

Abstract
Supramolecular nanoassemblies, which are capable of binding and delivering either lipophilic small molecules or hydrophilic molecules, are of great interest. Concurrently binding and delivering this combination of molecules is cumbersome, because of the opposing supramolecular host requirements. We describe the development of a versatile nanoassembly system that is capable of binding and delivering both, a protein and a lipophilic small molecule, simultaneously inside the cells.
Self-assembled nanostructures, with the capability of sequestering guest molecules, have garnered great interests for applications in drug delivery and biomedical diagnostics.1 These nanocarriers can be broadly classified into two categories depending on their cargo: carriers for lipophilic guest molecules2 and those for hydrophilic guests.3 With the surge in interest in areas such as nanotheranostics,4 which require the presence of a therapeutic and a diagnostic agent in a nanocarrier, there have been recent developments in nanoscale vehicles that can concurrently sequester and deliver two different molecules.4,5 One can envision reasonably satisfying this requirement for two lipophilic or two hydrophilic molecules through co-encapsulation strategies. However, the molecular design requirements become more complicated, when it is a combination of a water-soluble hydrophilic molecule and a water-insoluble lipophilic molecule. It is especially cumbersome when one of the cargos is a protein. While concurrent encapsulation involving macromolecules that lack well-defined secondary structures is feasible,6 the propensity of proteins to irreversibly denature under non-native conditions presents a significant challenge to develop such a strategy for protein-small molecule combinations. We outline a versatile strategy for such nanocarriers, especially for the combination of water-soluble globular proteins and lipophilic small molecules.
We envisaged the possibility of utilizing the hydrophobic interior of a polymer assembly to stably encapsulate lipophilic guest molecules, while utilizing the complementary electrostatic interaction between the surface of the polymer assembly with a peptide to bind proteins (see Figure 1 for a schematic illustration). We use β-galactosidase (β-gal, pI: 4.8, from E.coli) as the model protein for this study, because the activity of this enzyme can be studied conveniently both inside and outside the cells, a feature essential to test the versatility of our strategy. We use 1,1′-dioctadecyl-3,3,3′,3′-tetramethyl-indcarbocyanine perchlorate (DiI) dye as the model small molecule, since it is lipophilic and exhibits fluorescence characteristics (complementary to the fluorescein labeling of the protein) allowing for concurrent monitoring of the protein and the small molecule in solution and inside the cells. Finally, we use our recently developed self-crosslinked nanogels as the nanocarrier, because these tunable vehicles have shown the capabilities for stably encapsulating lipophilic dye molecules.7
Figure 1.
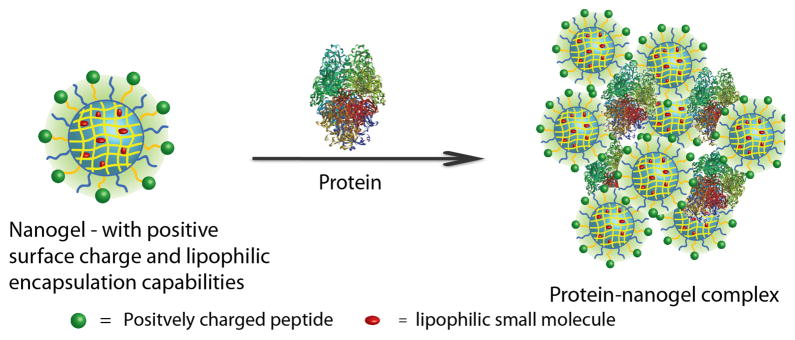
Nanogel-protein complexation by complementary electrostatic interactions
We prepared two nanogels, NG1 and NG2, with 6% and 14% crosslinking densities respectively. The nanogel was prepared from a random copolymer, obtained using a hydrophilic oligoethyleneglycol methacrylate and a lipophilic methacrylate monomer containing a pyridyldisulfide (PDS) moiety. The self-organized amphiphilic nanoassembly formed from this polymer was utilized to non-covalently sequester the lipophilic DiI molecule. The self-assembled structure was “locked in” with the lipophilic guest molecules by initiating a thiol-disulfide exchange reaction among the PDS units using dithiothreitol (DTT).7b The extent of crosslinking was simply controlled by the amount of DTT added to the reaction mixture.
