

Summary
Cilia are evolutionarily conserved, membrane-bound, microtubular projections emanating from the cell surface. They are assembled on virtually all cell types in the human body, with very few exceptions, and several recent reviews have covered the topic in great detail [1–3]. The cilium is assembled from mature (mother) centrioles or basal bodies, which serve to nucleate growth of axonemes that give rise to two structurally distinct variants, motile and non-motile cilia. Whereas motile cilia are typically found in large bundles and beat synchronously to generate fluid flow, primary cilia (with the exception of those found at the embryonic node) are generally immotile and are found as solitary organelles [3, 4]. Remarkably, until recently, the primary cilium was considered a vestigial organelle without apparent biological function. However, research over the past decade has established that the primary cilium is capable of transducing essential signaling information from the extracellular milieu [1, 5]. Defects in the cilium, and the structure from which it arises, the basal body, have been shown to cause a spectrum of diseases, ranging from developmental defects to obesity, diabetes, and cancer [6]. Many of these diseases, or ciliopathies, are manifested as genetic syndromes, such as Joubert syndrome, Bardet-Biedel (BBS), Meckel-Gruber (MKS), and Nephronophthisis (NPHP) [6], illustrating the importance of understanding cilium structure and function and the mechanisms required for its assembly. This review focuses primarily on recent advances in our understanding of the regulatory controls governing the assembly and maintenance of the primary cilium.
Basal body assembly: priming the next stage
Primary cilia typically form during quiescence or the G1 phase of the cell cycle. The mother centriole, which, prior to ciliation, serves as a component of the centrosome and microtubule organizing center, differentiates into a basal body to nucleate a cilium. Multi-ciliated cells, however, only assemble cilia upon terminal differentiation, and the basal bodies accompanying the motile cilia are rapidly generated de novo or using the pre-existing centrioles as templates [7, 8]. The conversion of centrioles to basal bodies and initiation of ciliogenesis occur through a well-orchestrated, but poorly understood process. Several features distinguish a basal body from a mother centriole. First, basal feet, which are anchored to cytoplasmic microtubules, assemble from sub-distal appendages as the mother centriole differentiates into basal bodies (Figure 1). It has been shown that eliminating the sub-distal appendage (basal foot) leads to defects in polarized alignment of basal bodies of motile cilia, which in turn compromises ciliary beating, while cilia formation per se is largely unimpaired [9]. In addition, pinwheel-shaped transition fibers originate from the distal appendages of mother centrioles [7, 8, 10, 11] (Figure 1). The earliest stage of ciliogenesis is marked by the association of basal bodies with membranous compartments, such as ciliary vesicles or the plasma membrane, after which axonemal growth commences [12, 13]. Such 'docking' is facilitated by the transition fiber/distal appendage, of which Cep164 has been identified as a major structural component [14, 15]. Cep164, which is required for ciliogenesis, helps maintain the structural integrity of the distal appendages and promotes an association with ciliary vesicles via interaction with a small GTPase, Rab8a, and its guanine nucleotide exchange factor (GEF), Rabin8 [14, 15], essential vesicular trafficking components that build the incipient ciliary membrane.
Figure 1.
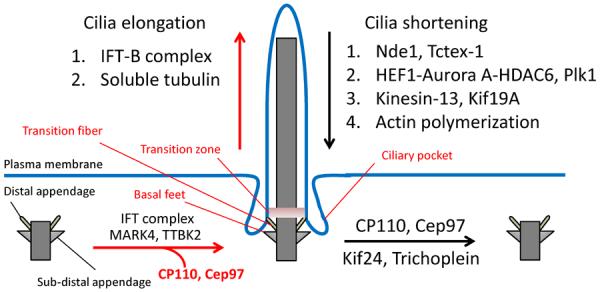
Summary of factors that regulate the assembly or disassembly of cilia. Upon cilia formation and elongation (red arrow), IFT components, MARK4, and TTBK2 are recruited to the basal body, whereas ciliary inhibitors, such as CP110, Cep97, Kif24, and Trichoplein are displaced. Ciliary elongation is directly promoted by the abundance and motility rate of the IFT-B complex and the availability of soluble tubulin as axonemal precursors. Several structural modifications also occur during cilia formation; the distal and sub-distal appendages become transition fibers and basal feet, respectively, and the formation of transition zones and ciliary pockets for their role in ciliary trafficking. Conversely, during ciliary disassembly (black arrow), CP110, Cep97, Kif24, and Trichoplein are expressed at the basal body. Additionally, several factors, including components involved in the IFT-A complex, axonemal deacetylation, axonemal microtubule depolymerization, and actin polymerization negatively regulate cilium assembly.
