal cell Derived Factor-1, with alternative splicing alpha) had been previously shown to retain much of the binding and signaling activity of the full-length molecule17. We speculated that peptides of Nef might retain some of the apoptotic activity of the full protein, and that these peptides would necessarily contain the Nef apoptotic domain(s). To identify these peptides, and consequently the Nef apoptotic domain(s), we obtained a set of 20 HIV-1 Nef scanning peptides from the NIH AIDS Research and Reference Reagent Program. These 20-aa peptides, each overlapping 10 amino acids of its neighbor, are named by the number of their last amino acid, i.e. N20 spans Nef amino acids 1-20, N30 spans Nef amino acids 11-30, etc16. We found that exposing T-cells extracellularly to specific peptides overlapping two distinct 10-aa domains in the full-length Nef protein induced apoptosis in these cells. Subsequent analysis of these Nef-derived apoptotic peptides revealed their ability to physically interact with the chemokine receptor CXCR4 on the surface of T-cells, with binding kinetics that allowed these peptides to competitively inhibit binding between CXCR4 and its natural ligand SDF-1α. Finally, the Nef-derived apoptotic peptides' interaction with CXCR4 was found to induce a stress response in these T-cells leading to apoptosis. This evidence allowed us to quickly map Nef's functional domains; a process that would have taken much longer using standard DNA mutagenic techniques such as alanine scanning mutagenesis. It also showed that these short Nef-derived peptides retained the biological function of the apoptotic domains in the full-length protein.
Having identified a role for extracellular Nef, i.e. apoptosis of T-cells, we sought a better understanding of how Nef was secreted from cells. Using a series of mutated Nef constructs, we mapped highly conserved Nef protein motifs in the N-terminal regions of both HIV Nef, and its Rhesus macaque equivalent SIVmac (Simian Immunodeficiency Virus) Nef, that are critical for exNef secretion13 . One of these motifs, the Secretion Modification Region (SMR; 66VGFPV70), was particularly critical, as alanine replacement of any of its five amino acids either greatly reduced or abolished exNef secretion. When delivered to cells via Active Motif's Chariot Protein Delivery Reagent, a peptide containing the SMR attached to a FLAG peptide sequence (SMRwt) was found to inhibit exNef secretion from both Nef-transfected and HIV-infected cells 18. Based on our previous experience with peptides, we decided to use this peptide to elucidate the molecules and mechanisms underlying the Nef SMR's role in exNef secretion.
Using the SMRwt peptide as our "bait protein", we co-immunoprecipitated cellular binding partners of the SMR from uninfected T-cell lysates18. The FLAG peptide sequence provided a convenient handle for capturing the SMRwt peptide using anti-FLAG affinity resin. Our previous finding that a single valine to alanine mutation in the SMRwt peptide was sufficient for abolishing its inhibition of exNef secretion identified a convenient, highly specific control peptide (SMRmut) that we used to rule out co-immunoprecipitated proteins not specific for the SMR. Using a peptide with the specific domain of interest rather than the full-length protein allowed us to bypass the screening of dozens of cellular factors that bind to other domains in Nef19.
Once we identified the SMR-binding partners, a logical next step was to show that the identified cellular binding partners are important for the biological function18. The standard procedure to accomplish this is to knockdown protein levels using target protein-specific miRNA or siRNA, and subsequently assay the effect on the biological function. We performed miRNA knockdown, which inhibits translation of the target mRNA, reducing production of that target, in this case a SMR-binding protein. This reduction of the target protein is an indirect effect, and possibly has a delayed effect on the biological function the targeted protein plays a role in. Consequently, we also employed the less common antibody inhibition technique to determine if directly disrupting the activity of the target protein reduces or eliminates the biological function. In this procedure, antibodies raised against the targeted protein are transfected into the cell using Chariot Reagent, and interact directly with the target protein either sequestering it from its site of function, or blocking its relevant binding domain. Inhibition of the target protein through this procedure directly disrupts its function, and can complement RNA knockdown procedures by further confirming the importance of the target protein to the biological function.
While Chariot Reagent is effective at delivering peptides and proteins into cells; this process is time-consuming and limits the types of experiments, e.g. prolonged or repeated exposures and in vivo animal studies that can be performed. Consequently, we added a cell-penetrating peptide (CPP) sequence to the SMRwt peptide (SMRwt-CPP)18 to generate a peptide that could be taken up by cells passively from the culture media. This version was as effective as the former at inhibiting exNef secretion.
The evidence from these published experiments demonstrates the ability of small peptides containing specific functional motifs to antagonize the function of the full-length protein through competitive inhibition, and to isolate the proteins that bind these motifs. One would expect that these techniques should be useful in many experimental protocols. They should also be effective in engineering novel peptide inhibitors of many cellular processes; a function that can be further enhanced by linkage to CPP sequences.
Protocol
I. Use of Short Peptides in Biological Analysis
I.1. Mapping Biologically Functional Motifs Using Peptides
- Treating cells with Nef scanning peptides
- Procure a set of peptides scanning the region of interest. For our experiments, 20mer peptides, with 10 amino acid overlap, spanning the entire HIV-1 Nef protein (205 aa), were obtained from the AIDS Reagent Program. The peptides are assayed individually as follows:
- Add 10 ng/ml of peptide to the cell culture medium.
- Add 10 ml of culture medium per plate to Jurkat cell cultures.
- Incubate cultures for 24 hr at 37 °C.
