

Abstract
Pediatric rhegmatogenous retinal detachments are rare, accounting for less than ten percent of all rhegmatogenous retinal detachments. While most retinal detachments in the adult population are related to posterior vitreous detachment, pediatric retinal detachment are often related to trauma or an underlying congenital abnormalities or genetic syndrome. The anatomy of pediatric eyes, the often late presentation of the disease, and the high incidence of bilateral pathology in children all pose significant challenges in the management of these patients. We discuss the epidemiology of pediatric rhegmatogenous retinal detachment, review the genetic syndromes associated with a high incidence of retinal detachment, and examine other common causes of retinal detachment in this age group. We then outline an approach to evaluation and management and describe the expected outcomes of repair of retinal detachment in the pediatric population.
Keyword: Pediatric rhegmatogenous retinal detachment
Introduction
Pediatric rhegmatogenous retinal detachment (RRD) is an uncommon and challenging disease. Though often associated with poor visual outcomes, careful ophthalmologic evaluation and management of these children can preserve vision and profoundly impact these young patients’ lives. Pediatric retinal detachments are distinct from adult detachments in their anatomy, the tissue characteristics, and the ocular and systemic comorbidities. An understanding of these differences leads to appreciation of the poorer prognosis of retinal detachments in children and guides the vitreoretinal surgeon’s approach to restoring the anatomy. Here, we give a brief review of the epidemiology, clinical presentation and prognosis of pediatric RRD. Strategies for evaluating and managing the disease are then discussed.
Epidemiology
RRD has an annual incidence of approximately 12.4 cases per 100,000 population.1 Pediatric RRD is much less common, with an annual incidence only 0.38–0.69 per 100,000,2 making up only 0.5–8% of all retinal detachments.2–9 A number of case series of pediatric RRD have been reported (Table 1).2–18 Most of these case series include patients up to 18 years of age and the mean or median ages of presentation that are reported are 9–13 years of age. Across these series, trauma accounts for about 40% of all cases (Table 1), compared to just 11% in the adult population.1 Seventy to eighty percent of these patients are boys. While some of these series have shown an equal proportion of male and female detachments when cases of trauma are excluded,15 most show a significant male preponderance even of non-traumatic detachments. Unlike adults, congenital abnormalities commonly play a causative role in pediatric RRDs accounting for 35–56% of all cases in Western populations but only 12–17% in series from East Asia (Table 1). Higher rates of myopia may account for the regional difference. Associations with congenital abnormalities are even more common among patients less than ten years of age.4,10,14,15,17. In the more recent case series from Western populations, even excluding eyes with prior trauma, a high percentage of eyes, about 30%, also have a history of prior intraocular surgery, most often cataract surgery.2,11,18
Table 1.
Summary of case series of pediatric rhegmatogenous retinal detachment spanning from 1959 to 2006. Baseline characteristics, risk factors, surgical approach, and outcomes are detailed.