Recently, several additional transition fiber/distal appendage proteins have been identified, including Cep83, SCLT1, and FBF1, which work in a hierarchical and concerted manner to recruit Cep164 and Cep89 (or Ccdc123) to the distal appendage [16]. Failure to assemble these proteins impairs the recruitment and disappearance of two proteins, TTBK2 and CP110, respectively, resulting in the loss of basal body docking to the plasma membrane [16–18]. Indeed, TTBK2 recruitment and CP110 removal play critical roles during the earliest stages of basal body maturation and ciliogenesis (see below).
Triggering ciliogenesis: the positive and negative regulators
Since basal bodies originate from mother centrioles upon cell cycle exit, a set of robust regulatory controls is required to suppress the untimely conversion to basal bodies and cilia in actively dividing cells. This is achieved through coordinated destruction of negative regulators and recruitment of positive regulators of ciliogenesis to maternal centrioles during basal body formation (Figure 1). CP110 and its binding partners, Cep97 and the kinesin, Kif24, have been identified as negative regulators of cilia formation, and their abundance decreases dramatically during cilia assembly [18, 19]. Further, ectopic expression of these proteins suppresses cilia formation, whereas depletion leads to aberrant ciliogenesis [18, 19]. CP110 localizes to the distal end of centrioles, regulating the growth of centrioles and cilia, and CP110 stability is regulated by Cep97 [18, 20–22]. Indeed, CP110 and Cep97 are uniquely lost from maternal centrioles prior to ciliation [21]. Kif24, a member of the microtubule depolymerizing kinesin family of proteins, co-localizes with CP110 [19]. Kif24 is capable of depolymerizing centriolar microtubules, and its timely destruction helps trigger ciliary axoneme assembly [19]. Additional proteins that function as negative regulators of ciliogenesis, akin to CP110, are likely to exist. For example, trichoplein is also displaced from the basal body during cilia formation, and depletion of this protein promotes ciliation, whereas over-expression suppresses ciliary growth [23]. Conversely, other proteins, such as the Ser/Thr kinase, TTBK2, could act to promote cilia formation [17]. Upon serum deprivation, TTBK2 localizes to the distal end of the basal body, where it displaces CP110 and recruits intraflagellar transport (IFT) complexes (Figure 1). Interestingly, genetic ablation of TTBK2 abolishes ciliogenesis in mice and leads to spinocerebellar ataxia in humans, and null cells are distinguished by persistent CP110 at basal bodies [17]. Although the kinase domain of TTBK2 is required for cilia formation [17], further studies are required to identify key substrates and to determine whether the phosphorylation of such substrate(s) is directly involved in the earliest stages of ciliogenesis. Furthermore, a second Ser/Thr kinase, MARK4, positively regulates cilia assembly (Figure 1) [24]. Depletion of MARK4 impairs the loss of a CP110-Cep97 complex from basal bodies, reminiscent of TTBK2 ablation, and axoneme elongation does not occur in MARK4-depleted cells, whereas basal body attachment to ciliary vesicles was not impaired [24]. These intriguing observations suggest that MARK4 activity, as well as CP110 displacement, underlie a key regulatory process, which coincides with the period during which ciliary vesicles fuse, prior to the onset of axonemal growth.
Despite these advances, our understanding of the mechanisms by which CP110, distal appendage proteins, and other regulators promote basal body formation and ciliary vesicle attachment is rudimentary. For example, it is not known how distal appendages are anchored to ciliary vesicles or the plasma membrane. Nor is it clear how proteins asymmetrically associate with the mother centriole or basal body, catalyzing the accumulation of some proteins and the destruction of others. Nevertheless, these recent findings have shed considerable new light on the “parts lists” and pivotal events that convert a centriole to basal body capable of assembling a primary cilium. When combined with new innovations in super-resolution microscopy that will unveil spatial relationships between these proteins, these studies will catalyze an exciting wave of investigations into the early events of ciliogenesis.