- Terminal deoxynucleotidyltransferase-mediated dUTP biotin Nick End Labeling (TUNEL) assay of apoptosis
- Wash cells with 1X PBS, and fix for 30 min at room temperature with 4% paraformaldehyde in 1X PBS, pH 7.4.
- Wash with 1X PBS, and permeabilize with 0.1% Triton X-100 for 10 min at room temperature.
- Rinse the cells twice with 1X PBS.
- Count stained cells by epifluorescence on a computer-controlled microscopy system.
I.2. Competitive Inhibition Analysis using Peptides
- Co-transfection of Peptides using the Chariot Protein Delivery Reagent Kit
- Per transfection reaction mixture, dilute 1 μg of wtNef-GFP plasmid DNA and 100 ng of either wildtype peptides derived from the SMR domain, or peptides containing alanine-replacement of individual amino acids in the SMR motif, to a final volume of 100 μl with 1X PBS.
- Dilute 6 μl of a 1/10 dilution of Chariot Reagent to a final volume of 100 μl with sterile water, and combine with the diluted DNA-peptide mix.
- Incubate at room temperature for 30 min to allow formation of the Chariot/DNA-peptide complex.
- Pellet 3 x 105 Jurkat cells by centrifugation at 600 x g for 5 min.
- Wash cells twice with 1X PBS, and resuspend in the Chariot/DNA-peptide complex.
- Add 400 μl of serum-free RPMI 1640 medium to the suspension and incubate the cells in culture plates at 37 °C for 2 hr.
- Add 1 ml of fresh RPMI 1640 medium containing 10% FBS to each plate, and incubate the cells for 48 hr at 37 °C.
- Harvest cells and determine transfection efficiency by fluorescent microscopy.
- Nef-GFP secretion fluorescent plate reader assay of peptide inhibition
- Collect the cell-free conditioned media from the harvested cells, and transfer 100 μl to individual wells of a 96-well black microtiter plate.
- Measure GFP fluorescence in the media on a Tecan GENEios fluorimeter with excitation wavelength 485 nm and emission wavelength 515 nm in relative fluorescent units.
- Set the level of GFP fluorescence in the control (media from cells co-transfected with Nef-GFP and a scrambled peptide) at 100%.
- Compare this to the level of GFP fluorescence in the media from cells co-transfected with various SMR peptides to determine changes in Nef-GFP secretion.
I.3. Isolating Motif Binding Partners Using Peptides as Bait
- Co-immunoprecipitation with SMR peptides
- Grow Jurkat cells in RPMI 1640 medium containing 10% FBS at 37 °C in a T-75 tissue culture flask, and pellet cells by centrifugation at 1,000 x g for 20 min at 4 °C. Determine the transfection efficiency, by placing the plate under a fluorescent microscope, capture images, and subsequently count the total and labeled cells and determine the percent labeled cells.
- Resuspend the cell pellet in 1 ml 1X PBS, and transfer it to a 1.5-ml Eppendorf tube. Microcentrifuge at 1,000 x g for 1 min. Resuspend the pellet in 1 ml α-FLAG Lysis buffer by gentle pipetting. Lysis buffer: Mix 50 mM Tris•HCl pH 7.4, 150 mM sodium chloride [NaCl], 1 mM EDTA, and 1% Triton X-100, with 1 mM phenylmethylsulfonyl fluoride [PMSF], and 10 μl/ml Protease Inhibitor Cocktail for use with mammalian cell and tissue extracts [PIC; Sigma]. Make immediately before using.
- Differentially microcentrifuge the suspensions at 2,000 x g for 5 min, and at 13,000 x g for 10 min to pellet the cell debris and DNA, respectively. Collect the supernatant (hereafter referred to as the "lysate").
- Add 1 mg of lysate protein and 1 μg of either the SMRwt or SMRmut peptide to 20 μl of anti-FLAG M2 Affinity Gel (hereafter called "resin"; Sigma). Note that both SMRwt and SMRmut used here have a FLAG tag sequence embedded in the peptides. Bring the mixture to a final volume of 1 ml in Co-IP buffer (α-FLAG Lysis buffer minus PMSF and PIC). Incubate the mixture at 4 °C overnight with end-over-end rotation. As a negative control, incubate the lysate with the resin in the absence of either peptide.
- Pellet the resin by microcentrifugation at 5,000 - 8,000 x g for 30 sec. Allow resin to settle for 2 min before handling the sample and then aspirate the supernatant. Remove the supernatant with a narrow-end pipette tip, or a Hamilton syringe, being careful not to transfer any resin. Narrow-end pipette tips can be made using forceps to pinch the opening of a plastic pipette tip until it is partially closed. Wash the resin four times with 500 μl of wash buffer (50 mM Tris•HCl pH 7.4, and 150 mM NaCl). Elute the bound cellular proteins with 15 μg of 3X FLAG peptide (Sigma) in 50 μl of wash buffer and incubate on rocker/shaker for 30 min at room temperature.
- Concentrate the eluates by acetone precipitation, followed by boiling in Laemmli sample buffer (Bio-Rad). Separate the proteins by SDS-PAGE on a 4-20% Tris-HCl Criterion precast gel (Bio-Rad). Stain the gel with Coomassie Brilliant Blue R-250 (Bio-Rad). Note: for mass spectrometry identification experiments (Figure 4A), each condition (Co-IP with SMRwt, SMRmut, or no peptide) was performed in quadruplicate, and the eluates from all four replicates were pooled prior to acetone precipitation for a total of 4 mg of total protein input/condition).