| Case series | Years | Location of case series | Number of eyes (patients) | Age cutoff | Age distribution | % Male | Trauma (%) | Congenital (%)h | Prior surgery (%) | Myopia (%) | Uveitis (%) | Bilateral detachment (%) | Fellow eye pathology (%), non-trauma | Initial acuity | Macula involving (%) | GRT (%) | PVR at least grade C (%) | Initial SB (%) | Initial SB/PPV (%) | Initial PPV (%) | Total lensectomy (%) | Total PPV (%) | % Of total eyes with SO placement | % of PPV eyes with SOl | Mean surgeries | Single surgery anatomic success (%) | Final anatomic success % | Final acuity |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Hudson5 | 1959–1964 | London, UK | 35 (32) | 15 | 72% >10 | NR | 51 | NR | NR | 34 | 0 | 9 | NR | 50% <20/200 | NR | 3 | NR | 45 Total procedures | 0 | 0 | 0 | 0 | No PPV | No PPV | 1.9 | NR | 71 | 71% 20/200 or better |
| Scharf and Zonis7 | 1964–1973 | Haifa, Israel | 27 (27) | 20 | 74% >10 | 81 | 44 | NR | NR | 30 | 3.7 | 28 | 73 | NR | NR | 0 | NR | 28 Total procedures | 0 | 0 | 0 | 0 | No PPV | No PPV | 1.3 | NR | 78 | NR |
| Winslow and Tasman8 | 1966–1974 | Philadelphia, USA | 187 (179)d | 16 | NR | 73 | 44 | NR | 12 | 15 | 2.7 | 30% of myopes | 56.5% of myopes | 73.4 <20/200 | 77 | NR | NR | 100 | 0 | 0 | 0 | 0 | No PPV | No PPV | 1.2 For successful eyes | 66 | 80 | 58% 20/200 or better in reattached eyes |
| Okinami et al.14 | 1966-1985 | Moriguchi, Kyoto and Suita Japan | 908 (802) | 19 | 7% <10, 29% 10–14, 64% 15–19 | 74 | 26 | NR | NR | 53 | NR | 14.6 | NR | NR | NR, 21% involved 4 quadrants | NR | NR | 87 | NR | 2.7j | NR | NR | NR | NR (NR) | NR | NR | 83 | NR |
| Haring and Wiechansa,12 | 1980–1994 | Kiel, Germany | 33 (31) | 18 | 1 Patient <10 | 54 | 24f | Excluded | 9 | 42 | Excluded | 8 | NR | 39% >20/40 | 45 | 0 | PVR excluded from study | 100 | 0 | 0 | 0 | 3 | NR | No PPV (NR) | 1.2 | 88 | 100 | 61% >20/40 |
| Rumelt et al.b,6 | 1980-2000 | Jerusalem, Israel | 144 (127)e | 18 | Mean 10.8 | 73 | 42 | 62 | 10 | 14 | NR | 16 | NR | 70% <20/400 | 77 | 13 | NR | NR | NR | NR | NR | NR | NR | NR (NR) | NR | NR | 52 | 37% >20/400 |
| Wang et al.17 | 1983-2003 | Taoyuan, Taiwan | 296 (278) | 18 | 13% 0–10, 36% 11–15, 51% 16–18 | 74 | 30.7 | 16.6 | 5.1 | 79.1 | 0 | NR | NR | Median 20/600 | 81 | 5 | 46 | 76 | 19 | 5 | NR | 30.7 | 9 | 25 (29) | 1.34 | 72 | 85 | Median 20/100 |
| Yokoyama et al.9 | 1988-2001 | Hiroshima, Japan | 55 (49) | 15 | Mean 12 | 86 | 27f | 16 (13% FEVR) | 2 | 25 | 0 | NR | NR | Median 0.3 | NR | 1.8 | 22 | 76 | 0 | 24 | NR | 27 | NR | NR (NR) | NR | 78 | 87 (42 in cases with PVR) | Median 0.7 |
| Chang et al.14 | 1989-2003 | Taipei, Taiwan | 152 (146) | 18 | Mean 13.1 | 70 | 32.9 | 11.8 | 5.9 | 37.5 | 3.9 | 6 | NR | NR | 73 | 6.6 | 18.4 | 61.2 | NR | 38.8j | NR | 44.1 | 32.2 | 71.2 (73.1) | 1.5 | 58.5 | 78.3 | 42.8% improved by 2 or more lines |
| Akabane et al.3 | 1991–1996 | Sakura, Japan | 32 (28) | 15 | Mean 12.8 | 65 | 21.9 | 15.6 (all FEVR) | 9.4 | 37.5 | 0 | NR | NR | 65.6% >20/400 | 6.3 | 37.5 | 78 | 0 | 22 | NR | 31 | NR | NR (NR) | NR | 88 | 94 | 90.6% 20/200 or better | |