Ciliogenesis and cilia length control
Primary ciliogenesis proceeds through two distinct pathways, depending upon the cell type. In epithelial cells, such as those found in the lung or kidney, the basal body relocates to, and docks with, the apical surface of the plasma membrane, whereupon the axoneme assembles, protruding into the extracellular space [12, 25, 26]. In contrast, in mesenchymal cells, fibroblasts, and neuronal precursors, ciliary vesicles associate with the distal appendages of mother centrioles, and membrane biogenesis occurs in parallel with axoneme elongation as secondary vesicles are recruited, and subsequently fuse, with the plasma membrane, exposing the cilium [13, 27]. In either pathway, membrane biogenesis and vesicular trafficking are required.
Several key proteins have been identified recently. The discovery of genes mutated in diverse ciliopathies led to the identification of a role for the small GTPase, Rab8a, and its GEF, Rabin8, in ciliary vesicle formation and extension [28]. Rabin8 is a downstream effector of the GTP-bound Rab11, which regulates vesicle transport from the trans-Golgi network and recycling endosomes, implicating a Rab11-Rabin8-Rab8 pathway in ciliary vesicle formation during cilia assembly [29, 30]. Furthermore, Rabin8 interacts with the TRAPPII complex, which regulates intra-Golgi transport through vesicle tethering, and together, this complex targets Rab11-Rabin8 to the basal body, prior to localization of Rab8 [30, 31]. Recruitment of Rab8a to the basal body is also regulated by Ahi1 (or Jouberin), a protein which, when mutated, leads to Joubert syndrome [32, 33]. The sequential activation of the small G-proteins, Rab11 and Rab8a, has been widely studied in the context of exocytosis [34]. Activated Rab11 and Rab8 recruit Sec15, a component of the Exocyst complex, and the actin-based motor protein, Myosin, which participate in the tethering and transport of exocytic vesicles, respectively [34–36]. It also thought that specific SNARE proteins, syntaxin 3 and SNAP-25, work in conjunction with Rab8 to promote expansion of the ciliary membrane by vesicular fusion during the development of outer segments of photoreceptor cells [37]. This suggests that the Rab11-Rabin8-Rab8 complex facilitates ciliary vesicle formation and enlargement by sequentially promoting the processes of vesicle formation, transport, tethering, and fusion, which accompany the exocytic pathway during early cilia formation. Given that ciliary membrane expansion and fusion with the plasma membrane is topologically identical to exocytosis, it will be most interesting to identify the complete set of factors that work in conjunction with the aforementioned Rab proteins during the early stages of ciliogenesis.
Once the basal body is formed, ciliary axoneme extension occurs exclusively at the distal end of the outer doublets [38]. Since cilia are compartmentalized from the cell body and lack protein synthesis machinery, materials required for proper growth and maintenance must be transported from the cell body [1, 2, 39]. Proteins destined for the cilium are transported into the axoneme by the IFT machinery [39]. IFT is composed of two complexes, namely, IFT-A and IFT-B, which direct the retrograde (cilia to cell body) and anterograde (cell body to cilia) movement of ciliary proteins, respectively [39]. In general, the IFT-B complex loads and transports the cargo to the ciliary tip, thereby providing the building blocks for axonemal growth, whereas turnover products or signaling components destined for internalization (such as Hedgehog signaling components) are returned to the cell body via the IFT-A complex [39]. Consequently, defects in the IFT-B complex lead to loss of ciliary axoneme assembly, while ciliary growth is largely unimpaired by defects in the IFT-A complex [3, 39].
It is thought that in all ciliated cells, the regulation of ciliary growth is dependent upon IFT. Studies of the flagella of Chlamydomonas indicate that there is continuous assembly and disassembly at the distal tip, suggesting that a balance in the uptake of axonemal precursors (tubulins) and the rate of axonemal turnover governs overall ciliary length [40, 41]. Increasing the abundance or activity of the IFT-B complex leads to further elongation of the cilia, whereas decrease in the mobility of the IFT-B complex will generate a shorter cilium [40, 41]. This observation is recapitulated in mammalian cells, as inducing elongated cilia accelerates the motility of the IFT-B complex [42]. Further, restricting the motility of the IFT-A complex by, ablating, for example, Tctex-1, a putative component of the IFT-associated dynein, also leads to longer cilia [43]. Another protein, Nde1, has been shown to interact with a dynein light chain subunit, LC8, in the regulation of cilia length [44]. Loss of Nde1 promotes extension of cilia, whereas its over-expression results in shortened, bulbous cilia, reminiscent of cilia with defective retrograde IFT, suggesting that Nde1 may negatively regulate IFT-A function [44]. In addition to the regulation of IFT, over-production of soluble tubulin as axonemal 'building blocks' has been shown to promote ciliary length, while limiting the supply of 'free'-tubulin by treating cells with Taxol (microtubule stabilizing drug) resulted in cilia loss or shortening (Figure 1) [45, 46].
