

Abstract
Mitochondrial disorders are a group of clinically heterogeneous diseases, commonly defined by lack of cellular energy due to genetic defects of oxidative phosphorylation (OXPHOS). Ocular involvement is a prominent clinical feature of mitochondrial disease. This can manifest as optic nerve dysfunction specifically involving retinal ganglion cells as typified by Leber hereditary optic neuropathy (LHON), or progressive external ophthalmoplegia (PEO) and ptosis involving the extraocular muscles which is commonly associated with either primary mitochondrial DNA (mtDNA) mutations or acquired mtDNA defects secondary to a nuclear genetic disorder of mtDNA maintenance. In this short review, we will outline the unique characteristics of mitochondrial genetic disease and its investigation with reference to the clinical features and molecular genetic abnormalities underlying mitochondrial ophthalmological disease.
Keywords: mtDNA, Mitochondrial disease, Progressive external ophthalmoplegia, Optic atrophy, LHON, Retinopathy, Extraocular muscle
1. Introduction
Mitochondrial diseases are a clinically multifarious group of genetic disorders that affect organs heavily dependent on aerobic metabolism with extensive phenotypic and disease burden variability (McFarland et al., 2010). Neuro-ophthalmic manifestations of mitochondrial disorders are common and include retinal, macular and optic nerve dysfunctions, external ophthalmoplegia with ptosis and retrochiasmal visual loss. Ocular features are rarely in isolation and may be associated with neurological and/or systemic symptoms. The differential diagnosis of ocular muscle dysmotility is broad and includes ocular myasthenia, ocular myositis, thyroid associated orbitopathy, congenital cranial dysinnervation disorders, oculopharyngeal muscular dystrophy and other neurodegenerative or dystrophic disorders (Schoser and Pongratz, 2006). This review article highlights the salient clinical and molecular features of ocular manifestations of mitochondrial diseases.
1.1. Basic mitochondrial genetics
Mitochondria are responsible for producing >90% of a cell’s ATP through the pathways of electron transfer and oxidative phosphorylation (OXPHOS) that comprise the respiratory chain. The only location of extrachromosomal DNA within the cell, mitochondria are under the dual genetic control of both nuclear DNA and the mitochondrial genome (mtDNA), a small (16.6 kb) multicopy, double-stranded circular DNA molecule which encodes 13 essential polypeptides of the respiratory chain and the necessary RNA machinery for their translation (Fig. 1). The remaining protein subunits that make up the respiratory chain complexes, together with many hundreds of other proteins found within the organelle – many of which are required for mtDNA maintenance, replication and the translation of mitochondrial proteins – are synthesised on cytoplasmic ribosomes and specifically targeted and sorted to their correct mitochondrial location.
Figure 1.
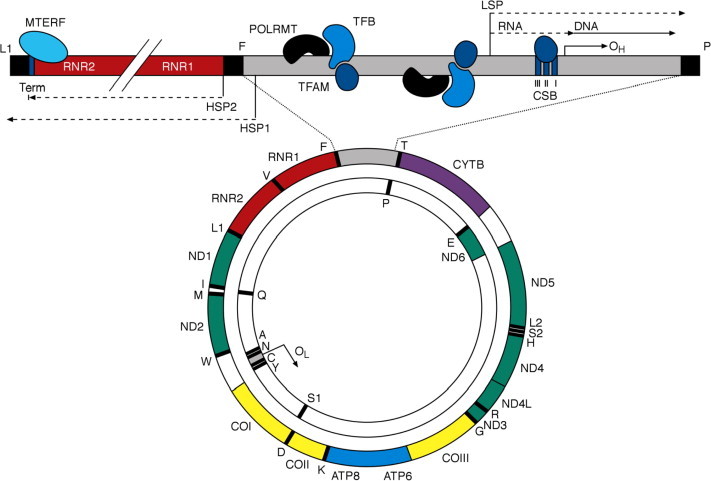
The human mitochondrial genome. Represented is a schematic diagram of the 16.6 kb circular, double-stranded human mitochondrial genome with an enhanced, linearised view of the D-loop and transcription termination regions. The outer circle represents the heavy (H) strand of the genome and the inner circle the light (L) strand. Human mtDNA encodes the two mt-rRNA genes (shown in red) RNR1 (12S rRNA) and RNR2 (16S rRNA), 22 mt-tRNAs (black bars) identified by their single letter abbreviation, and 13 essential respiratory chain polypeptides: seven subunits (ND1–ND6 and ND4L) of complex I (green), CYTB of complex III (purple), three catalytic subunits (COI–COIII) of complex IV (yellow) and ATP6 and ATP8 of complex V (blue). Major non-coding regions of the genome (grey) include the origin of L-strand replication (OL) and the 1.1 kb D-loop in which the origin of H-strand replication (OH) and regulatory elements and binding sequences for key factors involved in mtDNA transcription initiation and termination are located.
The mitochondrial genome possesses unique characteristics that distinguish it from Mendelian genetic rules (Taylor and Turnbull, 2005). It is strictly maternally-inherited, and is polyploid, i.e. multiple copies of mtDNA are present within each mitochondrion with several thousands present in individual cells. Normally, all of the mitochondrial genomes within an individual are identical, a situation termed homoplasmy. However, a mutation occurring in one copy of mtDNA can eventually lead to dual populations of wild type and mutated mtDNA coexisting within the same cell – heteroplasmy. At mitosis, both wild-type and mutated mtDNA are randomly segregated to each daughter cell, thereby affecting both disease expression and inheritance, further contributing to the wide range of clinical presentations seen in mtDNA disorders. The majority of deleterious mtDNA mutations are heteroplasmic, with clinical manifestations only becoming evident when the number of mutated mtDNA molecules exceeds a critical ‘threshold’. This threshold varies between organs depending upon their energy requirements, and reflects the incapacity of the remaining wild type mtDNA to compensate for the mutated mtDNA, leading to OXPHOS impairment and consequently cellular (routinely demonstrated by the histocytochemical assessment of cytochrome c oxidase (COX) activity) and organ dysfunction (see later). Although many important factors such as nuclear genetic background and mtDNA genotype can influence the phenotypic effect of particular mtDNA mutations, the relative abundance and specific tissue distribution of mutated mtDNAs are key determinants of the severity of the resulting clinical phenotype.