Note that the surface of this nanogel contains charge-neutral functional groups. We were interested in introducing positively charged functional groups to bind to the negatively charged surface of β-gal. We targeted tri-arginine as the functional group for this purpose, since this tripeptide can perform the dual role of providing the positive charge and also exhibiting cell-penetrating peptide characteristics.8 Since only a small percentage of the PDS groups were used for the crosslinking reaction, we took advantage of the residual PDS moieties to conjugate the tri-arginine moiety using a Cys-Arg-Arg-Arg (CRRR) peptide. Here, the thiol moiety in cysteine reacts with the PDS units to provide the targeted nanogel. The structures of the nanogel’s polymer precursor and the CRRR peptide is shown in Scheme 1.
Scheme 1.
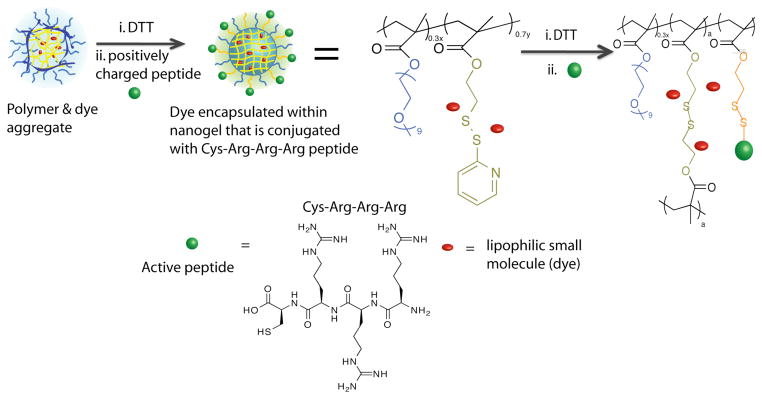
Structures of the nanogel’s polymer precursor and tri-arginine peptide.
In the nanogel formation process, NG1 showed only a small amount of production of pyridiothione (the byproduct of disulfide crosslinking) at 330–340 nm. On the other hand, NG2 showed a large amount of pyridiothione release indicating that NG2 is more densely crosslinked (see Supporting Information for details - Figure S3 and crosslink density estimation). Subsequent CRRR addition showed a further increase in the absorption peak of pyridiothione, indicative of peptide conjugation onto the surface of the nanogels. The same amount of CRRR addition showed a smaller increase for NG2 compared to NG1. This means that the degree of modification with CRRR peptide is much more for NG1 than for NG2. This is because the remaining PDS groups for surface modification after nanogel formation are less at high crosslinking densities. Thus, the two nanogels have different degrees of surface functionalization, which should have implications in protein binding and delivery. We also found that the absorption intensity of hydrophobic dye, DiI, did not change during the surface modification step. This suggests that peptide modification process doesn’t affect the encapsulation stability of the hydrophobic guest in the nanogel.
Next, we investigated the ability of these nanogels to bind β-gal. For this, we utilized β-gal functionalized with fluorescein (FITC- β-gal). The absorption and emission spectra of DiI and fluorescein suggest that these two dye molecules can be ideal fluorescence resonance energy transfer (FRET) partners. FRET is one of the most efficient techniques to evaluate the proximity of two different molecules. Using FRET, we found that the optimum ratios of NG1-CRRR and NG2-CRRR complexation with β-gal were 2:1 and 4:1, respectively. Notably, NG1-CRRR showed stable complexation at a lower concentration than NG2-CRRR, suggesting stronger binding by the former nanogel. We attribute this to the greater amount of the peptide on the NG1-CRRR surface.