- Co-immunoprecipitation with Nef-GFP protein
- Mix 50 μl of Dynabeads Protein G magnetic beads (Invitrogen) with 2 μg of murine monoclonal anti-GFP in 1 ml of 1X PBS with 0.02% Tween 20 (PBS-T). Incubate the mixture with end-over-end rotation in a 1.5-ml Eppendorf tube for 1 hr at 4 °C.
- Separate the beads from the buffer by placing the tube on a 1.5-ml MagnaSphere Technology Magnetic Separation Stand (Promega Corp., Madison, WI) allowing magnetic pulldown followed by removal of the supernatant. Wash the anti-GFP coated beads with 1 ml of PBS-T using gentle pipetting.
- Lyse Nef-GFP transfected Jurkat cells with 1 ml of 1X RIPA+ buffer by gentle pipetting and incubation for 15 min on ice. RIPA+ buffer: 10X RIPA Lysis Buffer, pH 7.4 [Millipore, Bedford, MA] diluted 1:10 in sterile water, 0.02% sodium azide [Sigma], and 0.1% sodium dodecyl sulfate [SDS], with 1 mM PMSF and 10 μl/ml PIC added immediately before using.
- Pellet the cell debris by microcentrifugation at 2,000 x g for 10 min. Collect the supernatant (hereafter referred to as the "lysate").
- Add 1 mg of lysate protein to the anti-GFP coated beads, bring the final volume to 1 ml with 1X RIPA+ buffer, and incubate the mixture with end-over-end rotation for 1 hr at 4 °C. Note: For peptide competition studies, 1 mg of Lysate protein may be pre-incubated with 2 μg of SMRwt or SMRmut peptide in 1 ml of 1X RIPA+ buffer, prior to being added to the anti-GFP coated beads.
- Wash the beads through resuspension in 1 ml of wash buffer (50 mM Tris•HCl pH 7.4, 150 mM NaCl, and 0.1% Tween 20). Incubate with end-over-end rotation for 5 min at 4 °C. Separate the beads from the wash using the Magnetic Separation Stand. Wash the beads a second and third time with wash buffers containing 250 mM and 300 mM NaCl, respectively.
- Resuspend the beads in 200 μl of PBS, transfer the mixture to a clean tube, and separate the beads from the wash on the magnetic stand. Boil the beads in 50 μl of Laemmli sample buffer 5 min at 95 °C, allow the mixture to cool for 10 min at room temperature, and then place the mixture on the magnetic stand for separation, and collect the supernatants ("eluates").
- Separate the proteins of the eluate by SDS-PAGE on an 8-16% Tris-HCl Criterion precast gel.
- Mass spectrometry
- Excise the protein bands of interest from Coomassie-stained gels.
- Reduce the samples in 10 mM 1,4-dithiothreitol (DTT) for 45 min at 37 °C.
- Alkylate the samples in 55 mM iodoacetamide for 45 min at 25 °C.
- Digest the samples overnight with sequencing grade trypsin (Promega) at 37 °C.
- Desalt the sample peptides with C-18 ZipTip (Millipore). Mix the sample peptides with alpha-CHC Matrix (Agilent Technologies, Santa Clara, CA), and spot them onto a MTP target frame III (Bruker Daltonics Inc., Billerica, MA).
- Perform tandem matrix-assisted laser desorption/ionization time of-flight (MALDI-TOF/TOF) Mass Spectrometry analysis using a Bruker Daltonics ultraflex III TOF/TOF.
- Compare the MS/MS data to the NCBI non-redundant and Swiss-Prot databases using the Mascot algorithm (www.matrixscience.com).
- Immunoblot Analysis
- Separate the protein samples by SDS-PAGE on 4-20% or 8-16% Tris-HCl Criterion precast gels (Bio-Rad). Transfer the bands electrophoretically to a nitrocellulose membrane.
- Wash the membrane in Tris Buffered Saline (TBS; Bio-Rad) for 5 min. Block with 5% nonfat milk in TTBS (TBS with 0.1% Tween 20) for 1 hr by shaking at room temperature.
- Process the membrane for immunoblotting using a specific primary antibody in 5% nonfat milk with shaking at 4 °C overnight. Wash in TTBS for 20 min followed by a secondary HRP conjugated IgG (H+L) antibody in 5% nonfat milk for 1 hr at room temperature. Wash in TTBS for 30-60 min.
- Detect the protein bands by Western Blotting Luminol Reagent (Santa Cruz Biotechnology, Inc., Santa Cruz, CA). Expose the filter to photographic film. Note: In some experiments, a stripping reagent was used to strip the membrane (Pierce, Rockford, IL) with subsequent hybridization with a different primary and secondary antibody.
- Scan the X-ray films into Adobe Photoshop 5.0.2. Perform densitometry analysis using ImageJ software (National Institutes of Health, Bethesda, MD).
II. Use of Antibody Inhibition in Functional Analysis
II.1. Co-transfection of Antibodies using the Chariot Protein Delivery Reagent Kit
Per transfection reaction mixture, dilute 1 μg of wtNef-GFP plasmid DNA and 1 μg of either anti-mortalin antibody or anti-α-tubulin antibody, to a final volume of 100 μl with 1X PBS. Note: It was previously found that co-transfection of the anti-α-tubulin antibody has no detectable effect on exNef secretion, and thus, it serves as an effective negative control in these experiments.
Dilute 6 μl of undiluted Chariot Reagent to a final volume of 100 μl with sterile water, and combine with the diluted DNA-antibody mix.
Incubate at room temperature for 30 min to allow formation of the Chariot/DNA-antibody complex.
Pellet 3 x 105 Jurkat cells by centrifugation at 600 x g for 5 min.