1.2. The laboratory diagnosis of mitochondrial disease
The laboratory diagnosis of mitochondrial disease is far from straightforward; mtDNA heteroplasmy, the lack of clear genotype–phenotype correlations in many patients, selective organ involvement and the complex interactions between the nuclear and mitochondrial genome are all contributing factors. The majority of mitochondrial diagnostic reference laboratories will often adopt a multidisciplinary approach, piecing together information from clinical, histochemical and respiratory chain enzyme testing to inform molecular genetic screening as guided by rational diagnostic algorithms (McFarland et al., 2010). Some patients with mitochondrial disease and ocular involvement (e.g. Leber hereditary optic neuropathy (LHON)) can be easily diagnosed by a simple DNA test using an EDTA-blood sample. For others, however, as in the case of Chronic Progressive External Ophthalmoplegia (CPEO or PEO), the causative mtDNA mutation – or in the case of Mendelian PEO, acquired mtDNA abnormalities (e.g. multiple mtDNA deletions) which are secondary to the primary nuclear genetic defect – may require the investigation of muscle biopsy tissue. Many patients with mtDNA disease show histological and histochemical changes indicative of OXPHOS dysfunction. Classically, these are ragged-red fibres – so called because of the appearance of fibres on Gomori trichrome staining showing subsarcolemmal accumulation of abnormal mitochondria – and COX-deficient fibres which signal mtDNA involvement (Fig. 2). mtDNA rearrangements (deletions and duplications) are assessed by Southern blotting or PCR-based strategies, whilst sequencing the entire mitochondrial genome can be undertaken relatively easily to identify rare or novel mtDNA mutations when common mtDNA mutations have been excluded (McFarland et al., 2010). The phenotypic and genotypic variability associated with primary mtDNA mutations (both point mutations and single, large-scale mtDNA deletion) and ocular phenotypes is illustrated in Fig. 3.
Figure 2.
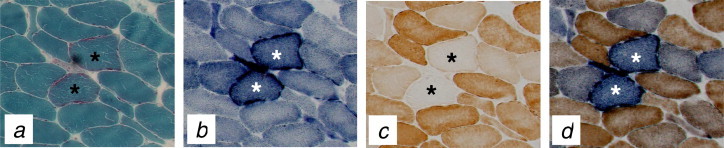
Skeletal muscle histopathological abnormalities associated with pathogenic mtDNA mutations. The histological and histochemical assessments of skeletal muscle may provide useful clues in determining mtDNA involvement in disease pathology. Shown is a sequential series of transverse-orientated muscle biopsy sections from a patient with CPEO due to a mitochondria tRNA gene point mutation, reacted for the following stains or enzyme activities: (a) modified Gomori trichome stain highlighting classical ragged-red muscle fibres, in which abnormal mitochondrial accumulate around the subsarcolemmal region; (b) SDH, which further accentuates the subsarcolemmal accumulation of mitochondrial enzyme activity; (c) COX histochemistry showing pale-reacting COX-deficient fibres within a population of normal fibres, a typical “mosaic” distribution; (d) sequential COX/SDH histochemistry highlighting individual COX-deficient fibres which retain SDH activity (blue) against a background of fibres with normal COX activity (brown).
Figure 3.
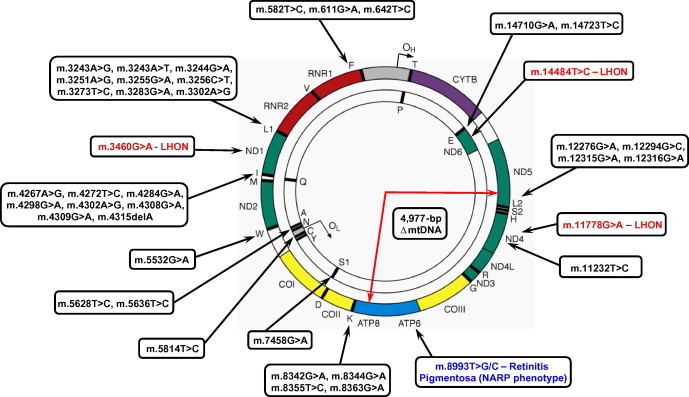
Pathogenic mtDNA mutations associated with ocular phenotypes. The circular, double-stranded human mitochondrial genome is depicted with sites of common mtDNA mutations highlighted associated with LHON (the three primary mtDNA mutations in the MTND1, MTND4 and MTND6 genes are shown in red), retinitis pigmentosa as a feature of the NARP phenotype (m.8993T>G/C mutations shown in blue) or PEO and ophthalmoparesis (shown in black). These include single, large-scale mtDNA deletions (as highlighted by the site of the 4977-bp “common” deletion), rare mutations in mitochondrial RNA genes (e.g. m.11232T>C in MTND4) or mitochondrial tRNA point mutations. Whilst these appear to be distributed amongst the tRNA genes, mutations in genes encoding mt-tRNALeu(UUR), mt-tRNALeu(CUN), mt-tRNAIle and mt-tRNALys are most prevalent.
2. Ocular manifestations of mitochondrial disease
2.1. Chronic Progressive External Ophthalmoplegia (PEO) with ptosis
CPEO, first described in the late 19th century (Von Graefe, 1867) is characterised by bilateral, slowly progressive loss of extraocular muscle mobility, orbicularis oculi weakness and ptosis. Due to the insidious and often symmetrical nature of onset, patients with PEO seldom complain of diplopia and are often unaware of ocular restriction. In the early stages of PEO, restriction is not always equal; however, as the disorder progresses and the muscles become fibrotic, near complete ophthalmoplegia may ensue with relative sparing of downgaze (Fig. 4) (Schoser and Pongratz, 2006). Pupillary response and accommodation remain intact, and unlike supranuclear gaze palsies, PEO remains unchanged on oculocephalic maneuver (doll’s eyes) and caloric testing.
Figure 4.