| Fivgas and Capone2 | 1991–1997 | Atlanta, USA | 29 (27) | 18 | Mean 9.6 | 70 | 42g | 45 | 34 | 34i, 4 with myopia alone | 3.5 | 22 | 89 | 24% >20/800 | 79 | 14 | 45 | 28 | NR | 72j | NR | 92 | 72 | 44 (78) | 2.2 | NR | 72 | 41% >20/800 |
| Weinberg et al. 18 | 1991–2000 | Chicago, USA | 39 (34) | 18 | Median 10 | 79 | 36 | 53 | 51 total, 28 excluding trauma related surgery | NR, none with myopia alone | 15 | 26 (includes trauma) | NR | Median CF | 74 | 15 | 31 | 41 | 46 | 13k | NR | 67 | 23 | NR (35) | 1.6 | 53 | 79 | Median 20/400 |
| Chen et al. 10 | 1995–2004 | Changhua City, Taiwan | 35 (32) | 15 | Median 13 | 75 | 23 | 49 (20% FEVR) | 2.9 | 60i, 23 myopia alone | 0 | 22 (29% excluding trauma) | NR | NR | 91 | 8.6 | 5.7 | 65.7 | 22.9 | 0 | 26 | 28.6 | 5.7 | 25 (20) | 1.4 | 65.7 | 80 | 37% 20/50 or better, 17% NLP |
| Soheilian et al.15 | 1997–2005 | Tehran, Iran | 127 (108) | 18 | Mean 12.1 | 81 | 44.3 | 39.3 (11% Stickler) | 7.9 | 9.4 | 0 | 18 (27.4 excluding trauma) | 85 | 44% ⩾20/200 | 80 | NR | 45 | 31 | 63 | 0 | 44 | 74.8 | 65.4 | NR (86.5) | 1.55 | 58 | 74.9 | 37% ⩾20/200 |
| Kocaoglan et al.13 | 2003c | Ankara, Turkey | 30 (29) | 18 | Mean 13.6 | NR | 43.3 | 6.6 | 3.3 | 29 | 0 | NR | NR | NR | NR | NR | NR, excluded >C2 | 100 | 0 | 0 | NR | NR | NR | NR (NR) | NR | 83.3 | 96.6 | NR |
| Gonzales et al.11 | 2001–2006 | Los Angeles, USA | 46 (45) | 18 | Median 9 | 71 | 43 | 35 | 61 total, 30 excluding trauma related surgery | 17i | 4.3 | 18, (includes trauma) | 37 | Median CF | 83, 58% total RD | 20 | 59 | 26 | 30 | 44 | 55 | NR | 56.5 | NR (88) | NR | 52 | 78 (88) | 44% ⩾20/200 |
| Wadhwa et al.16 | 2006c | New Delhi, India | 230 (216) | 18 | Mean 11.12 | 81.9 | 34.3 | 24.3i | 3.9 | 14.3i | 0.40% | 16.2 (includes trauma) | NR | 27% ⩾20/200 | 98 | NR | 44.8 | 37 | NR | 63j | NR | 69 | 69 | 100 (100) | 1.98 | NR | 88.7 | 30% ⩾20/200 |
NR: not reported; GRT: giant retinal tear; PVR: proliferative vitreoretinopathy; SB: scleral buckle (any type); SB/PPV: scleral buckle and pars plana vitrectomy; PPV: pars plana vitrectomy; SO: silicone oil.
Study assessed scleral buckle only in non-complex retinal detachment.
Series includes exudative detachments.
Date of publication or submission, dates of surgeries not included.
Outcomes reported for only 117 eyes of 109 patients, who were followed for >6 months.
115 eyes with RRD.
Perforating trauma excluded from study.
Traumatic cases excluded from other analysis.
Excluding congenital disorders leading to surgery with no vitreoretinal disorder; unless otherwise noted, includes cicatricial ROP and inherited syndromes such as Stickler and Marfan.
Stickler, Marfan, and ROP included in myopia group as well as congenital except for Wadhwa et al. where these cases were considered only in the myopia group.
Not clear if SB/PPV done in some of these cases.
Surgeons performed scleral buckle as part of initial surgery except for one case of macular hole as etiology of RRD, scleral buckle not placed in other cases because they were deemed inoperable.
Percentage of eyes undergoing PPV as part of initial surgery with placement of silicone oil during initial surgery (percentage of eyes undergoing any vitrectomy with any placement of silicone oil).
