

Abstract
Trisomy 21 in humans causes cognitive impairment, craniofacial dysmorphology, and heart defects collectively referred to as Down syndrome. Yet, the pathophysiology of these phenotypes is not well understood. Craniofacial alterations may lead to complications in breathing, eating, and communication. Ts65Dn mice exhibit craniofacial alterations that model Down syndrome including a small mandible. We show that Ts65Dn embryos at 13.5 days gestation (E13.5) have a smaller mandibular precursor but a normal sized tongue as compared to euploid embryos, suggesting a relative instead of actual macroglossia originates during development. Neurological tissues were also altered in E13.5 trisomic embryos. Our array analysis found 155 differentially expressed non-trisomic genes in the trisomic E13.5 mandible, including 20 genes containing a homeobox DNA binding domain. Additionally, Sox9, important in skeletal formation and cell proliferation, was upregulated in Ts65Dn mandible precursors. Our results suggest trisomy causes altered expression of non-trisomic genes in development leading to structural changes associated with DS. Identification of genetic pathways disrupted by trisomy is an important step in proposing rational therapies at relevant time points to ameliorate craniofacial abnormalities in DS and other congenital disorders.
Keywords: Trisomy 21, experimental animal models, developmental delay disorders, genotype-phenotype correlation
INTRODUCTION
Trisomy of human chromosome 21 (Hsa21) results in Down syndrome (DS) and occurs in ~1/700 live births [Parker et al., 2010]. Individuals born with Trisomy 21 (Ts21) exhibit phenotypes with varying severity including craniofacial, heart and central nervous system malformations [Epstein, 2001]. Essentially all individuals with DS have distinguishing oral and facial features that persist from early prenatal to postnatal and adult stages, and include a small jaw and oral cavity, a shortened midface, and a flat nasal bridge [Allanson et al., 1993; Guihard-Costa et al., 2006]. These alterations influence orofacial structure and function which may predispose tongue hyperprotrusion, as well as impaired eating, speech and breathing in individuals with DS [Faulks et al., 2008; Shott, 2006]. Although the underlying cause of DS is known, the mechanisms by which trisomy alters development and leads to the craniofacial phenotypes of DS are not well defined.
Neural crest cells (NCC) contribute to the majority of the bone, cartilage, connective tissue and peripheral nervous tissue in the head [Knight and Schilling, 2006; Santagati and Rijli, 2003]. Subsets of cranial NCC migrate to the first pharyngeal arch (PA1) or mandibular precursor. The PA1 contributes to Meckel's cartilage and the formation of the lower jaw as well as the maxilla [Knight and Schilling, 2006]. Trisomy may disrupt genetic pathways and cellular constituents, including NCC, early in development and affect the structure and function of craniofacial and other tissues associated with DS [Roper et al., 2009].
Mouse models recapitulate traits associated with DS and have been used to examine the gene-phenotype relationship in DS [Lana-Elola et al., 2011; Wiseman et al., 2009]. The most widely used and well-studied model of DS is the Ts(1716)65Dn (hereafter Ts65Dn) mouse. This segmental trisomy model has a small translocation chromosome that contains about half of the gene orthologs found on Hsa21 [Reeves et al., 1995; Sturgeon and Gardiner, 2011]. Ts65Dn mice show DS-related phenotypes including reduced birth weight, perinatal lethality, craniofacial abnormalities, cognitive and behavioral impairments, cardiovascular malformations, and neurological structural deficiencies [Belichenko et al., 2004; Reeves et al., 1995; Richtsmeier et al., 2000; Roper et al., 2006b; Williams et al., 2008]. Parallel craniofacial phenotypes have been observed in Ts65Dn mice and individuals with DS including a diminished craniofacial skeleton and brachycephaly; a flattened occiput and small midface; and a reduced interorbital breadth as well as maxilla and mandible [Richtsmeier et al., 2000]. Differences in the face, neurocranium, palate and mandible have been found in newborn Ts65Dn as compared to euploid mice [Hill et al., 2007]. The growth trajectory of the mandible from neonatal to adult mice was also significantly altered by trisomy. We showed that the PA1 was smaller and contained fewer NCC in Ts65Dn when compared to euploid offspring at embryonic day 9.5 (E9.5) [Roper et al., 2009].
Analyses of gene expression in tissues from humans with DS as well as DS mice have shown that trisomic as well as non-trisomic (euploid) genes are dysregulated [Hewitt et al., 2007; Laffaire et al., 2009; Rozovski et al., 2007; Sommer et al., 2008]. It has been hypothesized that trisomic craniofacial development is altered by two trisomic genes, Dyrk1a and Rcan1, through the Nfat pathway [Arron et al., 2006]. Yet, we still do not understand intermediate developmental and genetic steps affected by trisomy that result in DS-associated craniofacial structures. We report on a spatial and temporal specific analysis of developmental and genetic changes caused by trisomy in the mandibular precursor. Our results suggest the importance of non-trisomic genes in mandibular development and imply that trisomy alters genetic pathways early in development to cause DS phenotypes. Unlocking the relationship between trisomic and non-trisomic gene expression and developmental processes disrupted by trisomy may help understand additional DS phenotypes and other craniofacial abnormalities.
MATERIALS AND METHODS
Embryo generation and volume quantification
The generation of E13.5 offspring from Ts65Dn mice as well as genotyping, embedding and processing of E13.5 embryos were as described [Blazek et al., 2010]. Animal use and protocols were approved by the IUPUI School of Science Institutional Animal Care and Use Committee. Total embryo and tissue-specific volumes were quantified using unbiased stereology in accordance with established principles [Mouton, 2002]. Images were viewed using the Stereologer system and software (Stereology Resource Center, Chester, MD) on a Nikon Eclipse 80i microscope. To obtain volumes for each tissue of interest, systematic random sampling using the region volume fraction and a Cavalieri point grid was utilized. Sections containing the tissue of interest (e.g., mandible, tongue, Meckel's cartilage; See Supplementary eFigures S1 and S2 in Supporting Information online) were exhaustively sampled using a random start point at the beginning of each specific region and a defined number of sections thereafter. Section thicknesses were measured at 1000X magnification. Sections were examined at 40X magnification using the following parameters (except neocortex at 100X): Total volume: every fifteenth section using 100,000 μm2 per point; mandible: every fifth section and 10,000 μm2 per point; brain volume: every tenth section and 9,000 μm2 per point; neocortical precursor: every seventh section and 1500 μm2 per point; tongue volume: every fourth section and 10,000 μm2 per point; Meckel's and hyoid cartilages (Supplementary eFigure S2 in Supporting Information online), every third section and 1000 μm2 per point; heart: every fifth section and 3500 μm2 per point; and liver: every eighth section and 5000 μm2 per point. The variation between and within sections (sampling error) was controlled by limiting the coefficient of error (CE) for each tissue of each animal to less than 0.1 [Mouton 2002]. The CE values averaged around 0.01 for each region of interest in our analyses. Statistical significance was determined using one- (mandible, Meckel's and hyoid cartilages and neocortex) or two-tailed (tongue, brain, cardiac, hepatic tissues) t-tests that were dependent on previous testing in some tissues. A p-value of ≤ 0.05 was considered statistically significant.