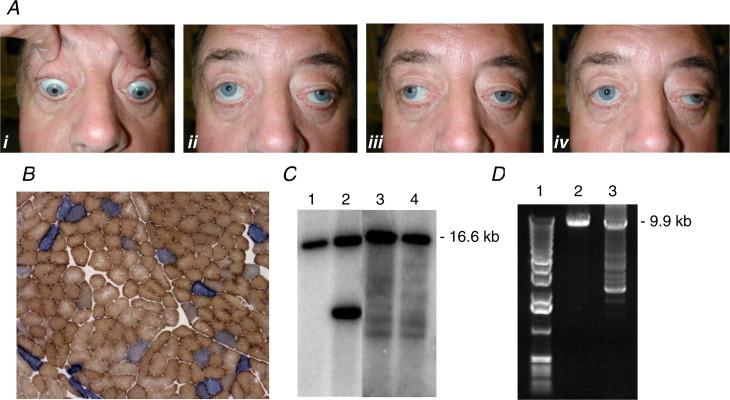
The clinical, histochemical and mtDNA abnormalities associated with mitochondrial PEO. (A) Typical clinical features of a patient with mitochondrial CPEO, extraocular motility in cardinal directions of gaze is severely reduced with relative preservation of downgaze as the patient is asked to look down (i), up (ii), right (iii) and left (iv). (B) Sequential COX-SDH histochemistry showing the presence of scattered COX-deficient fibres in a quadriceps muscle biopsy. (C) Southern blotting of muscle DNA indicates the patterns observed in a control (lane 1), a patient with a heteroplasmic, single large-scale mtDNA deletion (lane 2) and multiple mtDNA deletions (lanes 3 and 4). (D) Long-range PCR is also widely used to identify multiple mtDNA deletions (lane 3) which are amplified in addition to full-length, wild type mtDNA. Also shown is a control (lane 2) and DNA size markers (lane 1).
When PEO and ptosis are associated with other neurological or multisystem abnormalities, these may be termed ophthalmoplegia-plus (PEO+) syndromes. A myriad of systemic features have been reported and include limb, facial and bulbar muscle weakness, short stature, diabetes mellitus, deafness, cardiac conduction defects, central and peripheral respiratory dysfunction and endocrine and sex organ abnormalities (Biousse and Newman, 2001). Kearns–Sayre Syndrome (KSS) is the most common form of a PEO+ syndrome (Kearns and Sayre, 1958). Its cardinal features include early age of PEO onset (before the age of 20 years), degenerative pigmented retinopathy and cardiac conduction block with the risk of potentially life threatening arrhythmias, cerebellar dysfunction or elevated cerebrospinal fluid protein (>100 mg/dl) (Rowland et al., 1983). With disease progression, a proximal myopathy develops in addition to deafness, bulbar dysfunction, areflexia and stroke-like episodes. Although reports of sibling pairs exist (Rowland et al., 1988), most cases of KSS are sporadic.
2.1.1. Primary mtDNA mutations causing CPEO
Late onset-PEO is the most common form of extraocular mitochondrial myopathy and often arises spontaneously, characterised by heteroplasmic, single large-scale mtDNA rearrangements (predominantly mtDNA deletions but occasionally associated duplications) and COX-deficient fibres in skeletal muscle tissue (Fig. 4). The pathogenic mtDNA deletion accounts for 20–90% of the total skeletal muscle mtDNA (Biousse and Newman, 2001), although in some patients low levels of mtDNA deletion have been reported (Schaefer et al., 2005). Variability in the proportions of deleted mtDNA present in different tissues may in part account for some of the clinical aberration, but no clear correlation between the size and site of the mtDNA rearrangement and biochemical and phenotypic expression exists. Approximately 20% of patients with PEO demonstrate a Mendelian pattern of inheritance, implicating nuclear DNA abnormalities. Mendelian PEO can be dominantly- or recessively-inherited, with patients exhibiting COX-deficient fibres in muscle and a “laddering” of smaller-sized, variable mtDNA deletions on either Southern blot or long-range PCR analysis. In the absence of mtDNA rearrangements (single or multiple mtDNA deletions), mtDNA point mutations should also be investigated. The m.3243A>G MTTL1 gene mutation, typically associated with the MELAS phenotype, is a recognised cause of mitochondrial PEO (Kuncl and Hoffman, 1999; Moraes et al., 1993) whilst discreet, rarer mtDNA mutations can be found in mitochondrial-encoded mRNA genes but more typically tRNA genes (Fig. 4). In many cases, akin to single, large-scale mtDNA rearrangements, the causative tRNA mutation arises sporadically, is not transmitted and is often restricted to skeletal muscle (Fu et al., 1996; Chinnery et al., 1997; Taylor et al., 1998, 2002; Karadimas et al., 2002; Berardo et al., 2010).
Occasionally, the pathogenic tRNA mutation may be present at very low levels in a diagnostic skeletal muscle biopsy (Alston et al., 2010). The affected muscles in patients with PEO are the extraocular muscles, which exhibit major biological differences to other skeletal muscles. Recent studies have shown that extraocular muscles readily accumulate mtDNA mutations with age, indicative of an accelerated ageing process (Yu-Wai-Man et al., 2010a) whilst in CPEO patients with proven mtDNA mutations, higher proportions of COX-deficient fibres are present in extraocular muscle samples which have been removed during corrective strabismus and ptosis surgery compared to quadriceps muscle taken for diagnostic purposes (Greaves et al., 2010) (Fig. 5).
Figure 5.
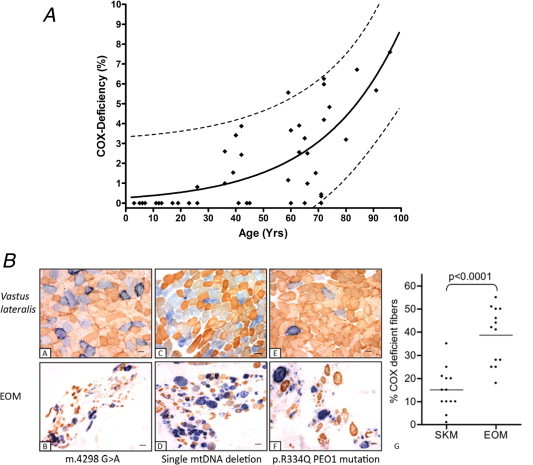
Vulnerability of extraocular muscles to mtDNA mutation accumulation. (A) The data shown in this panel highlight the distribution of COX-deficient extraocular muscle fibres with age in a series of 46 normal human subjects (age range 3–96 years). The simulated curve models an exponential increase in COX deficiency with normal aging (r2 = 0.5753) whilst the dotted lines accentuate the predicted upper and lower 95% confidence range within this group. Data are reproduced from Yu-Wai-Man et al. (2010a) with permission. (B) Enhanced mitochondrial histochemical abnormalities are evident in extraocular muscle compared to skeletal muscle (quadriceps) biopsies in patients with mitochondrial CPEO. Illustrated is sequential COX/SDH histochemistry on paired muscle samples from patients with a mitochondrial tRNA point mutation (m.4298G>A) (a and b), a single, large-scale 5.0 kb mtDNA deletion (c and d) and multiple mtDNA deletions secondary to a p.R334Q PEO1 (Twinkle) gene mutation (e and f). A comparison of the percentage of COX-deficient fibres between skeletal muscle and extraocular muscles in a panel of 13 mitochondrial CPEO patients reveals a significantly higher percentage of COX-deficient fibres in extraocular muscle compared to skeletal muscle (p < 0.0001) (g). Data are reproduced from Greaves et al. (2010) with permission.