We also evaluated the size change and charge alteration of the nanogel surface due to the complexation event using dynamic light scattering (DLS) and zeta potential measurements respectively. The hydrodynamic diameters of individual NG1-CRRR, NG2-CRRR, and β-gal were found to be approximately 20 nm, 12 nm, and 10 nm, respectively (Figure 2a and b). Once the nanogels and the protein were mixed, the size shifts to about 40 nm for the complex of NG1-CRRR/β-gal and 30 nm for NG2-CRRR/β-gal complex. Based on the sizes of the nanogels, β-gal, and the complexes, combined with their ratio, we estimate the average numbers of nanogel and β-gal per complex to be 7.6 and 3.8 respectively for NG1-CRRR and 13.6 and 3.4 for NG2-CRRR (see Supporting Information for details). Zeta-potential studies suggest that the apparent surface charge of the nanoparticles shift towards the negative values, from +25 mV to −5 mV for NG1-CRRR and from 0 mV to −20 mV for NG2-CRRR after β-gal incorporation (Figure 2c and d). Evidence of nanogel-protein complexation is also supported by gel electrophoresis studies (Figure S5). While, NG2-CRRR showed a large amount of unbound β-gal, NG1-CRRR showed a negligible amount of free β-gal. Put together, these results suggest that while both nanogels bind β-gal, the more positively charged NG1-CRRR exhibits stronger binding to the protein.
Figure 2.

DLS and ζ-potential graphs of: (a) β-gal, NG1-CRRR and β-gal:NG1-CRRR complex (1:2) and (b) β-gal, NG2-CRRR and β-gal:NG2-CRRR-β-gal complex (1:4).
The key motivation in developing this system is to achieve efficient intracellular delivery of the hydrophilic protein along with the lipophilic small molecules. It is important to maintain stability and activity of the delivered protein throughout the process.8 To this end, we tested the activity of the protein when complexed to the nanogel in solution before performing any intracellular release studies. An enzyme activity assay was done for β-gal complexed to NG1-CRRR and NG2-CRRR after one hour incubation with the nanogels. We observed that the protein retains its activity even when bound to the surface of the nanogels (Figure S6). These results demonstrate the robustness of the complex in maintaining protein activity.
Next, we tested the intracellular delivery of DiI and β-gal. As the nanogels consist of biodegradable disulfide crosslinkers, the release of the complexed protein and encapsulated lipophilic molecule can be triggered upon exposure to glutathione (GSH), a biological reducing agent which is present at higher concentrations inside the cells (mM), compared to the extracellular concentration (μM). To compare the release profile of these dual delivery systems, NG1-CRRR and NG2-CRRR containing DiI in its interior and complexed with FITC labeled β-gal, were added to HeLa cells with serum and without serum. If there is cell internalization of both encapsulated DiI and FITC labeled protein, a yellow color would be observed due to the co-localization and overlay of both red and green fluorophores for DiI and FITC, respectively. If there is no internalization of the nanogel, we would not see any color. For this experiment, HeLa cells were treated with a 2:1 ratio of NG1-CRR/FITC-β-gal complexes and a 4:1 ratio of NG2-CRRR/FITC-β-gal complexes. Fluorescence distribution of DiI and FITC was observed over time by confocal fluorescence microscopy with simultaneous excitation of both dye molecules at 488 nm and 543 nm for FITC and DiI, respectively. As shown in Figures 3a and b, green and red emissions were observed when the cells were treated with the nanogel-protein complexes. On the other hand, cells incubated with FITC-β-gal alone displayed no fluorescence, suggesting that the protein is not capable of cell penetration by itself (Figure S7). The green fluorescence of the FITC (label for protein) and red fluorescence for DiI (encapsulated small molecule) appear uniformly distributed. While co-localization of DiI dye and β-gal in cells both with and without serum was observed for NG1-CRRR (Figure 3a and 3c), NG2-CRRR showed almost no efficiency of intracellular delivery of either cargos in the presence of serum (Figure 3b and 3d). The differences in delivery efficiency appear to be due to the stronger interaction of the protein with NG1-CRRR compared to NG2-CRRR, where serum proteins appear to compete for the interaction with the nanogels. Also, note the presence of red fluorescence with the yellow fluorescence in the case of the NG1-CRRR complex. This suggests that the nanogel and the protein are not only internalized, but also are also starting to spatially separate from each other, presumably due to GSH-based degradation of the disulfide bonds.
Figure 3.

Protein and hydrophobic dye delivery using NG1-CRRR (a and c) and NG2-CRRR (b and d) without serum, and with serum, respectively. Within each image set, top left is the FITC channel which shows green color (β-gal) and top right is the DiI channel which shows red color (hydrophobic dye). Bottom left is the DIC image and bottom right is an overlap of all three. Yellow color is overlay of green and red. Scale bar is 20 μm. (e) X-gal activity assay of delivered β-gal into cells.