Gene expression analysis of E13.5 mandibular precursor
RNA was isolated from the mandibular precursor of 13 Ts65Dn and 11 euploid E13.5 embryos using TRIzol. RNA was quantified and converted to biotin labeled (BCL) cRNA using the Ambion Illumina TotalPrep RNA amplification kit. Input RNA and post in vitro transcription yields were quantified to normalize and equalize the amount of trisomic and euploid cRNA added to the arrays. These samples were hybridized on 24 arrays using four MouseWG-6 v2.0 bead (BCL) chips (Illumina Inc., San Diego, CA). Each probe was represented with an average of 30-fold redundancy (5-fold minimum). A total of 45,281 probes can be represented on each MouseWG-6 v2.0 bead array.
The resultant array data (probe level data) were preprocessed by Genomestudio software version 1.0.6 (Illumina Inc., San Diego, CA) to perform quantile normalization, background correction and give gene level data (30,774 genes). Data were stored and expression intensity values were adjusted to remove background noise (described in supplementary material).
Normalization and analysis of the gene level data was conducted in R 2.7.1, (www.r-project.org), using functions from Bioconductor 2.1 (www.bioconductor.org). The “lumi” Bioconductor package was employed to read in the data and complete the normalization procedures. MA plots indicated that additional normalization was appropriate and the loess function in R was used to normalize the data and reduce the non-linear relationship between arrays. A log2 transformation was also applied to stabilize the variance observed in the data. Two different analysis of variance (ANOVA) models were used to test for differentially expressed genes between trisomic and euploid samples: 1) All the loess-normalized data simultaneously for estimation and the treatment effect for each probe was tested using the pooled variance estimated in this model, and 2) Each probe was evaluated separately when estimating the difference in the samples (see supplementary information) and the treatment effect for each probe was tested using estimated variance for that probe. Because there were 30,774 genes (tests) in this analysis, the Type I error rate was adjusted for the large number of tests that were conducted. To accommodate the multiple testing issue in all analyses presented, both the false discovery rate (FDR) and the Holm's sequential Bonferroni correction procedure were used to adjust the Type I error rate. The experiment-wise significance level (α) was chosen to be 0.05. Nucleotide sequence data reported are available in the GenBank database under the accession number GSE24554.
Relative trisomic vs. euploid gene expression differences by qPCR
Excess RNA extracted to perform the microarray was converted to cDNA and real-time quantitative PCR was performed on 13 differentially expressed homeobox containing genes, and 15 Hsa21 homologs using methodology as previously described [Deitz and Roper, 2011]. Each cDNA sample was run in duplicate on each primer/probe sequence and six trisomic and euploid samples were used for each gene primer.
Trisomic and non-trisomic gene interaction analyses using PathGen
Potential interactions between trisomic genes found on Mus musculus chromosome 16 (Mmu16) in regions homologous to Hsa21 and the 20 homeobox genes (Table III) as well as Sox9 were determined by the Pathgen transitive gene pathway generator [Clement et al., 2010]. Hsa gene names were inserted in the gene finder using a search depth of ”3” to find potential interactions between trisomic and non-trisomic genes (Supplementary eTables S4a and 4b in Supporting Information online). If no Hsa gene identifier was found for the desired gene, the Mmu gene was used and potential linkage to both Hsa and Mmu homeobox genes were tested.
Table III.
Twenty differentially expressed homeobox containing genes in E13.5 trisomic vs normal embryos.
| Gene | Fold Change (microarray) | p-value | Fold change (qPCR) |
|---|---|---|---|
| Hoxa4 | 0.8331 | 1.50E-05 | N/A |
| Hoxa5 | 0.5501 | 0 | 0.5217 (0.0954) |
| Hoxa7 | 0.7460 | 4.26E-14 | 0.3045 (0.0912) |
| Hoxb2 | 0.8402 | 7.14E-06 | N/A |
| Hoxb4 | 0.6897 | 0 | 0.5613 (0.0404) |
| Hoxb5 | 0.6412 | 0 | 0.4487 (0.1032) |
| Hoxb6 | 0.6895 | 0 | 0.5758 (0.0778) |
| Hoxb7 | 0.5905 | 0 | 0.5890 (0.0853) |
| Hoxb9 | 0.8332 | 1.52E-05 | N/A |
| Hoxc6 | 0.5401 | 0 | 0.3704 (0.1050) |
| Hoxd4 | 0.7560 | 3.27E-11 | 0.4912 (0.1281) |
| Hoxd8 | 0.8454 | 1.49E-05 | N/A |
| En1 | 1.2119 | 5.18E-06 | 0.5407 (0.1045) |
| En2 | 1.6832 | 0 | 2.4048 (0.3073) |
| Nkx6-1 | 1.2110 | 5.66E-06 | 1.7457 (0.2314) |
| Otx1 | 1.1620 | 1.08E-04 | N/A |
| Otx2 | 1.4532 | 0 | 3.079 (0.3620) |
| Pou3f4 | 1.2062 | 1.35E-06 | 0.4027 (0.0994) |
| Six2 | 1.1703 | 5.04E-05 | N/A |
| UNC homeobox | 1.1689 | 2.16E-04 | N/A |
The directionality was confirmed for 11 (bolded) of 13 genes. Fold change is represented by the 2-ΔΔCP or the logarithmic change in the CP-value between trisomic and euploid samples where the level of expression of the actin control was subtracted from the level expression of the gene of interest. N/A = not assessed by qPCR. SEM for qPCR is listed in parentheses.