2.1.2. Autosomal dominant (ad)PEO
The majority of families with autosomal dominant PEO (adPEO) harbour mutations in one of the following genes: POLG encoding the catalytic subunit of mitochondrial pol γ (Van Goethem et al., 2001), PEO1 encoding mitochondrial helicase Twinkle (Spelbrink et al., 2001), SLC25A4 encoding adenine nucleotide translocator 1 (Kaukonen et al., 2000) and RRM2B encoding a subunit of the p53-inducible ribonucleotide reductase protein required for maintaining balanced mitochondrial dNTP pools (Tyynismaa et al., 2009). Other mutated genes include POLG2, encoding the accessory beta-subunit of pol γ (Longley et al., 2006) and OPA1 which encodes a dynamin-related GTPase required for mitochondrial dynamics and also mtDNA maintenance (Hudson et al., 2008; Amati-Bonneau et al., 2008). Mitochondrial Twinkle helicase mutations, which also underlie autosomal recessive infantile onset spinocerebellar ataxia (IOSCA) and mtDNA depletion syndromes, are a common cause of late-onset adPEO, characterised by the accumulation of variable, large-scale mtDNA deletions in muscle over time. PEO1 gene mutations are associated with a predominantly myopathic phenotype with mild multisystem involvement (Fratter et al., 2010), somewhat in contrast to POLG mutations which are the most common cause of PEO and multiple mtDNA deletions but are associated with a broad clinical spectrum (Horvath et al., 2006).
2.1.3. Autosomal recessive (ar)PEO
Autosomal recessive PEO cases have been reported in pedigrees harbouring mtDNA rearrangements in skeletal muscle, and are particularly associated POLG mutations (reviewed in Copeland, 2008). Recessive mutations in the POLG gene also cause the sensory ataxia neuropathy, dysarthria and ophthalmoparesis (SANDO) phenotype (Fadic et al., 1997) and mitochondrial recessive ataxia syndrome (MIRAS) (Winterthun et al., 2005) in addition to arPEO.
2.2. Other mitochondrial myopathies associated with CPEO
2.2.1. Mitochondrial encephalomyopathy with lactic acidosis and stroke-like episodes (MELAS)
Rarely, CPEO may occur in patients with MELAS (mitochondrial encephalopathy, lactic acidosis, stroke-like episodes). As mentioned briefly, there is a subset of patients with PEO who harbour the m.3243A>G point mutation typically associated with MELAS but who manifest no other clinical features of this condition (Kuncl and Hoffman, 1999). MELAS (Pavlakis et al., 1984), which is classically associated with headaches, vomiting, stroke-like episodes, focal neurological deficits, systemic lactic acidosis and evidence of mitochondrial dysfunction on muscle biopsy is usually associated with significant morbidity. MELAS may also be associated with other systemic features including short stature, deafness, diabetes and seizures. Additional ocular manifestations may include pigmentary retinopathy and maculopathy. MELAS symptoms commonly begin before the age of 15 years but reports of disease manifestations as late as the fifth decade have been documented. Brain imaging often shows basal ganglia calcification with white matter lesions apparent in cases of repeated episodes of headaches, vomiting, seizures or focal neurological symptoms. Muscle biopsy can show classical mitochondrial abnormalities (ragged-red fibres, COX-deficient fibres) although that may be normal. Molecular confirmation of the m.3243A>G mutation can be made through mtDNA analysis of blood, muscle or urine with urinary m.3243A>G heteroplasmy levels shown to be a good clinical correlate of central disease manifestations (Whittaker et al., 2009).
2.2.2. Myoclonic epilepsy, myopathy with ragged-red fibres (MERRF)
CPEO has also been described in myoclonic epilepsy, myopathy with ragged-red fibres (MERRF), a mitochondrial cytopathy characterised by myoclonic epilepsy, myopathy and a progressive cerebellar syndrome (Fukuhara et al., 1980). Other clinical features may include deafness, optic atrophy, Wolf Parkinson White Syndrome, peripheral neuropathy and dementia. Creatine kinase and serum lactate may be moderately elevated, whilst brain imaging may simply show mild diffuse atrophy only. Molecular confirmation can be made by screening the m.8344A>G mutation in blood.
2.2.3. Mitochondrial neurogastrointestinal encephalomyopathy (MNGIE)
Mutations in the nuclear-encoded TYMP gene encoding thymidine phosphorylase may result in mitochondrial neurogastrointestinal encephalomyopathy (MNGIE), a recessively-inherited disorder that is associated with PEO and ptosis. It is defined clinically by cachexia, severe gastrointestinal dysmotility, sensorimotor neuropathy, skeletal myopathy and leukoencephalopathy which may be associated with cognitive decline (Nishino et al., 2000). MNGIE is often associated with high morbidity and mortality with death usually ensuing in the 3rd or 4th decades of life. Deafness is common but ataxia and seizures, which are common clinical associates of other mitochondrial myopathies, are typically absent. Ragged-red and COX-deficient fibres are reported in muscle biopsies, which also reveal multiple mtDNA deletions.
2.3. Neurogenic weakness, ataxia and retinitis pigmentosa (NARP)
NARP is a clinical syndrome characterised by developmental delay, pigmentary retinopathy, dementia, seizures, sensory neuropathy, proximal myopathy, ataxia, migraine and deafness. Serum pyruvate and lactate may be normal, as are muscle biopsy findings. Brain imaging, if abnormal, may only show non-specific cerebral or cerebellar atrophy in patients with high disease burden. The majority of cases exhibit mutation at m.8993 in the MTATP6 gene, easily detectable in the blood DNA given there is an excellent correlation between mutation load and clinical severity.