Wash cells twice with 1X PBS, and resuspend in the Chariot/DNA-antibody complex.
Add 400 μl of serum-free RPMI 1640 medium to the suspension and incubate the cells in culture plates at 37 °C for 2 hr.
Add 1 ml of fresh RPMI 1640 medium containing 10% FBS to each plate, and incubate the cells for 48 hr at 37 °C.
II.2. Nef-GFP Secretion Fluorescent Plate Reader Assay of Antibody Inhibition
Collect the cell-free conditioned media from the harvested cells, and transfer 100 μl to individual wells of a 96-well black microtiter plate.
Measure GFP fluorescence in the media on a Tecan GENEios fluorimeter with excitation wavelength 485 nm and emission wavelength 515 nm in relative fluorescent units.
Set the level of GFP fluorescence in the control (media from cells co-transfected with Nef-GFP and anti-α-tubulin antibody) at 100%.
Compare this to the level of GFP fluorescence in the media from cells co-transfected with anti-mortalin antibody to determine changes in Nef-GFP secretion.
Representative Results
Mapping Biologically Functional Motifs using Peptides. Two regions were identified on Nef proteins that induce apoptosis. Peptide-driven apoptosis was observed (Figure 2) beginning with peptide N50 (aa30-50), peaking at N60 (aa40-60) and N70 (aa50-70), and diminishing to background levels at peptide N100 (aa80-100). The major Motif 1 (M1) peak, centering on aa50-60, induces >80% of the apoptotic levels of the full-length Nef protein. A second, smaller apoptotic peak was observed spanning peptides N180 (aa160-180) and N190 (aa170-190). This minor Motif 2 (M2) peak, centered on aa170-180, induces ~30% of the apoptotic levels of full-length Nef protein16.
Competitive Inhibition Analysis using Peptides. In Figure 3, peptides containing an intact SMR significantly inhibited exNef secretion. This was true whether the motif was located at the N-terminus (Wildtype-1) or middle (Wildtype-2) of the peptide, or was duplicated in the peptide (Wildtype-3). Alternatively, alanine-substitution of any amino acid of the motif, irrespective of the motif's position or number in the peptide, abrogated the peptide's function18. An 11-amino-acid scrambled version of Nef's apoptotic Motif 1 (sM1), previously described14 and obtained from Sigma Genosys, was used as a negative control and had no effect on exNef secretion. See Table 1 for a list of the peptide sequences.
Isolating Motif Binding Partners Using Peptides as Bait. Figure 4A demonstrates the co-immunoprecipitation of four proteins by the SMRwt peptide (60, 65, 75, & 250 kDa). The negative control SMRmut peptide did not capture these proteins, and these proteins did not non-specifically interact with the affinity resin in the absence of peptide, suggesting that the co-immunoprecipitated proteins were interacting specifically with the SMR motifs of the SMRwt peptide. These proteins were subsequently identified by MALDI-TOF tandem mass spectrometry (see Figure 4B for a representative sample), and verified by immunoblot analysis18.
Use of Antibody Inhibition in Functional Analysis. Inhibition with anti-mortalin antibody completely abolished exNef secretion (Figure 5). This confirmed that an intact Nef/mortalin interaction is required for exNef secretion. Furthermore, it strongly suggests that the mechanism by which the SMRwt peptide inhibits exNef secretion is through disruption of this interaction by direct competition for mortalin18.
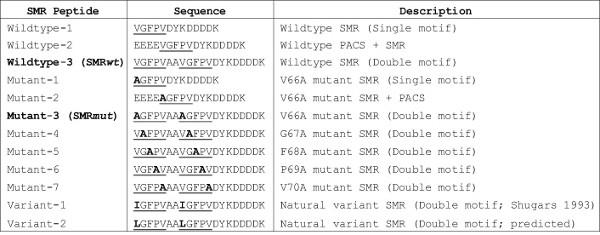 Table 1. Panel of SMR peptides developed and tested for their effect on Nef-induced exNef secretion. This table was originally published in Shelton et al., JVI, 201218. In previous studies, alanine-replacement of single amino acids of the SMR motif significantly reduced, or abolished, Nef-induced secretion 18. Virus isolated from one long-term nonprogressor expressed a variant of Nef with an isoleucine in place of V66; one of the few reported variations in the highly-conserved SMR motif 20. Click here to view larger table.
Table 1. Panel of SMR peptides developed and tested for their effect on Nef-induced exNef secretion. This table was originally published in Shelton et al., JVI, 201218. In previous studies, alanine-replacement of single amino acids of the SMR motif significantly reduced, or abolished, Nef-induced secretion 18. Virus isolated from one long-term nonprogressor expressed a variant of Nef with an isoleucine in place of V66; one of the few reported variations in the highly-conserved SMR motif 20. Click here to view larger table.