Sox9 expression in Meckel's and hyoid cartilages
Sox9 expression was examined in E13.5 embryos that were sectioned, deparaffinized, rehydrated in ethanol, subjected to antigen retrieval using sodium citrate, and blocked with 10% donkey serum (MP Biomedicals, Solon, OH) containing 1% BSA (Invitrogen, Carlsbad, CA) in PBS. Sections were incubated overnight at 4 °C with Sox9 (H-90) anti-mouse rabbit polyclonal IgG, (Santa Cruz Biotechnology, Santa Cruz, CA; 1:500) and stained with Alexa Fluor 594 donkey anti-rabbit IgG (H+L), (Invitrogen, Eugene, OR; 1:500) for 1 hr at room temperature. Slides were coated with antifade/DAPI and cover-slip sealed with clear nail polish.
Images were taken with an Olympus FV 1000 confocal microscope using lasers emitting light with 405 nm (DAPI) and 548 nm (Alexafluor 594) at 50x magnification. An average of nine sections from five euploid and eight sections from five trisomic embryos for Meckel's cartilage and average of eight sections from four euploid and eight sections from five trisomic embryos for hyoid cartilage were used. An average of four scans per image was taken. Images were quantified with Adobe Photoshop CS4 based on previous analyses [Kirkeby and Thomsen 2005; Matkowskyj et al., 2000]. Briefly, all cells within, but not including the perichondrium, from Meckel's or hyoid cartilages were selected and placed onto a separate layer with an artificial black background. The black background was selected and inversed to objectively indicate stained cells. Channels representing either DAPI or Sox9 intensity on the histogram were selected and the values for mean intensity, standard deviation, median intensity, and pixel area recorded. To control for experimental variations such as sectioning differences and signal fading, each Sox9 value was divided by DAPI intensity (Supplementary eFigure S5 in Supporting Information online). Differences between relative values were determined with a one-tailed Student's t-test (p ≤ 0.05).
RESULTS
Reduced mandibular but not tongue volume in E13.5 Ts65Dn embryos
The mandibular precursor in E13.5 Ts65Dn embryos was significantly reduced when compared to euploid littermates (Table I and Supplementary eFigure S1 in Supporting Information online) and appeared to result from the small PA1 at E9.5 [Roper et al., 2009]. Meckel's cartilage, largely derived from PA1 NCC and the template for mandibular growth [Ramaesh and Bard, 2003], and hyoid cartilage, derived from PA2 and PA3 NCC and important in craniofacial function [Knight and Schilling 2006], were also significantly reduced in trisomic embryos (Table I and Supplementary eFigure S2 in Supporting Information online). In contrast, the tongue precursor volume (also containing NCC derivatives) was not significantly different between trisomic and euploid E13.5 embryos (Table I).
Table I.
Tissue specific volumes from euploid and Ts65Dn E13.5 embryos
| Euploid (n) | Ts65Dn (n) | P value | % of Euploid | |
|---|---|---|---|---|
| Mandible | 0.94 ± 0.05 (21) | 0.78 ± 0.02 (19) | 0.005a | 83% |
| Tongue | 0.36 ± 0.01 (21) | 0.33 ± 0.02 (19) | 0.33b | 94% |
| Meckel's | 0.022 ± 0.002 (21) | 0.018 ± 0.002 (18) | 0.05a | 80% |
| Hyoid | 0.0071 ± 0.0005 (21) | 0.0048 ± 0.0003 (19) | 0.0002a | 68% |
| Heart | 0.88 ± 0.04 (21) | 0.88 ± 0.05 (19) | 0.90b | 100% |
| Liver | 3.63 ± 0.23 (20) | 2.87 ± 0.23 (17) | 0.03b | 79% |
| Brain | 10.00 ± 0.42 (20) | 9.63 ± 0.43 (19) | 0.25a | 96% |
| Neocortex | 0.53 ± 0.02 (19) | 0.48 ± 0.02 (17) | 0.02a | 90% |
Volume measurements are listed in mm3. The ± value is SEM.
one-tailed Student's t-test.
two-tailed Student's t-test.
Numbers of euploid and trisomic embryos analyses are next to each value.
Total, cardiac, neurologic, and hepatic volumes in E13.5 embryos
Trisomic E13.5 Ts65Dn embryos displayed a significantly smaller total volume than euploid littermate embryos (Table II). Due to the differences between mandibular but not tongue precursors in E13.5 trisomic embryos, additional developmental structures were evaluated. Because heart defects have been observed in the Ts65Dn mouse model [Williams et al., 2008], we measured the volume of the developing cardiac tissue and found it not significantly different between trisomic and euploid embryos (Table I). Due to the thickness of the sections used to obtain the volumetric information, we were unable to accurately resolve developing structures closely linked to DS cardiac deficits. Brains of Ts65Dn as compared to euploid mice are reported to exhibit no gross volumetric differences at adult and early postnatal stages [Baxter et al., 2000; Chakrabarti et al., 2007]. Although the size of E13.5 Ts65Dn and euploid brains was not different, a smaller neocortex was found in trisomic E13.5 embryos that corroborated previous research (Table I) [Chakrabarti et al., 2007]. Additionally, the hepatic precursor tissue was significantly smaller in trisomic compared to euploid embryos (Table I). Relative analysis using overall embryo volume to scaled tissue-specific volumes revealed that not all tissues were reduced on grossly smaller trisomic E13.5 embryos (Table II).
Table II.
Total volume and scaled tissue sizes to total or other embryonic structural volumes
| Euploid (n) | Ts65Dn (n) | P value | % of Euploid | |
|---|---|---|---|---|
| Total Volume (TV) | 44.1 ± 2.4 (21) | 38.3 ± 1.5 (19) | 0.02 | 87% |
| Heart/TV | 0.021 ± 0.001 (21) | 0.023 ± 0.001 (19) | 0.02 | 113% |
| Brain/TV | 0.23 ± 0.01 (20) | 0.25 ± 0.01(19) | 0.05 | 108% |
| Mandible/TV | 0.022 ± 0.001 (21) | 0.021 ± 0.0001 (19) | 0.17 | 95% |
| Hyoid/Mandible | 0.0076 ± 0.0004 (21) | 0.0062 ± 0.0005 (29) | 0.02 | 82% |
| Meckel's/Mandible | 0.023 ± 0.001 (21) | 0.023 ± 0.002 (18) | 0.34 | 96% |
| Neocortex/Brain | 0.053 ± 0.003 (19) | 0.046 ± 0.003 (17) | 0.06 | 87% |
The ± value is SEM. All Student's t-tests are one-tailed. Numbers of euploid and trisomic embryos analyses are next to each value.