2.4. Pigmentary retinopathy
Pigmentary retinopathy is not infrequent in mitochondrial disease with classical ‘salt and pepper’ appearances on fundoscopic evaluation (Isashiki et al., 1998; Kerrison et al., 2000; Ortiz et al., 1993). Frequently this is only present as fine pigment dusting in the periphery, necessitating formal indirect ophthalmoscopy for accurate detection and evaluation. The early features of mitochondrial-related pigmentary retinopathy are somewhat different from classical retinitis pigmentosa which is associated with a bony spicule pattern of pigmentation; however, with disease and patient age progression, this latter pattern of retinopathy may emerge (Kerrison et al., 2000). Vascular attenuation is common with mild visual loss occurring in up to half of those affected. Although pigmentary retinopathy is recognised as a cardinal feature of KSS, it can also occur in MELAS, MIDD, OPA1 mutation (Hudson et al., 2008) and NARP (Kerrison et al., 2000).
Bilateral macular dystrophy has been recognised in MIDD (maternally-inherited diabetes and deafness) and MELAS associated with the m.3243A>G mutation (Ambonville et al., 2008; Adjadj et al., 2008; Latkany et al., 1999; Latvala et al., 2002; Massin et al., 1995, 1999; Michaelides et al., 2008; Smith et al., 1999) (Fig. 6A). Fundus autofluorescence characteristics are distinctive from most other dystrophies, with a notable discrepancy between the size of abnormal autofluorescence and that which would be suggested from fundoscopic appearance. The most common pattern of m.3243A>G mutation-associated macular dystrophy is circumferentially-distributed perifoveal atrophy with central fovea sparing. Pale deposits and granularity at the level of the retinal pigment epithelium and subretinal pigment clumping is evident adjacent to the areas of atrophy. A subtype of maculopathy consistent with a pattern dystrophy is described with no perifoveal atrophy and more diffuse speckled appearance of the macula (Michaelides et al., 2008; Rath et al., 2008).
Figure 6.
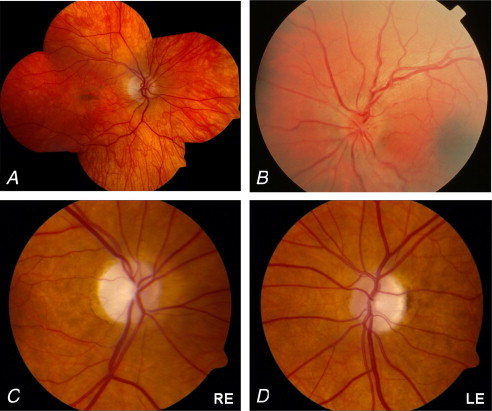
Ocular examination in mitochondrial disorders. (A) Macular pigmented retinopathy associated with the m.3243A>G mutation. (B) The acute fundal appearance in Leber hereditary optic neuropathy is characterised by parapapillary nerve fibre layer swelling, tortuosity of the retinal vessels and disc hyperaemia (Yu-Wai-Man et al., 2009). (C and D) Optic disc appearance in DOA highlighting total disc pallor and prominent temporal wedge of pallor typically observed in patients with OPA1 mutation (Yu-Wai-Man et al., 2010b). RE, right eye; LE, left eye.
2.5. Leber hereditary optic neuropathy (LHON)
Leber hereditary optic neuropathy (LHON) is the commonest primary mitochondrial disease associated with bilateral optic neuropathies (Newman, 2005; Riordan-Eva et al., 1995; Yu-Wai-Man et al., 2009). It is recognised as the most frequent cause of isolated blindness in young men with estimated point prevalence of 1 in 31,000 (Man et al., 2003). Maternally-transmitted LHON pedigrees show incomplete penetrance with a male preponderance for visual loss (3:1 male to female ratio). The reason for this remains unknown but an X-linked nuclear modifier has been proposed. Age of onset is variable ranging from the first to eighth decade, but most cases present in their late teens and twenties.
Visual loss associated with LHON is characteristically sub acute, central and painless. In most cases visual loss occurs in one eye, followed sequentially in the other eye with a median interim period of 8 weeks but this can range from days to years (Riordan-Eva et al., 1995). Typically visual loss occurs in isolation – pedigrees are otherwise clinically asymptomatic – and it may be severe, deteriorating in most patients to acuities of 20/200 (criteria to register blind) or worse. Visual field defects consistent with central or cecocentral defects are typical. Colour vision dysfunction may be an early and severe feature of LHON-associated visual loss. Pupillary function is relatively preserved. Fundoscopic changes are characterised by peripapillary telangiectatic microangiopathy or circumpapillary nerve fibre layer swelling (pseudooedema) in the early stages (Fig. 6B), later followed by non-specific optic atrophy with nerve fibre layer drop out, especially in the papillomacular bundle (Savini et al., 2005).
Although LHON is typically associated with isolated visual loss, neurological and multisystem organ involvement has been recognised in association with a multiple-sclerosis-like illness (usually in women), dystonia, myelopathy, neuropathy, severe encephalomyelopathy and cerebellar ataxia (Bhatti and Newman, 1999; Horvath et al., 2000). One of three mtDNA point mutations account for up to 95% of known cases of LHON: m.3460G>A, m.11778G>A, m.14484T>C all of which occur in mitochondrial-encoded structural subunits of complex I. Younger age-at-onset (less than 15 years) and mutation type appear to dictate visual outcome; patients with the m.14484T>C MTND6 mutation have a better visual prognosis with 60% attaining some visual improvement compared to only 5% of those harbouring the m.11778G>A MTND4 gene mutation.