Cell shape and contractility also contribute to ciliary length regulation [47]. For example, retinal pigment epithelial (RPE1) cells grown in a spatially confined manner assembled an ezrin-rich cortical actin network on the apical surface, which facilitated cilia elongation [47]. In contrast, cells that were more sparsely distributed developed highly contractile actin stress fibers at the baso-lateral surface and lacked cilia [47]. The involvement of actin cytoskeleton dynamics in cilia formation has been further demonstrated by a high-throughput RNA interference screen, which demonstrated that the polymerization of actin filaments suppresses cilia formation [48]. Two gelsolin family members, GSN and AVIL, which sever actin filaments, have been shown to promote cilia formation. In contrast, ACTR3, which polymerizes actin filaments, inhibits ciliation, and its depletion leads to increased cilium length [48]. Likewise, treatment of cells with cytochalasin D, a drug that inhibits actin filament polymerization, increased the length of the cilia [48]. Interestingly, it has been shown that treatment with Jasplakinolide, a drug that induces the formation of actin filaments, also led to an increase in ciliary length [45]. Although cytochalasin D and jasplakinolide have seemingly opposing affects on the actin cytoskeleton, it is notable that both drugs suppress the formation of stress fibers by limiting actin filament formation or by depleting monomeric actin by uncontrolled formation of actin polymers, respectively [49, 50]. This collectively suggests that the orientation of the actin cytoskeleton and the level of stress fiber formation have a pronounced impact on ciliation (Figure 1). Further studies regarding the regulatory machinery underlying cilia formation may provide an explanation to as why a subset of cells found in the human body lack cilia.
Cilia disassembly
Ciliary tubulins are distinguished by a set of post-translational modifications, including acetylation, detyrosination, polyglutamylation, and glycylation, which determine the stability and biochemical properties of the axoneme [51]. Acetylation occurs inside the lumen of the tubulin polymers, and this is thought to increase the cohesion between tubulin subunits and therefore the rigidity of microtubular strands [52–54]. Detyrosination refers to removal of the tyrosine residue at the C-terminus of α-tubulin subunits, which is thought to stabilize the axonemal fiber. Polyglutamylation and poly-glycylation, on the other hand, could modulate the recruitment of proteins to the axoneme [55].
These post-translational modifications, especially ones that govern axoneme stability, are eliminated as the cilium disassembles during the process of cell cycle re-entry [56]. Cilia assembled in G0/G1 phase must disassemble as cells re-enter the cell cycle, since the structure is not compatible with mitotic spindle formation [56]. It has been shown that growth factor stimulation of ciliated cells triggers the stabilization of HEF1, which in turn activates Aurora A kinase [57]. Aurora A subsequently stimulates histone deacetylase 6 (HDAC6) found at the basal body and ciliary stalk, resulting in deacetylation of axonemal microtubules, rendering them unstable [57]. In addition to HEF1, Pitchfork (Pifo), which is specifically expressed at the basal body of the embryonic node, has been shown to interact with Aurora A and facilitate its activity during the course of cilia disassembly [58]. Recently, it has also been shown that Plk1, which stabilizes HEF1, may directly activate HDAC6 by phosphorylation [57, 59–61]. This suggests that in mammalian cells, destabilization of the ciliary axoneme by deacetylation promotes ciliary shortening, contributing an additional mode of regulating ciliary length (Figure 1).
In lower eukaryotes, such as Chlamydomonas, flagella, which possess acetylated microtubules, undergo deacetylation as they resorb upon mitotic entry, and during this process, a microtubule depolymerizing kinesin, Crkinesin-13 (homologous to mammalian MCAK), is recruited to the axoneme (Figure 1) [62–66]. Given the highly conserved process of ciliation or flagellation, the mammalian cilium may also require a microtubule depolymerizing enzyme(s), similar to Kinesin-13, during active ciliary disassembly. Two such microtubule depolymerizing kinesins, namely, Kif19A and Kif24 may participate in this process. In mice, Kif19A was recently shown to localize to the ciliary tip and regulate ciliary length [67], whereas Kif24 restricts aberrant ciliation at the basal body [19]. In vitro studies show that Kif19A can depolymerize axonemal microtubules that lack any post-translational modifications [67], suggesting the possibility that Kif19A actively disassembles cilia as they become deacetylated through the aforementioned HEF1-AuroraA-HDAC6 pathway (Figure 1). Further studies will be required to identify additional proteins and molecular mechanisms involved in Kif19A- and Kif24-mediated regulation of cilia assembly and disassembly.