Finally, we interrogated whether the protein activity is in tact after internalization into the cells. X-gal assays were performed on cells, which had been incubated with the nanogel/β-gal complex, to probe the intracellular activity of the delivered β-gal (Figure 3e). The observed blue color is due to the conversion of the 5-bromo-3-indoyl-β-D-galactopyranoside substrate to an intensely blue colored indigo derivative, by β-gal. From the optical microscope images, we see that the cells treated with protein conjugated NG1-CRRR show enhanced blue coloration, due to greater β-gal internalization compared to NG2-CRRR and the control cells. These studies suggest that the delivered β-gal is active inside the cells. The observation that protein bound to NG1-CRRR has better intracellular activity than NG2-CRRR is consistent with the internalization studies using confocal microscopy. Considering that the protein itself is not permeable through the cellular membrane and the hydrophobic dye is not soluble in water, the results above suggest that concurrent delivery of a protein and a hydrophobic dye simultaneously is indeed feasible with the approach outlined here.
In summary, we have developed a nanogel that is capable of encapsulating lipophilic small molecules within its crosslinked interiors and binding proteins on its surface through electrostatic interactions. We have shown that: (i) the nanogels can be functionalized with cell penetrating peptides efficiently; (ii) the nanogels bind oppositely charged proteins and that the charge density on the nanogel surface affects the efficiency of binding of the complementarily charged proteins; (iii) complexation of the protein with the nanogel does not alter the activity of the protein; (iv) the complex exhibits efficient uptake by cells, where both the lipophilic small molecule and the protein are concurrently taken up by the cells; (v) the enzyme retains its activity even upon cellular entry. The design strategy outlined here could have broad implications in a variety of areas including therapeutics, diagnostics, and a combination of the two by way of nanotheranostics.
Supplementary Material
Acknowledgments
This work was supported by DARPA and the NSF-MRSEC.
Footnotes
Author Contributions
The manuscript was written through contributions of all authors.
Supporting Information Available: Experimental details, fluorescence spectroscopy, Gel Electrophoresis and X-gal assay data. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.(a) Peer D, Karp JM, Hong S, Farokhzad OC, Margalit R, Langer R. Nat Nanotechnol. 2007;2:751–760. doi: 10.1038/nnano.2007.387. [DOI] [PubMed] [Google Scholar]; (b) Haag R. Angew Chem, Int Ed. 2004;43:278–282. doi: 10.1002/anie.200301694. [DOI] [PubMed] [Google Scholar]; (c) Lee CC, MacKay JA, Fréchet JMJ, Szoka FC. Nat Biotechnol. 2005;23:1517–1526. doi: 10.1038/nbt1171. [DOI] [PubMed] [Google Scholar]; (d) Jeong B, Bae YH, Lee DS, Kim SW. Nature. 1997;388:860–862. doi: 10.1038/42218. [DOI] [PubMed] [Google Scholar]