Gene expression differences in E13.5 trisomic vs. euploid mandibular precursors
To further understand the genetic alterations associated with trisomic mandibular development, we performed a microarray analysis of RNA from 13 Ts65Dn and 11 euploid mandibular precursors. The results from our ANOVA models comparing trisomic and euploid embryos revealed 155 statistically significant probe sets after a false discovery rate (FDR) multiple testing correction [Benjamini and Hochberg 1995] and 75 statistically significant probe sets after Holm's sequential multiple testing correction [Holm 1979] (Supplementary eTables S1 and S2 in Supporting Information online). It was not surprising that the intersection of the two sets is equal to the set resulting from the Holm's procedure due to the conservative nature of the Holm's procedure. Of the 155 genes that were significantly differentially expressed in the Ts65Dn mandibular precursor, 75 genes exhibited increased expression and 80 genes were down regulated (Supplementary eTables S1 and S2 in Supporting Information online). Biological roles and functional clustering of the differentially expressed genes from our microarray analysis were performed using the DAVID [Dennis et al., 2003; Huang da et al., 2009] (http://david.abcc.ncifcrf.gov/home.jsp) and GeneTools (http://www.genetools.microarray.ntnu.no/adb/index.php) databases. Of the 155 differentially expressed genes, 126 had a DAVID ID, and annotation clusters included enrichment categories for embryonic skeletal system development, DNA-binding region:Homeobox, embryonic organ morphogenesis, and transcription factor activity (Supplementary eTable S3 in Supporting Information online). GeneTools found 53 and 40 of the 155 genes were involved in cellular and developmental processes, respectively (Supplementary eFigure S3 in Supporting Information online).
The top 20 most dysregulated genes in the Ts65Dn mandibular precursor included six Hox genes as well as two additional homeobox genes (En2 and Otx2) that have both been linked to altered mandibular development [Degenhardt et al., 2002; Matsuo et al., 1995]. Of the 20 differentially expressed genes with the homeobox DNA binding region, all 12 Hox genes were down regulated in the Ts65Dn E13.5 mandibular precursor (Table III). Using qPCR, we verified the altered expression found in the microarray in 11 of 13 homeobox genes tested.
Altered expression of Sox9 in the mandibular precursor
Our functional analyses indicated that Sox9 was potentially involved in a number of biological processes relevant to mandible formation. Our gene expression analysis revealed a ~1.2 fold increase in Sox9 and Col2a1 (a direct target of Sox9) in the Ts65Dn mandibular precursor. Using immunohistochemistry, we localized Sox9 expression to the cells of Meckel's and hyoid cartilages of both Ts65Dn and euploid E13.5 embryos (Supplementary eFigure S4 in Supporting Information online). Relative intensity analysis confirmed the increase in Sox9 expression, previously identified in the microarray study, in Ts65Dn Meckel's and hyoid cartilages (Figure 1 and Supplementary eFigure S5 in Supporting Information online), suggesting that Sox9 may be a critical contributor to the Ts65Dn mandibular phenotype.
Fig. 1. Sox9 expression in the E13.5 Meckel's and hyoid cartilages.
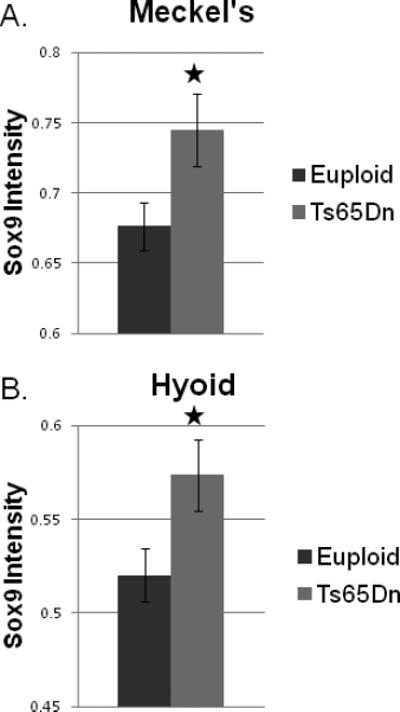
Sox9 expression was increased in theTs65Dn Meckel's cartilage (A) and hyoid cartilage (B) when compared to euploid littermate embryos. Average of nine sections from five euploid and eight sections from five trisomic embryos for Meckel's cartilage; and average of eight sections from four euploid and eight sections from five trisomic embryos for hyoid cartilage. Error bars indicate standard error (*p ≤ 0.05).
Linkage of dysregulated non-trisomic genes to trisomic genes
Although our microarray analysis found only differentially expressed non-trisomic genes, other microarray studies have shown that both trisomic and non-trisomic genes are important in tissue specific phenotypes in DS mouse models [Hewitt et al., 2007; Laffaire et al., 2009]. Expression analysis of 15 trisomic genes on Mmu16 found seven had a > 1.5 fold change by qPCR, suggesting altered expression of these genes in the Ts65Dn mandibular development that was not detected in the microarray analysis (Table IV). We hypothesized that the most important dysregulation of trisomic genes occurred early in mandibular development and resulted in gene expression differences in homeobox containing and other non-trisomic genes in the trisomic E13.5 mandibular precursor. To find potential pathway linkages between trisomic genes and homeobox genes as well as Sox9, we used the PathGen transitive gene pathway generator (http://psoda4.cs.byu.edu/pathgen/pathgen.cgi) [Clement et al., 2010] and found that 15 of the 20 homeobox genes (as well as Sox9) showed potential parsimonious linkages to trisomic genes found on Mmu16 that are homologous to those on Hsa21 (Supplementary eTables 4a,b in Supporting Information online). Of 99 Mmu16 genes in the analysis (93 of which were tested on the array), 45 were linked to one or more of the non-trisomic homeobox genes with an average linkage to six of the non-trisomic genes tested. Trisomic genes that were linked to greater than ten non-trisomic homeobox genes included App, Atp5O, Cct8, Dyrk1a, Ets2, Gabpa, Hmgn1, Ifnar1, Ifnar2 Ifngr2, Itsn1, Mx1, Olig2, Runx1, and Tiam1. These trisomic genes may be the genes that initiate cellular and genetic changes in the trisomic mandibular precursor.