2.6. Dominant optic atrophy (DOA)
Autosomal dominant optic atrophy, with an overall prevalence of 1:35,000 (Yu-Wai-Man et al., 2010b) is the most common autosomal hereditary optic neuropathy and unlike LHON exhibits no gender bias. DOA is a slowly progressive, painless, bilateral, symmetrical visual loss. Onset is insidious and usually within the first two decades of life. Visual loss is detected usually between the ages of 4 and 6 years, with up to 84% reporting visual disability by 11 years. There is a wide spectrum of visual acuity ranging from 20/20 to light perception; however, the majority of patients retain vision of 20/200 or better. Visual acuity decline may be progressive in up to half of the patients. Defects in colour vision result from a generalised dyschromatopsia. The most frequent visual field abnormalities in DOA are central, cecocentral, and paracentral scotomas consistent with early involvement of the papillomacular bundle, and may show a predilection for the superotemporal quadrant (Fig. 6C and D). Optic disc atrophy may be subtle, diffuse or present as a ‘temporal wedge’ defect (Yu-Wai-Man et al., 2009, 2010b). Mutations in the OPA1 gene – which is located on chromosome 3q28–29 – account for 60–70% of all cases of DOA. To date, over 200 pathogenic mutations in the OPA1 gene have been identified including missense, nonsense, deletion/insertion, and splicing mutations. In addition to OPA1, other loci have been identified. Optic atrophy may be a feature of other mitochondrial syndromes and has been reported in both MERRF and MELAS.
2.7. Retrochiasmal visual loss
Mitochondrial disease-related retrochiasmal visual loss is most commonly associated with the MELAS syndrome. Stroke-like episodes in MELAS have a propensity to affect primarily the parietal and occipital lobes, resulting in damage to the visual pathways and giving rise to homonymous hemianopia or cortical blindness. Fundoscopic examination and pupillary responses are invariably normal.
3. Concluding remarks
The clinical spectrum of mitochondrial disorders is vast and new patterns of disease are increasingly recognised. Diagnostic accuracy has significantly improved over the last decade with our ability to detect many of the common mtDNA pathogenic mutations non-invasively. However, muscle biopsy is often required to detect primary and secondary mtDNA abnormalities and as such it remains the cornerstone of laboratory diagnostic algorithms, particularly in patients with PEO. We suggest that the diagnosis of mitochondrial disease should be considered in all cases of ocular muscle dysmotility and in patients with visual failure of acute or insidious onset.
Disclosure statement
The authors have no financial interests to disclose. Each author has equally contributed in the writing and preparation of this manuscript.
Acknowledgements
We thank Dr. Patrick Yu-Wai-Man and Dr. Andrew Schaefer for their help in preparing clinical photographs. G.S.G. is funded by the UK NIHR Biomedical Research Centre for Ageing and Age-related disease award to the Newcastle upon Tyne Foundation Hospitals NHS Trust. R.W.T. is supported by the Wellcome Trust and the MRC Centre for Translational Research in Neuromuscular Disease Mitochondrial Disease Patient Cohort (UK). The mitochondrial diagnostic laboratory in Newcastle is funded by the UK NHS Specialised Services to provide the “Rare Mitochondrial Disease of Adults and Children” service (http://www.mitochondrialncg.nhs.uk/).
References
- Adjadj E., Mansouri K., Borruat F.X. Mitochondrial DNA (mtDNA) A3243G mutation associated with an annular perimacular retinal atrophy. Klin. Monatsbl. Augenheilkd. 2008;225:462–464. doi: 10.1055/s-2008-1027257. [DOI] [PubMed] [Google Scholar]
- Alston C.L., Lowe J., Turnbull D.M. A novel mitochondrial tRNAGlu (MTTE) gene mutation causing chronic progressive external ophthalmoplegia at low levels of heteroplasmy in muscle. J. Neurol. Sci. 2010;298:140–144. doi: 10.1016/j.jns.2010.08.014. [DOI] [PubMed] [Google Scholar]
- Amati-Bonneau P., Valentino M.L., Reynier P. OPA1 mutations induce mitochondrial DNA instability and optic atrophy ‘plus’ phenotypes. Brain. 2008;131:338–351. doi: 10.1093/brain/awm298. [DOI] [PubMed] [Google Scholar]
- Ambonville C., Meas T., Lecleire-Collet A. Macular pattern dystrophy in MIDD: long term follow-up. Diabetes Metab. 2008;34:389–391. doi: 10.1016/j.diabet.2008.05.002. [DOI] [PubMed] [Google Scholar]
- Berardo A., Coku J., Kurt B. A novel mutation in the tRNAIle gene (MTTI) affecting the variable loop in a patient with chronic progressive external ophthalmoplegia (CPEO) Neuromuscul. Disord. 2010;20:204–206. doi: 10.1016/j.nmd.2010.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhatti M.T., Newman N.J. A multiple-sclerosis-like illness in a man harbouring the mtDNA 14484 mutation. J. Neuroophthalmol. 1999;19:28–33. [PubMed] [Google Scholar]
- Biousse V., Newman N.J. Neuro-ophthalmology of mitochondrial diseases. Semin. Neurol. 2001;21:275–291. doi: 10.1055/s-2001-17945. [DOI] [PubMed] [Google Scholar]
- Chinnery P.F., Johnson M.J., Taylor R.W. A novel mitochondrial tRNA isoleucine mutation causing chronic progressive external ophthalmoplegia. Neurology. 1997;49:1166–1168. doi: 10.1212/wnl.49.4.1166. [DOI] [PubMed] [Google Scholar]
- Copeland W.C. Inherited mitochondrial diseases of DNA replication. Annu. Rev. Med. 2008;59:131–146. doi: 10.1146/annurev.med.59.053006.104646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fadic R., Russell J.A., Vedanarayanan V.V. Sensory ataxic neuropathy as the presenting feature of a novel mitochondrial disease. Neurology. 1997;49:239–245. doi: 10.1212/wnl.49.1.239. [DOI] [PubMed] [Google Scholar]