Conclusion
During the past decade, our knowledge of the cilium has grown exponentially. The use of genome-wide RNAi screens, forward genetic screens, and proteomic analyses of components of the basal body, isolated ciliary axonemes, and centrosomes will further expand our understanding of this essential organelle. In addition to uncovering novel proteins with potential roles in cilia assembly and disassembly, these studies will point to a multitude of human disease connections, enabling us to devise rational therapeutic strategies.
Acknowledgements
We thank members of the Dynlacht laboratory for helpful comments on the review, and we apologize to our colleagues for omissions due to space limitations. Work in our laboratory was supported by the March of Dimes (#1-FY11-51) and NIH (1R01HD069647-01).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- [1].Satir P, Christensen ST. Overview of structure and function of mammalian cilia. Annu Rev Physiol. 2007;69:377–400. doi: 10.1146/annurev.physiol.69.040705.141236. [DOI] [PubMed] [Google Scholar]
- [2].Kobayashi T, Dynlacht BD. Regulating the transition from centriole to basal body. J Cell Biol. 2011;193:435–44. doi: 10.1083/jcb.201101005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Ishikawa H, Marshall WF. Ciliogenesis: building the cell's antenna. Nat Rev Mol Cell Biol. 2011;12:222–34. doi: 10.1038/nrm3085. [DOI] [PubMed] [Google Scholar]
- [4].Nonaka S, Tanaka Y, Okada Y, et al. Randomization of left-right asymmetry due to loss of nodal cilia generating leftward flow of extraembryonic fluid in mice lacking KIF3B motor protein. Cell. 1998;95:829–37. doi: 10.1016/s0092-8674(00)81705-5. [DOI] [PubMed] [Google Scholar]
- [5].Tsiokas L, Kim S, Ong EC. Cell biology of polycystin-2. Cell Signal. 2007;19:444–53. doi: 10.1016/j.cellsig.2006.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Gerdes JM, Davis EE, Katsanis N. The vertebrate primary cilium in development, homeostasis, and disease. Cell. 2009;137:32–45. doi: 10.1016/j.cell.2009.03.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Anderson RG, Brenner RM. The formation of basal bodies (centrioles) in the Rhesus monkey oviduct. J Cell Biol. 1971;50:10–34. doi: 10.1083/jcb.50.1.10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Dawe HR, Farr H, Gull K. Centriole/basal body morphogenesis and migration during ciliogenesis in animal cells. J Cell Sci. 2007;120:7–15. doi: 10.1242/jcs.03305. [DOI] [PubMed] [Google Scholar]
- [9].Kunimoto K, Yamazaki Y, Nishida T, et al. Coordinated ciliary beating requires Odf2-mediated polarization of basal bodies via basal feet. Cell. 2012;148:189–200. doi: 10.1016/j.cell.2011.10.052. [DOI] [PubMed] [Google Scholar]
- [10].Anderson RG. The three-dimensional structure of the basal body from the rhesus monkey oviduct. J Cell Biol. 1972;54:246–65. doi: 10.1083/jcb.54.2.246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Fisch C, Dupuis-Williams P. Ultrastructure of cilia and flagella - back to the future! Biol Cell. 2011;103:249–70. doi: 10.1042/BC20100139. [DOI] [PubMed] [Google Scholar]
- [12].Sorokin SP. Reconstructions of centriole formation and ciliogenesis in mammalian lungs. J Cell Sci. 1968;3:207–30. doi: 10.1242/jcs.3.2.207. [DOI] [PubMed] [Google Scholar]
- [13].Sorokin S. Centrioles and the formation of rudimentary cilia by fibroblasts and smooth muscle cells. J Cell Biol. 1962;15:363–77. doi: 10.1083/jcb.15.2.363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Graser S, Stierhof YD, Lavoie SB, et al. Cep164, a novel centriole appendage protein required for primary cilium formation. J Cell Biol. 2007;179:321–30. doi: 10.1083/jcb.200707181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Schmidt KN, Kuhns S, Neuner A, et al. Cep164 mediates vesicular docking to the mother centriole during early steps of ciliogenesis. J Cell Biol. 2012;199:1083–101. doi: 10.1083/jcb.201202126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Tanos BE, Yang HJ, Soni R, et al. Centriole distal appendages promote membrane docking, leading to cilia initiation. Genes Dev. 2013;27:163–8. doi: 10.1101/gad.207043.