Table IV.
Relative expression of 15 Hsa21 gene homologs by qPCR
| Gene | Fold Change | Gene | Fold Change |
|---|---|---|---|
| Adamts1 | 1.6507 (0.1298) | Grik1 | 1.5278 (0.1069) |
| Adamts5 | 1.9837 (0.2866) | Itsn1 | 1.3524 (0.1165) |
| App | 2.1743 (0.1936) | Kcne2 | 3.1708 (0.4188) |
| Cbr3 | 0.6022 (0.0543) | Mrps6 | 1.2899 (0.0833) |
| Clic6 | 1.1490 (0.1108) | Olig2 | 0.7288 (0.0866) |
| Dyrk1a | 2.1397 (0.1603) | Rcan1 | 3.2163 (0.4548) |
| Erg | 0.4453 (0.1057) | Ripk4 | 0.4526 (0.0733) |
| Hunk | 1.1758 (0.0590) |
Fold change is represented by the 2-ΔΔCP or the logarithmic change in the CP-value between trisomic and euploid samples where the level of expression of the actin control was subtracted from the level expression of the gene of interest. SEM is listed in parentheses. None of the genes listed above were found to be significantly up or down regulated in our microarray analyses.
DISCUSSION
Cellular, genetic and morphological changes identified during trisomic development are important to understand the etiology and potential treatment of DS manifestations. Developmental alterations in cardiac, neurological and craniofacial precursors of Ts65Dn mice have been linked to postnatal DS-associated phenotypes [Chakrabarti et al., 2007; Roper et al., 2006a; Roper et al., 2009; Williams et al., 2008]. Ts65Dn E9.5 embryos display NCC generation, migration and proliferation deficits [Roper et al., 2009] and these alterations appear to affect the developing mandible and associated tissues including Meckel's and hyoid cartilages in E13.5 embryos. The resultant alterations in craniofacial precursors appear to lead to craniofacial abnormalities in neonatal and adult Ts65Dn mice [Hill et al., 2007; Richtsmeier et al., 2000]. Ts65Dn mice are trisomic for only about half of the genes homologous to Hsa21, yet structural and developmental similarities in humans and mice suggest trisomy for these genes causes similar developmental changes that lead to pre- and postnatal craniofacial abnormalities in humans with DS [Allanson et al., 1993; Faulks et al., 2008; Guihard-Costa et al., 2006].
We expected to see a reduction in size of developing tissues with a large NCC component, but the reduced trisomic mandibular and normal tongue precursors in E13.5 trisomic embryos demonstrated the complex development of trisomic phenotypes. A study of humans with DS demonstrated that the tongue hyperprotrusion was a result of “relative macroglossia” [Guimaraes et al., 2008]. Our results suggest that the origin of the relative macroglossia in individuals with DS begins in development with a normal sized tongue precursor in a small developing oral cavity. Though the tongue and the mandible both contain NCC derivatives, we hypothesize that trisomy may have a larger effect on some resultant tissue subsets of NCC through differential regulation of specific gene pathways. Such spatially and temporally specific gene regulation has been observed with Hoxa2 in NCC derived skeletal elements [Santagati et al., 2005]. Although it may be assumed that a generalized delay in the development and size of all tissues would likely follow, our and other data suggest that smaller embryonic volume does not cause a reduction in size of all developing tissues but may be altered in temporal and tissue-specific manners.
The significant differential expression of 20 genes with DNA homeobox regions including the downregulation of 12 Hox genes was prominent among our results (Table III). Six, 12, and one homebox genes were dysregulated in the postnatal day 0 (P0), P15, and P30 trisomic cerebellum, respectively [Dauphinot et al., 2005]. At P0, all six homeobox genes were downregulated including Hoxa5, b5, and d4 (also downregulated in the trisomic E13.5 mandibular precursor). Hoxa5, a regulator of cell growth and apoptosis [Chen et al., 2007], also was downregulated in the cerebellum in adult Ts65Dn mice [Saran et al., 2003]. Hoxb2 knockout mice are runted and have altered facies [Barrow and Capecchi, 1996] and in mouse motor neurons, Hoxb2 has been shown to interact with Sod1, the product of a trisomic gene in individuals with DS and Ts65Dn mice [Zhai et al., 2005]. In our interaction analysis, Hoxa5, b2 and d4 were predicted to be linked with 16, 28 and 26 trisomic genes, respectively, as was a linkage between Hoxb2 and Sod1. Additionally, ectopic expression of Hoxb6 in mice has been linked to craniofacial abnormalities [Kaur et al., 1992]. Although the patterning roles of Hox genes are well characterized, our results combined with work done by others indicate that dysregulation of Hox genes may play an essential role in multiple DS phenotypes and may also influence mandibular development in general.
In addition to the downregulation of Hox genes, other genes with a homeobox DNA binding domain that have a significant role in craniofacial development are upregulated in the Ts65Dn mandibular precursor. The genes En2 and Otx2 are expressed in the mandibular arch as early as E8.25 and E9.5, respectively, and Otx2 heterozygous mutant mice displayed a smaller jaw and other craniofacial malformations [Degenhardt et al., 2002; Kimura et al., 1997; Matsuo et al., 1995]. En1 has been shown to interact with the Fgf signaling pathway to regulate osteoblast differentiation and signaling in skull development [Deckelbaum et al., 2006] and is expressed in cells of the mandibular precursor at E13.5 [Zhong et al., 2010]. Our results suggest that trisomy causes altered expression of downstream homeobox containing genes that affect the development of the mandibular precursor.
Sox9 is a transcription factor that regulates chondrogenesis and is essential to endochondral bone formation [Akiyama et al., 2004; Mori-Akiyama et al., 2003] and a 1.2 fold overexpression of Sox9 has been shown to lead to decreased chondrocyte proliferation, delayed transition of proliferating chondrocytes to hypertrophy, and dwarfism in non-trisomic mice [Akiyama et al., 2004]. Because Meckel's and hyoid cartilages are formed from chondrocytes, the increased expression of Sox9 in our study may delay the proliferation and hypertrophy of the chondrocytes resulting in the smaller Meckel's and hyoid cartilages found in E13.5 Ts65Dn embryos. Meckel's cartilage undergoes endochondral ossification and affects the development of the mandible, and it is possible that Sox9 overexpression may play a significant role in the hypoplastic mandible of the Ts65Dn DS mouse model.