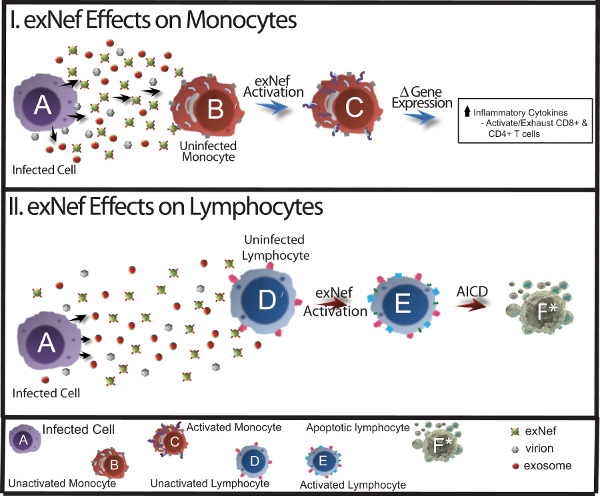 Figure 1. Effects of Nef exosomes on major immune cell types. This model displays exNef released from infected cells, and it's predicted effects on monocytic cells and lymphocytic cells that would lead to AIDS pathogenesis. AICD, activation induced cell death. I. Infected cell releases virus, normal exosomes, and exNef. exNef activates unactivated, uninfected monocytes (B). The activation of the monocytes (C) results in changes in gene expression leading to increases in the inflammatory state and result in activation and exhaustion of CD4+ and CD8+ T-cells. II. Infected cell releases virus, normal exosomes, and exNef. exNef activates unactivated, uninfected lymphocytes (D). The activated lymphocytes (E) undergo gene expression changes that result in AICD resulting in an apoptotic cell (F). Click here to view larger figure.
Figure 1. Effects of Nef exosomes on major immune cell types. This model displays exNef released from infected cells, and it's predicted effects on monocytic cells and lymphocytic cells that would lead to AIDS pathogenesis. AICD, activation induced cell death. I. Infected cell releases virus, normal exosomes, and exNef. exNef activates unactivated, uninfected monocytes (B). The activation of the monocytes (C) results in changes in gene expression leading to increases in the inflammatory state and result in activation and exhaustion of CD4+ and CD8+ T-cells. II. Infected cell releases virus, normal exosomes, and exNef. exNef activates unactivated, uninfected lymphocytes (D). The activated lymphocytes (E) undergo gene expression changes that result in AICD resulting in an apoptotic cell (F). Click here to view larger figure.
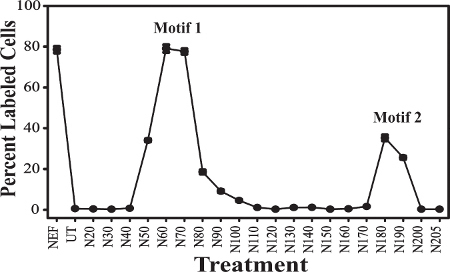 Figure 2. Peptide scanning analysis of the HIV-1 Nef protein for apoptotic motif(s). A set of 20mer peptides, each with 10 amino acid overlap, spanning the 205 amino acids of the KNFS Nef protein, was obtained from the AIDS Reagent Program and used in mapping the Nef apoptotic domains. This figure was originally published in Huang et al., JVI, 200413. The Y-axis shows percentage of cells that are TUNEL labeled, and the X-axis denotes the treatment condition. Full Nef protein [Nef], untreated cells [UT], or the peptides starting with aa1-20 [N20], and ending with aa190-205 [N205]. Error bars denote the standard error of measurement, and the results are a compilation of at least 3 independent experiments. The peaks of apoptosis are denoted by Motif 1 and Motif 2.
Figure 2. Peptide scanning analysis of the HIV-1 Nef protein for apoptotic motif(s). A set of 20mer peptides, each with 10 amino acid overlap, spanning the 205 amino acids of the KNFS Nef protein, was obtained from the AIDS Reagent Program and used in mapping the Nef apoptotic domains. This figure was originally published in Huang et al., JVI, 200413. The Y-axis shows percentage of cells that are TUNEL labeled, and the X-axis denotes the treatment condition. Full Nef protein [Nef], untreated cells [UT], or the peptides starting with aa1-20 [N20], and ending with aa190-205 [N205]. Error bars denote the standard error of measurement, and the results are a compilation of at least 3 independent experiments. The peaks of apoptosis are denoted by Motif 1 and Motif 2.
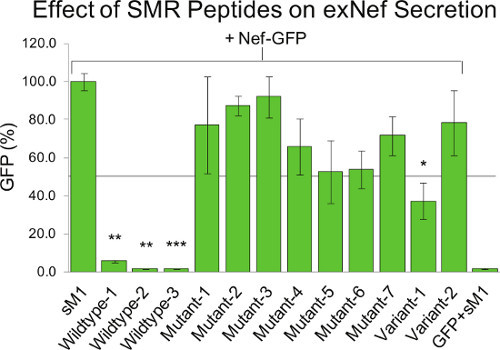 Figure 3. SMRwt peptide inhibits exNef. Peptides containing one or more wildtype Nef SMR sequence motifs inhibited the secretion of exNef, while alanine-replacement of any animo acid of this region greatly reduced the peptide's effectiveness. This figure was originally published in Shelton et al., JVI, 201215. Culture media from Jurkat T cells, co-transfected with a Nef-GFP clone and various SMR peptides, were assayed for exNef secretion. The amount of GFP fluorescence measured is equivalent to the amount of exNef-GFP in the extracellular media. Statistical significance was determined using an unpaired t-test. The effect on exNef secretion of various peptides was compared to that of a randomly scrambled peptide control (sM1; * p-value < 0.05; ** p-value < 0.01; *** p-value < 0.001).
Figure 3. SMRwt peptide inhibits exNef. Peptides containing one or more wildtype Nef SMR sequence motifs inhibited the secretion of exNef, while alanine-replacement of any animo acid of this region greatly reduced the peptide's effectiveness. This figure was originally published in Shelton et al., JVI, 201215. Culture media from Jurkat T cells, co-transfected with a Nef-GFP clone and various SMR peptides, were assayed for exNef secretion. The amount of GFP fluorescence measured is equivalent to the amount of exNef-GFP in the extracellular media. Statistical significance was determined using an unpaired t-test. The effect on exNef secretion of various peptides was compared to that of a randomly scrambled peptide control (sM1; * p-value < 0.05; ** p-value < 0.01; *** p-value < 0.001).