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Goetz SC, Liem KF, Jr, Anderson KV. The spinocerebellar ataxia-associated gene Tau tubulin kinase 2 controls the initiation of ciliogenesis. Cell. 2012;151:847–58. doi: 10.1016/j.cell.2012.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Spektor A, Tsang WY, Khoo D, Dynlacht BD. Cep97 and CP110 suppress a cilia assembly program. Cell. 2007;130:678–90. doi: 10.1016/j.cell.2007.06.027. [DOI] [PubMed] [Google Scholar]
- [19].Kobayashi T, Tsang WY, Li J, et al. Centriolar kinesin Kif24 interacts with CP110 to remodel microtubules and regulate ciliogenesis. Cell. 2011;145:914–25. doi: 10.1016/j.cell.2011.04.028. [DOI] [PubMed] [Google Scholar]
- [20].Kleylein-Sohn J, Westendorf J, Le Clech M, et al. Plk4-induced centriole biogenesis in human cells. Dev Cell. 2007;13:190–202. doi: 10.1016/j.devcel.2007.07.002. [DOI] [PubMed] [Google Scholar]
- [21].Schmidt TI, Kleylein-Sohn J, Westendorf J, et al. Control of centriole length by CPAP and CP110. Curr Biol. 2009;19:1005–11. doi: 10.1016/j.cub.2009.05.016. [DOI] [PubMed] [Google Scholar]
- [22].Kohlmaier G, Loncarek J, Meng X, et al. Overly long centrioles and defective cell division upon excess of the SAS-4-related protein CPAP. Curr Biol. 2009;19:1012–8. doi: 10.1016/j.cub.2009.05.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Inoko A, Matsuyama M, Goto H, et al. Trichoplein and Aurora A block aberrant primary cilia assembly in proliferating cells. J Cell Biol. 2012;197:391–405. doi: 10.1083/jcb.201106101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Kuhns S, Schmidt KN, Reymann J, et al. The microtubule affinity regulating kinase MARK4 promotes axoneme extension during early ciliogenesis. J Cell Biol. 2013 doi: 10.1083/jcb.201206013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Latta H, Maunsbach AB, Madden SC. Cilia in different segments of the rat nephron. J Biophys Biochem Cytol. 1961;11:248–52. doi: 10.1083/jcb.11.1.248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Dalen H. An ultrastructural study of the tracheal epithelium of the guinea-pig with special reference to the ciliary structure. J Anat. 1983;136:47–67. [PMC free article] [PubMed] [Google Scholar]
- [27].Baudoin JP, Viou L, Launay PS, et al. Tangentially migrating neurons assemble a primary cilium that promotes their reorientation to the cortical plate. Neuron. 2012;76:1108–22. doi: 10.1016/j.neuron.2012.10.027. [DOI] [PubMed] [Google Scholar]
- [28].Nachury MV, Loktev AV, Zhang Q, et al. A core complex of BBS proteins cooperates with the GTPase Rab8 to promote ciliary membrane biogenesis. Cell. 2007;129:1201–13. doi: 10.1016/j.cell.2007.03.053. [DOI] [PubMed] [Google Scholar]
- [29].Knodler A, Feng S, Zhang J, et al. Coordination of Rab8 and Rab11 in primary ciliogenesis. Proc Natl Acad Sci U S A. 2010;107:6346–51. doi: 10.1073/pnas.1002401107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Westlake CJ, Baye LM, Nachury MV, et al. Primary cilia membrane assembly is initiated by Rab11 and transport protein particle II (TRAPPII) complex-dependent trafficking of Rabin8 to the centrosome. Proc Natl Acad Sci U S A. 2011;108:2759–64. doi: 10.1073/pnas.1018823108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Hutagalung AH, Novick PJ. Role of Rab GTPases in membrane traffic and cell physiology. Physiol Rev. 2011;91:119–49. doi: 10.1152/physrev.00059.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Ferland RJ, Eyaid W, Collura RV, et al. Abnormal cerebellar development and axonal decussation due to mutations in AHI1 in Joubert syndrome. Nat Genet. 2004;36:1008–13. doi: 10.1038/ng1419. [DOI] [PubMed] [Google Scholar]