- Fratter C., Gorman G.S., Stewart J.D. The clinical, histochemical, and molecular spectrum of PEO1 (Twinkle)-linked adPEO. Neurology. 2010;74:1619–1626. doi: 10.1212/WNL.0b013e3181df099f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu K., Hartlen R., Johns T. A novel heteroplasmic tRNALeu(CUN) mtDNA point mutation in a sporadic patient with mitochondrial encephalomyopathy segregates rapidly in skeletal muscle and suggests an approach to therapy. Hum. Mol. Genet. 1996;5:1835–1840. doi: 10.1093/hmg/5.11.1835. [DOI] [PubMed] [Google Scholar]
- Fukuhara N., Tokiguchi S., Shirakawa K. Myoclonic epilepsy associated with ragged-red fibres (mitochondrial abnormalities) disease-entity or a syndrome? J. Neurol. Sci. 1980;47:117–133. doi: 10.1016/0022-510x(80)90031-3. [DOI] [PubMed] [Google Scholar]
- Greaves L.C., Yu-Wai-Man P., Blakely E.L. Mitochondrial DNA defects and selective extraocular muscle involvement in CPEO. Invest. Ophthalmol. Vis. Sci. 2010;51:3340–3346. doi: 10.1167/iovs.09-4659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horvath R., Abicht A., Shoubridge E.A. Leber’s hereditary optic neuropathy presenting as multiple-sclerosis-like disease of the CNS. J. Neurol. 2000;247:65–67. doi: 10.1007/s004150050015. [DOI] [PubMed] [Google Scholar]
- Horvath R., Hudson G., Ferrari G. Phenotypic spectrum associated with mutations of the mitochondrial polymerase γ gene. Brain. 2006;129:1674–1684. doi: 10.1093/brain/awl088. [DOI] [PubMed] [Google Scholar]
- Hudson G., Amati-Bonneau P., Blakeley E.L. Mutation of OPA1 causes dominant optic atrophy with external ophthalmoplegia, ataxia, deafness and multiple mitochondrial DNA deletions: a novel disorder of mtDNA maintenance. Brain. 2008;131:329–337. doi: 10.1093/brain/awm272. [DOI] [PubMed] [Google Scholar]
- Isashiki Y., Nakagawa M., Ohba N. Retinal manifestations in mitochondrial diseases associated with mitochondrial DNA mutations. Acta Ophthalmol. Scand. 1998;76:6–13. doi: 10.1034/j.1600-0420.1998.760103.x. [DOI] [PubMed] [Google Scholar]
- Karadimas C.L., Salviati L., Sacconi S. Mitochondrial myopathy and ophthalmoplegia in a sporadic patient with the G12315A mutation in mitochondrial DNA. Neuromuscul. Disord. 2002;12:865–868. doi: 10.1016/s0960-8966(02)00072-x. [DOI] [PubMed] [Google Scholar]
- Kaukonen J., Juselius J.K., Tiranti V. Role of adenine nucleotide translocator 1 in mtDNA maintenance. Science. 2000;289:782–785. doi: 10.1126/science.289.5480.782. [DOI] [PubMed] [Google Scholar]
- Kearns T.P., Sayre G.P. Retinitis pigmentosa, external ophthalmoplegia, and complete heart block. Arch. Ophthalmol. 1958;60:280–289. [PubMed] [Google Scholar]
- Kerrison J.B., Biousse V., Newman N.J. Retinopathy of NARP syndrome. Arch. Ophthalmol. 2000;118:298–299. doi: 10.1001/archopht.118.2.298. [DOI] [PubMed] [Google Scholar]
- Kuncl R.W., Hoffman P.N. Myopathies and disorders of neuromuscular transmission. In: Miller N.R., Newman N.J., editors. Walsh and Hoyt’s Clinical Neuro-Ophthalmology. fifth ed. Williams & Wilkins; Baltimore: 1999. pp. 1351–1460. [Google Scholar]
- Latkany P., Ciulla T.A., Cacchillo P.F. Mitochondrial maculopathy: geographic atrophy of the macula in the MELAS associated A to G 3243 mitochondrial DNA point mutation. Am. J. Ophthalmol. 1999;128:112–114. doi: 10.1016/s0002-9394(99)00057-4. [DOI] [PubMed] [Google Scholar]
- Latvala T., Mustonen E., Uusitalo R. Pigmentary retinopathy in patients with the MELAS mutation 3243A → G in mitochondrial DNA. Graefes Arch. Clin. Exp. Ophthalmol. 2002;240:795–801. doi: 10.1007/s00417-002-0555-y. [DOI] [PubMed] [Google Scholar]
- Longley M.J., Clark S., Yu Wai Man C. Mutant POLG2 disrupts DNA polymerase gamma subunits and causes progressive external ophthalmoplegia. Am. J. Hum. Genet. 2006;78:1026–1034. doi: 10.1086/504303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Man P.Y., Griffiths P.G., Brown D.T. The epidemiology of Leber hereditary optic neuropathy in the North East of England. Am. J. Hum. Genet. 2003;72:333–339. doi: 10.1086/346066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Massin P., Guillausseau P.J., Vialettes B. Macular pattern dystrophy associated with a mutation of mitochondrial DNA. Am. J. Ophthalmol. 1995;120:247–248. doi: 10.1016/s0002-9394(14)72615-7. [DOI] [PubMed] [Google Scholar]
- Massin P., Virally-Monod M., Vialettes B. Prevalence of macular pattern dystrophy in maternally inherited diabetes and deafness: GEDIAM Group. Ophthalmology. 1999;106:1821–1827. doi: 10.1016/s0161-6420(99)90356-1. [DOI] [PubMed] [Google Scholar]
- McFarland R., Taylor R.W., Turnbull D.M. A clinical perspective on mitochondrial disease. Lancet Neurol. 2010;9:829–840. doi: 10.1016/S1474-4422(10)70116-2. [DOI] [PubMed] [Google Scholar]
- Moraes C.T., Ciacci F., Silvestri G. Atypical clinical presentations associated with the MELAS mutation at position 3243 of human mitochondrial DNA. Neuromuscul. Disord. 1993;3:43–50. doi: 10.1016/0960-8966(93)90040-q. [DOI] [PubMed] [Google Scholar]
- Michaelides M., Jenkins S.A., Bamiou D.-E. Macular dystrophy associated with the A3243G mitochondrial DNA mutation: distinct retinal and associated features, disease variability, and characterization of asymptomatic family members. Arch. Ophthalmol. 2008;126:320–328. doi: 10.1001/archopht.126.3.320. [DOI] [PubMed] [Google Scholar]
- Newman N.J. Hereditary optic neuropathies. In: Miller N.R., Newman N.J., Biousse V., Kerrison J.B., editors. Walsh and Hoyt’s Clinical Neuro-Opthalmology. sixth ed. Williams & Willkins; Baltimore: 2005. pp. 465–501. [Google Scholar]