- 2.Allen TM, Cullis PR. Science. 2004;303:1818–1822. doi: 10.1126/science.1095833. [DOI] [PubMed] [Google Scholar]; (b) Davis ME, Chen Z, Shin DM. Nat Rev Drug Discovery. 2008;7:771–782. doi: 10.1038/nrd2614. [DOI] [PubMed] [Google Scholar]; (c) Kataoka K, Harada A, Nagasaki Y. Adv Drug Delivery Rev. 2001;47:113–131. doi: 10.1016/s0169-409x(00)00124-1. [DOI] [PubMed] [Google Scholar]; (d) Savić R, Luo L, Eisenberg A, Maysinger D. Science. 2003;300:615–618. doi: 10.1126/science.1078192. [DOI] [PubMed] [Google Scholar]; (d) Formina N, McFearin C, Sermsakdi M, Edigin O, Almutairi A. J Am Chem Soc. 2010;132:9540–9542. doi: 10.1021/ja102595j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.(a) Torchilin VP. Nat Rev Drug Discovery. 2005;4:145–160. doi: 10.1038/nrd1632. [DOI] [PubMed] [Google Scholar]; (b) Abu Lila AS, Ishida T, Kiwada H. Expert Opin Drug Delivery. 2009;6:1297–1309. doi: 10.1517/17425240903289928. [DOI] [PubMed] [Google Scholar]; (c) Eliaz RE, Nir S, Marty C, Szoka FC. Cancer Res. 2004;64:711–718. doi: 10.1158/0008-5472.can-03-0654. [DOI] [PubMed] [Google Scholar]; (d) Paramonov S, Bachelder E, Beaudette T, Standley S, Lee C, Dashe J, Fréchet J. Bioconjugate Chem. 2008;19:911–919. doi: 10.1021/bc7004472. [DOI] [PubMed] [Google Scholar]; (e) Cohen JL, Almutairi A, Cohen JA, Bernstein M, Brady SL, Schuster DP, Fréchet JMJ. Bioconjugate Chem. 2008;19:876–881. doi: 10.1021/bc700414j. [DOI] [PubMed] [Google Scholar]; (f) Murthy N, Xu M, Schuck S, Kunisawa J, Shastri N, Fréchet JMJ. Proc Nat Acad Sci USA. 2003;100:4995–5000. doi: 10.1073/pnas.0930644100. [DOI] [PMC free article] [PubMed] [Google Scholar]; (g) Mahmoud EA, Sankaranarayanan J, Morachis JM, Kim G, Almutairi A. Bioconjugate Chem. 2011;22:1416–1421. doi: 10.1021/bc200141h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.(a) Kelkar SS, Reineke TM. Bioconjug Chem. 2011;22:1879–1903. doi: 10.1021/bc200151q. [DOI] [PubMed] [Google Scholar]; (b) Acc Chem Res. 2011;44(10) (special issue on Theranostic Nanomedicine) [Google Scholar]
- 5.(a) Jain RK. Nature Med. 2001;7:987–989. doi: 10.1038/nm0901-987. [DOI] [PubMed] [Google Scholar]; (b) Sengupta S, Eavarone D, Capila I, Zhao G, Watson N, Kiziltepe T, Sasisekharan R. Nature. 2005;436:568–572. doi: 10.1038/nature03794. [DOI] [PubMed] [Google Scholar]
- 6.(a) Kim BS, Smith RC, Poon Z, Hammond PT. Langmuir. 2009;25:14086–14092. doi: 10.1021/la9017618. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Kim C, Shah BP, Subramaniam P, Lee KB. Mol Pharmaceutics. 2011;8:1955–1961. doi: 10.1021/mp100460h. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Wiradharma N, Tong YW, Yang YY. Biomaterials. 2009;30:3100–3109. doi: 10.1016/j.biomaterials.2009.03.006. [DOI] [PubMed] [Google Scholar]; (d) Sun T-M, Du J-Z, Yao Y-D, Mao C-Q, Dou S, Huang S-Y, Zhang P-Z, Leong KW, Song E-W, Wang J. ACS Nano, ASAP. doi: 10.1021/nn103349h. [DOI] [PubMed] [Google Scholar]
- 7.(a) Ryu J-H, Chacko RT, Jiwpanich S, Bickerton S, Babu RP, Thayumanavan S. J Am Chem Soc. 2010;132:17227–17235. doi: 10.1021/ja1069932. [DOI] [PubMed] [Google Scholar]; (b) Jiwpanich S, Ryu JH, Bickerton S, Thayumanavan S. J Am Chem Soc. 2010;132:10683–10685. doi: 10.1021/ja105059g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.(a) Rothbard JB, Jessop TC, Lewis RS, Murray BA, Wender PA. J Am Chem Soc. 2004;126:9506–9507. doi: 10.1021/ja0482536. [DOI] [PubMed] [Google Scholar]; (b) Nakase I, Takeuchi T, Tanaka G, Futaki S. Adv Drug Deliv Rev. 2008;60:598–607. doi: 10.1016/j.addr.2007.10.006. [DOI] [PubMed] [Google Scholar]
- 9.Savić R, Eisenberg A, Maysinger D. J Drug Target. 2006;14:343–355. doi: 10.1080/10611860600874538. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.