- [33].Dixon-Salazar T, Silhavy JL, Marsh SE, et al. Mutations in the AHI1 gene, encoding jouberin, cause Joubert syndrome with cortical polymicrogyria. Am J Hum Genet. 2004;75:979–87. doi: 10.1086/425985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Das A, Guo W. Rabs and the exocyst in ciliogenesis, tubulogenesis and beyond. Trends Cell Biol. 2011;21:383–6. doi: 10.1016/j.tcb.2011.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Sahlender DA, Roberts RC, Arden SD, et al. Optineurin links myosin VI to the Golgi complex and is involved in Golgi organization and exocytosis. J Cell Biol. 2005;169:285–95. doi: 10.1083/jcb.200501162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Hammer JA, 3rd, Wu XS. Rabs grab motors: defining the connections between Rab GTPases and motor proteins. Curr Opin Cell Biol. 2002;14:69–75. doi: 10.1016/s0955-0674(01)00296-4. [DOI] [PubMed] [Google Scholar]
- [37].Mazelova J, Ransom N, Astuto-Gribble L, et al. Syntaxin 3 and SNAP-25 pairing, regulated by omega-3 docosahexaenoic acid, controls the delivery of rhodopsin for the biogenesis of cilia-derived sensory organelles, the rod outer segments. J Cell Sci. 2009;122:2003–13. doi: 10.1242/jcs.039982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Rosenbaum JL, Child FM. Flagellar regeneration in protozoan flagellates. J Cell Biol. 1967;34:345–64. doi: 10.1083/jcb.34.1.345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Pedersen LB, Rosenbaum JL. Intraflagellar transport (IFT) role in ciliary assembly, resorption and signalling. Curr Top Dev Biol. 2008;85:23–61. doi: 10.1016/S0070-2153(08)00802-8. [DOI] [PubMed] [Google Scholar]
- [40].Marshall WF, Rosenbaum JL. Intraflagellar transport balances continuous turnover of outer doublet microtubules: implications for flagellar length control. J Cell Biol. 2001;155:405–14. doi: 10.1083/jcb.200106141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Marshall WF, Qin H, Rodrigo Brenni M, Rosenbaum JL. Flagellar length control system: testing a simple model based on intraflagellar transport and turnover. Mol Biol Cell. 2005;16:270–8. doi: 10.1091/mbc.E04-07-0586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Besschetnova TY, Kolpakova-Hart E, Guan Y, et al. Identification of signaling pathways regulating primary cilium length and flow-mediated adaptation. Curr Biol. 2010;20:182–7. doi: 10.1016/j.cub.2009.11.072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Palmer KJ, MacCarthy-Morrogh L, Smyllie N, Stephens DJ. A role for Tctex-1 (DYNLT1) in controlling primary cilium length. Eur J Cell Biol. 2011;90:865–71. doi: 10.1016/j.ejcb.2011.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Kim S, Zaghloul NA, Bubenshchikova E, et al. Nde1-mediated inhibition of ciliogenesis affects cell cycle re-entry. Nat Cell Biol. 2011;13:351–60. doi: 10.1038/ncb2183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Sharma N, Kosan ZA, Stallworth JE, et al. Soluble levels of cytosolic tubulin regulate ciliary length control. Mol Biol Cell. 2011;22:806–16. doi: 10.1091/mbc.E10-03-0269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Wang L, Piao T, Cao M, et al. Flagellar regeneration requires cytoplasmic microtubule depolymerization and kinesin-13. J Cell Sci. 2013 doi: 10.1242/jcs.124255. [DOI] [PubMed] [Google Scholar]
- [47].Pitaval A, Tseng Q, Bornens M, Thery M. Cell shape and contractility regulate ciliogenesis in cell cycle-arrested cells. J Cell Biol. 2010;191:303–12. doi: 10.1083/jcb.201004003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Kim J, Lee JE, Heynen-Genel S, et al. Functional genomic screen for modulators of ciliogenesis and cilium length. Nature. 2010;464:1048–51. doi: 10.1038/nature08895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Bubb MR, Spector I, Beyer BB, Fosen KM. Effects of jasplakinolide on the kinetics of actin polymerization. An explanation for certain in vivo observations. J Biol Chem. 2000;275:5163–70. doi: 10.1074/jbc.275.7.5163. [DOI] [PubMed] [Google Scholar]