- Nishino I., Spinazzola A., Papadimitriou A. Mitochondrial neurogastrointestinal encephalomyopathy: an autosomal recessive disorder due to thymidine phosphorylase mutations. Ann. Neurol. 2000;47:792–800. [PubMed] [Google Scholar]
- Pavlakis S.G., Phillips P.C., DiMauro S. Mitochondrial myopathy, encephalopathy, lactic acidosis, and strokelike episodes: a distinctive clinical syndrome. Ann. Neurol. 1984;16:481–488. doi: 10.1002/ana.410160409. [DOI] [PubMed] [Google Scholar]
- Ortiz R.G., Newman N.J., Shoffner J.M. Variable retinal and neurologic manifestations in patients harboring the mitochondrial DNA 8993 mutation. Arch. Ophthalmol. 1993;111:1525–1530. doi: 10.1001/archopht.1993.01090110091031. [DOI] [PubMed] [Google Scholar]
- Rath R.P., Jenkins S., Michaelides M. Characterisation of the macular dystrophy inpatients with the A3243G mitochondrial DNA point mutation with fundus autofluorescence. Br. J. Ophthalmol. 2008;92:623–629. doi: 10.1136/bjo.2007.131177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riordan-Eva P., Sanders M.D., Govan G.G. The clinical features of Leber’s hereditary optic neuropathy defined by the presence of a pathogenic mitochondrial DNA mutation. Brain. 1995;118:319–337. doi: 10.1093/brain/118.2.319. [DOI] [PubMed] [Google Scholar]
- Rowland L.P., Hayes A.P. Diverse clinical disorders associated with morphological abnormalities in mitochondria. In: Scarlato G., Cerri C., editors. Mitochondrial Pathology in Muscle Diseases. Piccin; Padua: 1983. pp. 141–158. [Google Scholar]
- Rowland L.P., Hausmanowa-Petrusewicz I., Barduska B. Kearns-Syndrome in twins: lethal dominant mutation or acquired disease? Neurology. 1988;38:1399–1402. doi: 10.1212/wnl.38.9.1399. [DOI] [PubMed] [Google Scholar]
- Savini G., Barboni P., Valentino M.L. Retinal nerve fibre layer evaluation by optical coherence tomography in unaffected carriers with Leber’s hereditary optic neuropathy mutations. Ophthalmology. 2005;112:127–131. doi: 10.1016/j.ophtha.2004.09.033. [DOI] [PubMed] [Google Scholar]
- Schaefer A.M., Blakely E.L., Griffiths P.G. Ophthalmoplegia due to mitochondrial DNA disease: the need for genetic diagnosis. Muscle Nerve. 2005;32:104–107. doi: 10.1002/mus.20319. [DOI] [PubMed] [Google Scholar]
- Schoser B., Pongratz D. Extraocular mitochondrial myopathies and their differential diagnoses. Strabismus. 2006;14:107–113. doi: 10.1080/09273970600701218. [DOI] [PubMed] [Google Scholar]
- Smith P.R., Bain S.C., Good P.A. Pigmentary retinal dystrophy and the syndrome of maternally inherited diabetes and deafness caused by the mitochondrial DNA 3243 tRNALeu A to G mutation. Ophthalmology. 1999;106:1101–1108. doi: 10.1016/S0161-6420(99)90244-0. [DOI] [PubMed] [Google Scholar]
- Spelbrink J.N., Li F.Y., Tiranti V. Human mitochondrial DNA deletions associated with mutations in the gene encoding Twinkle, a phage T7 gene 4-like protein localised in mitochondria. Nat. Genet. 2001;28:223–231. doi: 10.1038/90058. [DOI] [PubMed] [Google Scholar]
- Taylor R.W., Chinnery P.F., Bates M.J.D. A novel mitochondrial DNA point mutation in the tRNAIle gene: studies in a patient presenting with chronic progressive external ophthalmoplegia and multiple sclerosis. Biochem. Biophys. Res. Commun. 1998;243:47–51. doi: 10.1006/bbrc.1997.8055. [DOI] [PubMed] [Google Scholar]
- Taylor R.W., Schaefer A.M., McFarland R. A novel mitochondrial DNA tRNAIle (A4267G) mutation in a patient with mitochondrial myopathy. Neuromuscul. Disord. 2002;12:659–664. doi: 10.1016/s0960-8966(02)00026-3. [DOI] [PubMed] [Google Scholar]
- Taylor R.W., Turnbull D.M. Mitochondrial DNA mutations in human disease. Nat. Rev. Genet. 2005;6:389–402. doi: 10.1038/nrg1606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tyynismaa H., Ylikallio E., Patel M. A heterozygous truncating mutation in RRM2B causes autosomal-dominant progressive external ophthalmoplegia with multiple mtDNA deletions. Am. J. Hum. Genet. 2009;85:290–295. doi: 10.1016/j.ajhg.2009.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Von Graefe A. Peters; Berlin: 1867. Symptomenlehre der Augenmuskellähmungen. [Google Scholar]
- Van Goethem G., Dermaut B., Lofgren A. Mutation of POLG is associated with progressive external ophthalmoplegia characterized by mtDNA deletions. Nat. Genet. 2001;28:211–212. doi: 10.1038/90034. [DOI] [PubMed] [Google Scholar]
- Whittaker R., Blackwood J.K., Alston C.L. Urine heteroplasmy is the best predictor of clinical outcome in the m.3243A>G mtDNA mutation. Neurology. 2009;72:568–569. doi: 10.1212/01.wnl.0000342121.91336.4d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winterthun S., Ferrari G., He L. Autosomal recessive mitochondrial ataxic syndrome due to mitochondrial polymerase gamma mutations. Neurology. 2005;64:1204–1208. doi: 10.1212/01.WNL.0000156516.77696.5A. [DOI] [PubMed] [Google Scholar]
- Yu-Wai-Man P., Griffiths P.G., Hudson G. Inherited optic neuropathies. J. Med. Genet. 2009;46:145–158. doi: 10.1136/jmg.2007.054270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu-Wai-Man P., Lai-Cheong J., Borthwick G.M. Somatic mitochondrial DNA deletions accumulate to high levels in aging human extraocular muscles. Invest. Ophthalmol. Vis. Sci. 2010;51:3347–3353. doi: 10.1167/iovs.09-4660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu-Wai-Man P., Griffiths P.G., Burke A. The prevalence and natural history of dominant optic atrophy due to OPA1 mutations. Ophthalmology. 2010;117:1538–1546. doi: 10.1016/j.ophtha.2009.12.038. [DOI] [PMC free article] [PubMed] [Google Scholar]