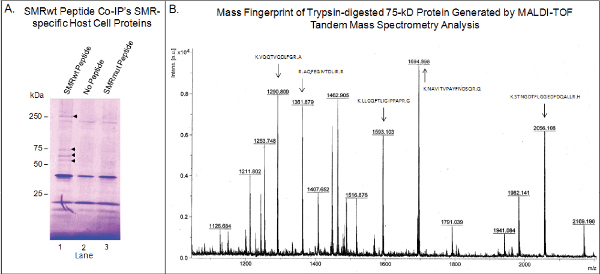 Figure 4. The SMRwt peptide interacts specifically with host cell proteins, including mortalin.(A) Proteins from Jurkat T cells were co-immunoprecipitated with either the SMRwt or SMRmut peptides using anti-FLAG M2 antibody-coupled affinity resin. Note that both SMRwt and SMRmut used here have a FLAG tag sequence embedded in the peptides. This figure was originally published in Shelton et al., JVI, 201215. This procedure was repeated in the absence of either peptide as a control for non-specific interactions with the affinity resin. The Coomassie Blue stained gel pictured displays proteins with molecular weights of 60, 65, 75, and 250 kDa (arrowheads) pulled down by SMRwt that were not pulled down by the SMRmut peptide or with no peptide. (B) The 75-kDa band was excised from the gel, trypsin-digested, and analyzed by MALDI-TOF tandem mass spectrometry. The MS/MS Ion Search identified the 75-kDa as mortalin. Arrows denote the sequences of the matching peptides. Click here to view larger figure
Figure 4. The SMRwt peptide interacts specifically with host cell proteins, including mortalin.(A) Proteins from Jurkat T cells were co-immunoprecipitated with either the SMRwt or SMRmut peptides using anti-FLAG M2 antibody-coupled affinity resin. Note that both SMRwt and SMRmut used here have a FLAG tag sequence embedded in the peptides. This figure was originally published in Shelton et al., JVI, 201215. This procedure was repeated in the absence of either peptide as a control for non-specific interactions with the affinity resin. The Coomassie Blue stained gel pictured displays proteins with molecular weights of 60, 65, 75, and 250 kDa (arrowheads) pulled down by SMRwt that were not pulled down by the SMRmut peptide or with no peptide. (B) The 75-kDa band was excised from the gel, trypsin-digested, and analyzed by MALDI-TOF tandem mass spectrometry. The MS/MS Ion Search identified the 75-kDa as mortalin. Arrows denote the sequences of the matching peptides. Click here to view larger figure
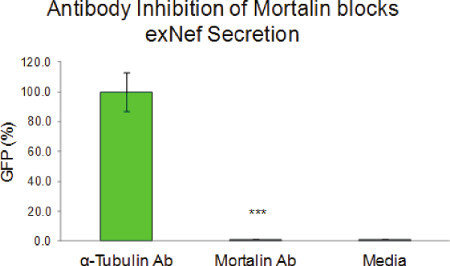 Figure 5. Mortalin antibody inhibition blocks exosome secretion. Extracellular culture media from Jurkat cells, co-transfected with a Nef-GFP clone and antibodies against either mortalin or α-tubulin, were assayed for exNef secretion. When compared to the effect of an antibody against a protein not involved in exNef secretion (α-tubulin), disruption of mortalin's activity by its antibody resulted in complete abolition of exNef secretion. Statistical significance was determined by unpaired t-test comparing exNef secretion in presence of the mortalin antibody versus the α-tubulin antibody (*** p-value < 0.001). This figure was originally published in Shelton et al., JVI, 201218.
Figure 5. Mortalin antibody inhibition blocks exosome secretion. Extracellular culture media from Jurkat cells, co-transfected with a Nef-GFP clone and antibodies against either mortalin or α-tubulin, were assayed for exNef secretion. When compared to the effect of an antibody against a protein not involved in exNef secretion (α-tubulin), disruption of mortalin's activity by its antibody resulted in complete abolition of exNef secretion. Statistical significance was determined by unpaired t-test comparing exNef secretion in presence of the mortalin antibody versus the α-tubulin antibody (*** p-value < 0.001). This figure was originally published in Shelton et al., JVI, 201218.
Discussion
Understanding the mechanisms underlying Nef's ability to manipulate the exosomal trafficking pathway will be useful in engineering novel inhibitors of exNef secretion. Inhibition of exNef secretion should diminish the CD4 T-cell depletion and CIA that drive HIV/AIDS pathogenesis. Towards this goal, we developed a number of novel reagents and methodologies that allowed us to analyze the genetics of exNef secretion, and begin to determine the cellular proteins involved. This approach also led to the development of the first inhibitor to directly target exNef secretion, the SMRwt peptide.
A key finding of our work is that specific short peptides derived from motifs found in full-length proteins, in our case HIV-1 Nef, not only retain their biological function, but can also competitively inhibit the function of the full-length protein. Because these peptides retain their biological function, they can be used to identify functional domains in the full-length protein. Two things are required: the generation of peptides that scan the protein of interest, either its entire length or a region of the protein thought to contain the functional domain; and an assay for testing the biological function in question. Since we lacked information that would allow us narrow our search for apoptosis-inducing motifs to specific regions of Nef, we used a set of peptides scanning its entire length. While this process was made simpler by Nef's relatively small size, we have also applied this approach to larger (>75 kDa) proteins. A well-established assay for measuring apoptosis (TUNEL) was employed to identify peptides retaining the biological function of interest, in this case, Nef-induced apoptosis of CD4 T-cells. Interestingly, the subsequent analysis in the original paper showed that the identified apoptotic peptides retained >80% of the biological activity of the full-length Nef protein, and competitively inhibited CXCR4 binding of its natural ligand SDF-1α.