- [50].Cooper JA. Effects of cytochalasin and phalloidin on actin. J Cell Biol. 1987;105:1473–8. doi: 10.1083/jcb.105.4.1473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Gaertig J, Wloga D. Ciliary tubulin and its post-translational modifications. Curr Top Dev Biol. 2008;85:83–113. doi: 10.1016/S0070-2153(08)00804-1. [DOI] [PubMed] [Google Scholar]
- [52].Akella JS, Wloga D, Kim J, et al. MEC-17 is an alpha-tubulin acetyltransferase. Nature. 2010;467:218–22. doi: 10.1038/nature09324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Nogales E, Whittaker M, Milligan RA, Downing KH. High-resolution model of the microtubule. Cell. 1999;96:79–88. doi: 10.1016/s0092-8674(00)80961-7. [DOI] [PubMed] [Google Scholar]
- [54].Cueva JG, Hsin J, Huang KC, Goodman MB. Posttranslational acetylation of alpha-tubulin constrains protofilament number in native microtubules. Curr Biol. 2012;22:1066–74. doi: 10.1016/j.cub.2012.05.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Konno A, Setou M, Ikegami K. Ciliary and flagellar structure and function--their regulations by posttranslational modifications of axonemal tubulin. Int Rev Cell Mol Biol. 2012;294:133–70. doi: 10.1016/B978-0-12-394305-7.00003-3. [DOI] [PubMed] [Google Scholar]
- [56].Kim S, Tsiokas L. Cilia and cell cycle re-entry: more than a coincidence. Cell Cycle. 2011;10:2683–90. doi: 10.4161/cc.10.16.17009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Pugacheva EN, Jablonski SA, Hartman TR, et al. HEF1-dependent Aurora A activation induces disassembly of the primary cilium. Cell. 2007;129:1351–63. doi: 10.1016/j.cell.2007.04.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Kinzel D, Boldt K, Davis EE, et al. Pitchfork regulates primary cilia disassembly and left-right asymmetry. Dev Cell. 2010;19:66–77. doi: 10.1016/j.devcel.2010.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Wang G, Chen Q, Zhang X, et al. PCM1 Recruits Plk1 to Pericentriolar Matrix to Promote Primary Cilia Disassembly before Mitotic Entry. J Cell Sci. 2013 doi: 10.1242/jcs.114918. [DOI] [PubMed] [Google Scholar]
- [60].Seeger-Nukpezah T, Liebau MC, Hopker K, et al. The centrosomal kinase Plk1 localizes to the transition zone of primary cilia and induces phosphorylation of nephrocystin-1. PLoS One. 2012;7:e38838. doi: 10.1371/journal.pone.0038838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Lee KH, Johmura Y, Yu LR, et al. Identification of a novel Wnt5a-CK1varepsilon-Dvl2-Plk1-mediated primary cilia disassembly pathway. EMBO J. 2012;31:3104–17. doi: 10.1038/emboj.2012.144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Piao T, Luo M, Wang L, et al. A microtubule depolymerizing kinesin functions during both flagellar disassembly and flagellar assembly in Chlamydomonas. Proc Natl Acad Sci U S A. 2009;106:4713–8. doi: 10.1073/pnas.0808671106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Blaineau C, Tessier M, Dubessay P, et al. A novel microtubule-depolymerizing kinesin involved in length control of a eukaryotic flagellum. Curr Biol. 2007;17:778–82. doi: 10.1016/j.cub.2007.03.048. [DOI] [PubMed] [Google Scholar]
- [64].L'Hernault SW, Rosenbaum JL. Reversal of the posttranslational modification on Chlamydomonas flagellar alpha-tubulin occurs during flagellar resorption. J Cell Biol. 1985;100:457–62. doi: 10.1083/jcb.100.2.457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].L'Hernault SW, Rosenbaum JL. Chlamydomonas alpha-tubulin is posttranslationally modified in the flagella during flagellar assembly. J Cell Biol. 1983;97:258–63. doi: 10.1083/jcb.97.1.258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Maruta H, Greer K, Rosenbaum JL. The acetylation of alpha-tubulin and its relationship to the assembly and disassembly of microtubules. J Cell Biol. 1986;103:571–9. doi: 10.1083/jcb.103.2.571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Niwa S, Nakajima K, Miki H, et al. KIF19A is a microtubule-depolymerizing kinesin for ciliary length control. Dev Cell. 2012;23:1167–75. doi: 10.1016/j.devcel.2012.10.016. [DOI] [PubMed] [Google Scholar]