Our microarray analysis found significantly differentially expressed non-trisomic genes in the trisomic mandibular precursor at E13.5. A subsequent qPCR analysis of 15 trisomic genes displayed more gene dysregulation than the array data. A limitation of our microarray study is that we may have failed to detect significant changes in trisomic genes because of high variability in gene expression. Increased gene expression variability specific to trisomic genes has been reported in other microarray analyses and may correlate with high phenotypic variability [Ait Yahya-Graison et al., 2007; Saran et al., 2003]. Alternatively, methodological differences including a potential 3’ bias due to poly T or random hexamer priming in the microarray and qPCR assays, respectively, as well as dissimilarities in probe location and binding efficiency between the two assays may have also caused the differential detection of gene expression. Trisomic genes may also be more uniformly dysregulated earlier in the developing mandible or in a specific cell type at this time point. A gene expression study of the pharyngeal arch and heart regions of E10.5 embryos trisomic for 17 Hsa21 homologs found altered expression of trisomic and non-trisomic genes [Liu et al., 2011]. Although the tissue analyzed by Liu et al., included more than the mandibular arch, some of the dysregulated genes in their study may be important for the early changes in the mandibular precursor in trisomic mice. Lastly, small changes in trisomic gene expression that are not found to be significant, as seen in this and other studies, may be very important in the resultant trisomic phenotypes.
Because non-trisomic genes are dysregulated during the development of DS phenotypes, trisomic genes may serve as an important initiator of differential gene expression found in multiple phenotypes identified in individuals with and without trisomy. Each of the individual phenotypes associated with DS occur to some extent in individuals without Ts21. The disruption of non-trisomic genes and their genetic pathways by trisomy point to the importance of these pathways in overall development. Our data suggest that trisomy is a good model to use to understand DS and genes that play an important role during development of all individuals.
Supplementary Material
ACKNOWLEDGMENTS
We thank Emily Merkel Thomas, Danny Carney, Sashana Gordon, Nicole Shepherd, Brady Harman, Nikki Duvall, and Justin VanHorn for assisting with the genotyping, dissecting, and processing E13.5 embryos. We are grateful to Pich Seekaw for helping to perform the unbiased stereology. This work was supported by a Research Support Funds Grant (RJR), and an Undergraduate Research Opportunity Grant (AN) from IUPUI; a gift from the Beaty-C/Tech Fund--Blue River Community Foundation (RJR); National Science Foundation GK-12 Urban Educators Program at IUPUI NSF DGE0742475 (JRA, JDB, and SLD); and National Institutes of Health DE021034 (RJR). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Footnotes
The authors declare that they have no conflict of interest.
REFERENCES
- Ait Yahya-Graison E, Aubert J, Dauphinot L, Rivals I, Prieur M, Golfier G, Rossier J, Personnaz L, Creau N, Blehaut H, Robin S, Delabar JM, Potier MC. Classification of human chromosome 21 gene-expression variations in Down syndrome: impact on disease phenotypes. Am J Hum Genet. 2007;81:475–491. doi: 10.1086/520000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akiyama H, Lyons JP, Mori-Akiyama Y, Yang X, Zhang R, Zhang Z, Deng JM, Taketo MM, Nakamura T, Behringer RR, McCrea PD, de Crombrugghe B. Interactions between Sox9 and beta-catenin control chondrocyte differentiation. Genes Dev. 2004;18:1072–1087. doi: 10.1101/gad.1171104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allanson JE, O'Hara P, Farkas LG, Nair RC. Anthropometric craniofacial pattern profiles in Down syndrome. Am J Med Genet. 1993;47:748–752. doi: 10.1002/ajmg.1320470530. [DOI] [PubMed] [Google Scholar]
- Arron JR, Winslow MM, Polleri A, Chang CP, Wu H, Gao X, Neilson JR, Chen L, Heit JJ, Kim SK, Yamasaki N, Miyakawa T, Francke U, Graef IA, Crabtree GR. NFAT dysregulation by increased dosage of DSCR1 and DYRK1A on chromosome 21. Nature. 2006;441:595–600. doi: 10.1038/nature04678. [DOI] [PubMed] [Google Scholar]
- Barrow JR, Capecchi MR. Targeted disruption of the Hoxb-2 locus in mice interferes with expression of Hoxb-1 and Hoxb-4. Development. 1996;122:3817–3828. doi: 10.1242/dev.122.12.3817. [DOI] [PubMed] [Google Scholar]
- Baxter LL, Moran TH, Richtsmeier JT, Troncoso J, Reeves RH. Discovery and genetic localization of Down syndrome cerebellar phenotypes using the Ts65Dn mouse. Hum Mol Genet. 2000;9:195–202. doi: 10.1093/hmg/9.2.195. [DOI] [PubMed] [Google Scholar]
- Belichenko PV, Masliah E, Kleschevnikov AM, Villar AJ, Epstein CJ, Salehi A, Mobley WC. Synaptic structural abnormalities in the Ts65Dn mouse model of Down Syndrome. J Comp Neurol. 2004;480:281–298. doi: 10.1002/cne.20337. [DOI] [PubMed] [Google Scholar]
- Benjamini Y, Hochberg Y. Controlling for false discovery rate: a practical and powerful approach to multiple testing. Journal of the Royal Statistical Society, Series B. 1995;57:289–300. [Google Scholar]
- Blazek JD, Billingsley CN, Newbauer A, Roper RJ. Embryonic and not maternal trisomy causes developmental attenuation in the Ts65Dn mouse model for Down syndrome. Dev Dyn. 2010;239:1645–1653. doi: 10.1002/dvdy.22295. [DOI] [PubMed] [Google Scholar]