Similarly, a short peptide containing the SMR motif of Nef retained its biological ability to interact with cellular proteins involved in exNef secretion. However, in this instance retention of activity did not lead to mimicry of a Nef function, as was the case with apoptosis induction, but led instead to inhibition. Because the SMRwt peptide effectively bound the SMR-binding sites of these cellular proteins, it disrupted their interactions with Nef, and consequently, prevented Nef's secretion in exosomes. As the Nef motif of interest was known from previous genetic mapping studies, we were able to limit peptide development to variations of the SMR. Instead of identifying novel functional motifs, we used these peptides, and a previously developed fluorescence-based assay for measuring exNef secretion, to demonstrate that the SMR's role in exNef secretion was linked directly to its specific primary sequence.
Additionally, the SMRwt peptide's ability to interact with SMR-binding cellular proteins was leveraged to isolate these proteins. Standard techniques could then be used to identify them. NCBI's HIV-1, Human Protein Interaction Database lists more than 60 cellular proteins that interact directly with Nef and possibly as many as 200 that interact directly or indirectly 19. By restricting the sequence of our bait protein to the motif of interest, we avoided having to sort through the dozens of proteins that would have been co-immunoprecipitated by the full-length Nef protein. The majority of these proteins, while specific for Nef, would not be binding partners for Nef's SMR, and would thus constitute background/noise. A conservative calculation is that we reduced the noise by 15x (60/4). In essence, our approach effectively increased the signal-to-noise ratio by minimizing background due to off-site, or in our case off-motif binding, allowing us to identify proteins that specifically bind the SMR. However, it is possible that, by using this method, we have failed to capture all SMR-binding cellular proteins, particularly those that require interaction with both the SMR and a second motif to bind Nef, or those whose binding is dependent on secondary or tertiary structure.
Mortalin, a cellular protein co-immunoprecipitated by the SMRwt peptide, was shown to be required for exNef secretion by miRNA knockdown and antibody inhibition. The former is a well-established technique that reduces or eliminates expression of the targeted protein. Conversely, antibody inhibition does not affect protein expression levels, but instead physically inhibits the targeted protein's interaction with other proteins. We used this second method to demonstrate the importance of the Nef-mortalin interaction for exNef secretion. Antibody inhibition is a straight-forward procedure that requires a method for introducing the antibody into the cell and an assay for testing its effect on the biological function in question. We used the Chariot Protein Delivery Reagent to shuttle anti-mortalin antibodies into the cell, and the aforementioned fluorescence-based assay to measure changes in exNef secretion. Like most antibody-based procedures, the effectiveness of antibody inhibition is limited by the affinity and specificity of the antibody for the targeted protein.
A major goal of HIV research is the development of novel therapeutics and the identification of potential targets for therapy. The procedures described here leverage specific short peptides, which retain their biological function and can competitively inhibit the function of full-length proteins, to accelerate this process. While the experiments described were directed at understanding a key HIV function, the techniques employed should be applicable to the study of protein-protein interactions and the development of peptide-based inhibitors in several fields.
Disclosures
The authors declare that they have no competing financial interests.
Acknowledgments
This work was supported by NIH/NIGMS/MBRS (Grant 58268), NIH/NCRR/RCMI (Grant G12-RR03034), Georgia Research Alliance funding grant GRA.VAC08.W, NIH/NIAID/NRSA grant F31AI091484, Emory CFAR grant P30 A1050409. This investigation was conducted in a facility constructed with support from Research Facilities Improvement Grant #C06 RR18386 from NIH/NCRR. The Jurkat cells, the set of 20 HIV-1 Nef peptides, as well as the rabbit anti-HIV-1 Nef antiserum were obtained from the NIH AIDS Research & Reference Reagent Program, (Rockville, MD).