- Chakrabarti L, Galdzicki Z, Haydar TF. Defects in embryonic neurogenesis and initial synapse formation in the forebrain of the Ts65Dn mouse model of Down syndrome. J Neurosci. 2007;27:11483–11495. doi: 10.1523/JNEUROSCI.3406-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen H, Zhang H, Lee J, Liang X, Wu X, Zhu T, Lo PK, Zhang X, Sukumar S. HOXA5 acts directly downstream of retinoic acid receptor beta and contributes to retinoic acid-induced apoptosis and growth inhibition. Cancer Res. 2007;67:8007–8013. doi: 10.1158/0008-5472.CAN-07-1405. [DOI] [PubMed] [Google Scholar]
- Clement K, Gustafson N, Berbert A, Carroll H, Merris C, Olsen A, Clement M, Snell Q, Allen J, Roper RJ. PathGen: a transitive gene pathway generator. Bioinformatics. 2010;26:423–425. doi: 10.1093/bioinformatics/btp661. [DOI] [PubMed] [Google Scholar]
- Dauphinot L, Lyle R, Rivals I, Dang MT, Moldrich RX, Golfier G, Ettwiller L, Toyama K, Rossier J, Personnaz L, Antonarakis SE, Epstein CJ, Sinet PM, Potier MC. The cerebellar transcriptome during postnatal development of the Ts1Cje mouse, a segmental trisomy model for Down syndrome. Hum Mol Genet. 2005;14:373–384. doi: 10.1093/hmg/ddi033. [DOI] [PubMed] [Google Scholar]
- Deckelbaum RA, Majithia A, Booker T, Henderson JE, Loomis CA. The homeoprotein engrailed 1 has pleiotropic functions in calvarial intramembranous bone formation and remodeling. Development. 2006;133:63–74. doi: 10.1242/dev.02171. [DOI] [PubMed] [Google Scholar]
- Degenhardt K, Rentschler S, Fishman G, Sassoon DA. Cellular and cis-regulation of En-2 expression in the mandibular arch. Mech Dev. 2002;111:125–136. doi: 10.1016/s0925-4773(01)00618-9. [DOI] [PubMed] [Google Scholar]
- Deitz SL, Roper RJ. Trisomic and allelic differences influence phenotypic variability during development of Down syndrome mice. Genetics. 2011;189:1487–1495. doi: 10.1534/genetics.111.131391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dennis G, Jr., Sherman BT, Hosack DA, Yang J, Gao W, Lane HC, Lempicki RA. DAVID: Database for Annotation, Visualization, and Integrated Discovery. Genome Biol. 2003;4:3. [PubMed] [Google Scholar]
- Epstein CJ. Down Syndrome (Trisomy 21). In: Scriver CR, Beaudet AL, Sly WS, Valle D, editors. The Metabolic & Molecular Bases of Inherited Disease. McGraw-Hill; New York: 2001. pp. 1223–1256. [Google Scholar]
- Faulks D, Collado V, Mazille MN, Veyrune JL, Hennequin M. Masticatory dysfunction in persons with Down's syndrome. Part 1: aetiology and incidence. J Oral Rehabil. 2008;35:854–862. doi: 10.1111/j.1365-2842.2008.01877.x. [DOI] [PubMed] [Google Scholar]
- Guihard-Costa AM, Khung S, Delbecque K, Menez F, Delezoide AL. Biometry of face and brain in fetuses with trisomy 21. Pediatr Res. 2006;59:33–38. doi: 10.1203/01.pdr.0000190580.88391.9a. [DOI] [PubMed] [Google Scholar]
- Guimaraes CV, Donnelly LF, Shott SR, Amin RS, Kalra M. Relative rather than absolute macroglossia in patients with Down syndrome: implications for treatment of obstructive sleep apnea. Pediatr Radiol. 2008;38:1062–1067. doi: 10.1007/s00247-008-0941-7. [DOI] [PubMed] [Google Scholar]
- Hewitt CA, Carmichael CL, Wilkins EJ, Cannon PZ, Pritchard MA, Scott HS. Multiplex ligation-dependent probe amplification (MLPA) genotyping assay for mouse models of down syndrome. Front Biosci. 2007;12:3010–3016. doi: 10.2741/2291. [DOI] [PubMed] [Google Scholar]
- Hill CA, Reeves RH, Richtsmeier JT. Effects of aneuploidy on skull growth in a mouse model of Down syndrome. J Anat. 2007;210:394–405. doi: 10.1111/j.1469-7580.2007.00705.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holm S. A simple sequentially rejective multiple test procedure. Scandanavian Journal of Statistics. 1979;6:65–70. [Google Scholar]
- Huang da W, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc. 2009;4:44–57. doi: 10.1038/nprot.2008.211. [DOI] [PubMed] [Google Scholar]
- Kaur S, Singh G, Stock JL, Schreiner CM, Kier AB, Yager KL, Mucenski ML, Scott WJ, Jr., Potter SS. Dominant mutation of the murine Hox-2.2 gene results in developmental abnormalities. J Exp Zool. 1992;264:323–336. doi: 10.1002/jez.1402640311. [DOI] [PubMed] [Google Scholar]
- Kimura C, Takeda N, Suzuki M, Oshimura M, Aizawa S, Matsuo I. Cis-acting elements conserved between mouse and pufferfish Otx2 genes govern the expression in mesencephalic neural crest cells. Development. 1997;124:3929–3941. doi: 10.1242/dev.124.20.3929. [DOI] [PubMed] [Google Scholar]
- Kirkeby S, Thomsen CE. Quantitative immunohistochemistry of fluorescence labelled probes using low-cost software. J Immunol Methods. 2005;301:102–113. doi: 10.1016/j.jim.2005.04.006. [DOI] [PubMed] [Google Scholar]
- Knight RD, Schilling TF. Cranial neural crest and development of the head skeleton. Adv Exp Med Biol. 2006;589:120–133. doi: 10.1007/978-0-387-46954-6_7. [DOI] [PubMed] [Google Scholar]
- Laffaire J, Rivals I, Dauphinot L, Pasteau F, Wehrle R, Larrat B, Vitalis T, Moldrich RX, Rossier J, Sinkus R, Herault Y, Dusart I, Potier MC. Gene expression signature of cerebellar hypoplasia in a mouse model of Down syndrome during postnatal development. BMC Genomics. 2009;10:138. doi: 10.1186/1471-2164-10-138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lana-Elola E, Watson-Scales SD, Fisher EM, Tybulewicz VL. Down syndrome: searching for the genetic culprits. Dis Model Mech. 2011;4:586–595. doi: 10.1242/dmm.008078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu C, Morishima M, Yu T, Matsui S, Zhang L, Fu D, Pao A, Costa AC, Gardiner KJ, Cowell JK, Nowak NJ, Parmacek MS, Liang P, Baldini A, Yu YE. Genetic analysis of Down syndrome-associated heart defects in mice. Hum Genet. 2011;130:623–632. doi: 10.1007/s00439-011-0980-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matkowskyj KA, Schonfeld D, Benya RV. Quantitative immunohistochemistry by measuring cumulative signal strength using commercially available software photoshop and matlab. J Histochem Cytochem. 2000;48:303–312. doi: 10.1177/002215540004800216. [DOI] [PubMed] [Google Scholar]
- Matsuo I, Kuratani S, Kimura C, Takeda N, Aizawa S. Mouse Otx2 functions in the formation and patterning of rostral head. Genes Dev. 1995;9:2646–2658. doi: 10.1101/gad.9.21.2646. [DOI] [PubMed] [Google Scholar]
- Mori-Akiyama Y, Akiyama H, Rowitch DH, de Crombrugghe B. Sox9 is required for determination of the chondrogenic cell lineage in the cranial neural crest. Proc Natl Acad Sci U S A. 2003;100:9360–9365. doi: 10.1073/pnas.1631288100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mouton PR. Principles and Practices of Unbiased Stereology: An Introduction for Bioscientists. Johns Hopkins University Press; Baltimore: 2002. [Google Scholar]
- Parker SE, Mai CT, Canfield MA, Rickard R, Wang Y, Meyer RE, Anderson P, Mason CA, Collins JS, Kirby RS, Correa A. Updated National Birth Prevalence estimates for selected birth defects in the United States, 2004-2006. Birth Defects Res A Clin Mol Teratol. 2010;88:1008–1016. doi: 10.1002/bdra.20735. [DOI] [PubMed] [Google Scholar]
- Ramaesh T, Bard JB. The growth and morphogenesis of the early mouse mandible: a quantitative analysis. J Anat. 2003;203:213–222. doi: 10.1046/j.1469-7580.2003.00210.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reeves RH, Irving NG, Moran TH, Wohn A, Kitt C, Sisodia SS, Schmidt C, Bronson RT, Davisson MT. A mouse model for Down syndrome exhibits learning and behaviour deficits. Nat Genet. 1995;11:177–184. doi: 10.1038/ng1095-177. [DOI] [PubMed] [Google Scholar]
- Richtsmeier JT, Baxter LL, Reeves RH. Parallels of craniofacial maldevelopment in Down syndrome and Ts65Dn mice. Dev Dyn. 2000;217:137–145. doi: 10.1002/(SICI)1097-0177(200002)217:2<137::AID-DVDY1>3.0.CO;2-N. [DOI] [PubMed] [Google Scholar]
- Roper RJ, Baxter LL, Saran NG, Klinedinst DK, Beachy PA, Reeves RH. Defective cerebellar response to mitogenic Hedgehog signaling in Down [corrected] syndrome mice. Proc Natl Acad Sci U S A. 2006a;103:1452–1456. doi: 10.1073/pnas.0510750103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roper RJ, St John HK, Philip J, Lawler A, Reeves RH. Perinatal loss of Ts65Dn Down syndrome mice. Genetics. 2006b;172:437–443. doi: 10.1534/genetics.105.050898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roper RJ, VanHorn JF, Cain CC, Reeves RH. A neural crest deficit in Down syndrome mice is associated with deficient mitotic response to Sonic hedgehog. Mech Dev. 2009;126:212–219. doi: 10.1016/j.mod.2008.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rozovski U, Jonish-Grossman A, Bar-Shira A, Ochshorn Y, Goldstein M, Yaron Y. Genome-wide expression analysis of cultured trophoblast with trisomy 21 karyotype. Hum Reprod. 2007;22:2538–2545. doi: 10.1093/humrep/dem214. [DOI] [PubMed] [Google Scholar]
- Santagati F, Minoux M, Ren SY, Rijli FM. Temporal requirement of Hoxa2 in cranial neural crest skeletal morphogenesis. Development. 2005;132:4927–4936. doi: 10.1242/dev.02078. [DOI] [PubMed] [Google Scholar]
- Santagati F, Rijli FM. Cranial neural crest and the building of the vertebrate head. Nat Rev Neurosci. 2003;4:806–818. doi: 10.1038/nrn1221. [DOI] [PubMed] [Google Scholar]
- Saran NG, Pletcher MT, Natale JE, Cheng Y, Reeves RH. Global disruption of the cerebellar transcriptome in a Down syndrome mouse model. Hum Mol Genet. 2003;12:2013–2019. doi: 10.1093/hmg/ddg217. [DOI] [PubMed] [Google Scholar]
- Shott SR. Down syndrome: common otolaryngologic manifestations. Am J Med Genet C Semin Med Genet. 2006;142:131–140. doi: 10.1002/ajmg.c.30095. [DOI] [PubMed] [Google Scholar]
- Sommer CA, Pavarino-Bertelli EC, Goloni-Bertollo EM, Henrique-Silva F. Identification of dysregulated genes in lymphocytes from children with Down syndrome. Genome. 2008;51:19–29. doi: 10.1139/g07-100. [DOI] [PubMed] [Google Scholar]
- Sturgeon X, Gardiner KJ. Transcript catalogs of human chromosome 21 and orthologous chimpanzee and mouse regions. Mamm Genome. 2011;22:261–271. doi: 10.1007/s00335-011-9321-y. [DOI] [PubMed] [Google Scholar]
- Williams AD, Mjaatvedt CH, Moore CS. Characterization of the cardiac phenotype in neonatal Ts65Dn mice. Dev Dyn. 2008;237:426–435. doi: 10.1002/dvdy.21416. [DOI] [PubMed] [Google Scholar]
- Wiseman FK, Alford KA, Tybulewicz VL, Fisher EM. Down syndrome--recent progress and future prospects. Hum Mol Genet. 2009;18:R75–83. doi: 10.1093/hmg/ddp010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhai J, Lin H, Canete-Soler R, Schlaepfer WW. HoxB2 binds mutant SOD1 and is altered in transgenic model of ALS. Hum Mol Genet. 2005;14:2629–2640. doi: 10.1093/hmg/ddi297. [DOI] [PubMed] [Google Scholar]
- Zhong SC, Chen XS, Cai QY, Luo X, Chen XH, Liu J, Yao ZX. Dynamic expression and heterogeneous intracellular location of En-1 during late mouse embryonic development. Cells Tissues Organs. 2010;191:289–300. doi: 10.1159/000260063. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.