References
- Forsman A, Weiss RA. Why is HIV a pathogen? Trends Microbiol. 2008;16(12):555–560. doi: 10.1016/j.tim.2008.09.004. [DOI] [PubMed] [Google Scholar]
- Moir S, Chun TW, Fauci AS. Pathogenic mechanisms of HIV disease. Annu. Rev. Pathol. 2011;6:223–248. doi: 10.1146/annurev-pathol-011110-130254. [DOI] [PubMed] [Google Scholar]
- Smith SM. The pathogenesis of HIV infection: Stupid may not be so dumb after all. Retrovirology. 2006;3(1):60. doi: 10.1186/1742-4690-3-60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levacher M, Hulstaert F, Tallet S, Ullery S, Pocidalo JJ, Bach BA. The significance of activation markers on CD8 lymphocytes in human immunodeficiency syndrome: staging and prognostic value. Clin. Exp. Immunol. 1992;90(3):376–382. doi: 10.1111/j.1365-2249.1992.tb05854.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giorgi JV, Liu Z, Hultin LE, Cumberland WG, Hennessey K, Detels R. Elevated levels of CD38+ CD8+ T cells in HIV infection add to the prognostic value of low CD4+ T cell levels: results of 6 years of follow-up. The Los Angeles Center, Multicenter AIDS Cohort Study. J. Acquir. Immune. Defic. Syndr. 1993;6(8):904–912. [PubMed] [Google Scholar]
- Bofill M, Mocroft A, Lipman M, Medina E, Borthwick NJ, Sabin CA, Timms A, Winter M, Baptista L, Johnson MA, Lee CA, Phillips AN, Janossy G. Increased numbers of primed activated CD8+CD38+CD45RO+ T cells predict the decline of CD4+ T cells in HIV-1-infected patients. AIDS. 1996;10(8):827–834. doi: 10.1097/00002030-199607000-00005. [DOI] [PubMed] [Google Scholar]
- Liu Z, Cumberland WG, Hultin LE, Prince HE, Detels R, Giorgi JV. Elevated CD38 antigen expression on CD8+ T cells is a stronger marker for the risk of chronic HIV disease progression to AIDS and death in the Multicenter AIDS Cohort Study than CD4+ cell count, soluble immune activation markers, or combinations of HLA-DR and CD38 expression. J Acquir. Immune. Defic. Syndr. Hum. Retrovirol. 1997;16(2):83–92. doi: 10.1097/00042560-199710010-00003. [DOI] [PubMed] [Google Scholar]
- Douek DC, Roederer M, Koup RA. Emerging concepts in the immunopathogenesis of AIDS. Annu. Rev. Med. 2009;60:471–484. doi: 10.1146/annurev.med.60.041807.123549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberts L, Passmore JA, Williamson C, Little F, Bebell LM, Mlisana K, Burgers WA, et al. Plasma cytokine levels during acute HIV-1 infection predict HIV disease progression. AIDS. 2010;24(6):819–831. doi: 10.1097/QAD.0b013e3283367836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mueller YM, Petrovas C, Bojczuk PM, Dimitriou ID, Beer B, Silvera P, Villinger F, Cairns JS, Gracely EJ, Lewis MG, Katsikis PD. Interleukin-15 increases effector memory CD8+ t cells and NK Cells in simian immunodeficiency virus-infected macaques. J. Virol. 2005;79(8):4877–4885. doi: 10.1128/JVI.79.8.4877-4885.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Picker LJ, Reed-Inderbitzin EF, Hagen SI, Edgar JB, Hansen SG, Legasse A, Planer S, Piatak M, Lifson JD, Maino VC, Axthelm MK, Villinger F. IL-15 induces CD4 effector memory T cell production and tissue emigration in nonhuman primates. J. Clin. Invest. 2006;116(6):1514–1524. doi: 10.1172/JCI27564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mueller YM, Do DH, Altork SR, Artlett CM, Gracely EJ, Katsetos CD, Legido A, Villinger F, Altman JD, Brown CR, Lewis MG, Katsikis PD. IL-15 treatment during acute simian immunodeficiency virus (SIV) infection increases viral set point and accelerates disease progression despite the induction of stronger SIV-specific CD8+ T cell responses. J. Immunol. 2008;180(1):350–360. doi: 10.4049/jimmunol.180.1.350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ali SA, Huang MB, Campbell PE, Roth WW, Campbell T, Khan M, Newman G, Powell F, Powell MD, Bond VC. Genetic Characterization of HIV Type 1 Nef-Induced Vesicle Secretion. AIDS Res. Hum. Retroviruses. 2010;26(2):173–192. doi: 10.1089/aid.2009.0068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raymond AD, Campbell-Sims TC, Khan M, Lang M, Huang MB, Bond VC, Powell MD. HIV Type 1 Nef Is Released from Infected Cells in CD45+ Microvesicles and Is Present in the Plasma of HIV-Infected Individuals. AIDS. 2011;27(2):167–178. doi: 10.1089/aid.2009.0170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- James CO, Huang M-B, Khan M, Garcia-Barrio M, Powell MD, Bond VC. Extracellular Nef Protein Targets CD4+ T Cells for Apoptosis by Interacting with CXCR4 Surface Receptors. J. Virol. 2004;78(6):3099–3109. doi: 10.1128/JVI.78.6.3099-3109.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang MB, Jin LL, James CO, Khan M, Powell MD, Bond VC. Characterization of Nef-CXCR4 Interactions Important for Apoptosis Induction. J. Virol. 2004;78(20):11084–11096. doi: 10.1128/JVI.78.20.11084-11096.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heveker N, Montes M, Germeroth L, Amara A, Trautmann A, Alizon M, Schneider-Mergener J. Dissociation of the signalling and antiviral properties of SDF-1-derived small peptides. Curr. Biol. 1998;8(7):369–376. doi: 10.1016/s0960-9822(98)70155-1. [DOI] [PubMed] [Google Scholar]
- Shelton MN, Huang MB, Ali SA, Powell MD, Bond VC. SMR-derived peptide disrupts HIV-1 Nef's interaction with mortalin and blocks virus and Nef exosome release. J. Virol. 2012;86(1):406–419. doi: 10.1128/JVI.05720-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ptak RG, Fu W, Sanders-Beer BE, Dickerson JE, Pinney JW, Robertson DL, Rozanov MN, Katz KS, Maglott DR, Pruitt KD, Dieffenbach CW. Cataloguing the HIV type 1 human protein interaction network. AIDS Res. Hum. Retroviruses. 2008;24(12):1497–1502. doi: 10.1089/aid.2008.0113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shugars DC, Smith MS, Glueck DH, Nantermet PV, Seillier-Moiseiwitsch F, Swanstrom R. Analysis of human immunodeficiency virus type 1 nef gene sequences present in vivo. J. Virol. 1993;67(8):4639–4650. doi: 10.1128/jvi.67.8.4639-4650.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]