Presenting characteristics
Pediatric retinal detachments are often diagnosed at a later stage of disease than adult retinal detachments tend to progress relatively slowly, and the visual loss is less acute than the typical retinal detachment associated with an acute posterior vitreous detachment. This fact, combined with children’s immature cognitive functioning, means that children are far less likely to recognize and report visual symptoms of an acute detachment. Only 40–70% of patients report symptoms at the time of diagnosis.2,11,12 As expected, this percentage is even less in younger age groups. Hence, children with retinal detachments are more likely to present with fluid under the macula and with proliferative vitreoretinopathy (PVR) (Table 1).6 Compared to the adult population, pediatric patients present with worse acuity, after greater delay, and with a higher percentage of macular involvement, 75–85% (Table 1) as opposed to about 50% in adults.6 Due to delayed presentation as well as increased cellular activity and proliferation, a higher percentage of pediatric patients present with PVR. Between 20% and 60% of patients present with PVR grade C or worse (Table 1). A relatively low clinical suspicion for this uncommon disease and the difficulty of evaluating the peripheral retina in children may also contribute to delayed diagnosis and referral.
In this population, patients frequently present for vitreoretinal care when the macula of the second eye becomes involved.2 Bilateral retinal detachments are more common in the pediatric population. Up to 30% of affected children either present with bilateral retinal detachment or develop the second detachment over a relatively short period of follow-up (Table 1). The percentage of bilateral detachments is highest in patients with underlying congenital abnormalities or syndromes. In two different case series, when considering patients presenting with or developing bilateral detachments, 60% of the more severe of the two detachments were deemed inoperable.2,16
Children with unilateral retinal detachments have very high rates of retinal tears and lattice degeneration in the contralateral eye that places them at risk for second detachment. When trauma is excluded, the rate of high-risk peripheral pathology is reported to be as high as 80–90% (Table 1).2,7,8,11,15
The configuration of pediatric retinal detachments differs from that of a typical adult retinal detachment. Often demarcation lines and retinal macrocysts are present, reflecting a chronic process. Giant retinal tears are more common, reported to be 15–20% in some case series (Table 1),11,18 and can complicate the surgical repair. Inferior retinal dialysis is commonly seen in the setting of severe blunt trauma.4,8,14
Specific causative factors
Trauma
A spontaneous retinal detachment in a child with a normal ocular anatomy is exceedingly rare. Trauma, on the other hand, is a leading cause of retinal detachments in children (Table 1). Thus, this possibility should be considered for all cases in which examination of the contralateral eye is unremarkable, regardless of the history. In children, traumatic retinal detachments occur relatively late due to the support of a well-formed vitreous. As noted above, congenital abnormalities, prior ocular surgery, and trauma are the predominant risk factors for RRD in the pediatric age range. Myopia alone is also an important factor, especially in the older pediatric patients and in East Asian populations (Table 1).3,4,17 In some series, one-third to over half of all patients have more than one of these risk factors. A number of congenital conditions are worthy of further discussion.
Inherited syndromes associated with high myopia and abnormal vitreous
Stickler syndrome
Stickler syndrome is the most common cause of inherited RRD in both the general and pediatric population. Stickler first described a pedigree with progressive arthro-ophthalmopathy in 1965. In this pedigree he described an autosomal dominant disorder consisting of congenital myopia with a high rate of retinal detachment, cleft palate, flat face with saddle nose, variable mandibular hypoplasia, and generalized arthropathy.19 He later noted associated hearing loss and spondyloepiphyseal dysplasia.
The ocular findings of Stickler syndrome include early cataract formation, high myopia, lattice degeneration, radial perivascular degeneration, and a high incidence of retinal tears, including giant retinal tears that lead to retinal detachment. Two main ocular phenotypes are recognized. Type I is characterized by a membranous vitreous phenotype with a fibrillar retrolenticular membrane extending to the pars plana and peripheral retina.20 This accounts for 75% of the cases Stickler syndrome21 and has the highest incidence of retinal detachment, reported as high as 73%, with a slight majority of patients having bilateral detachments.22 It is associated with dominant mutations in the COL2A1 gene, which encodes collagen type II.20,23 Kniest syndrome is also associated with autosomal dominant mutations in COL2A1 that are distinct from the mutations associated with type I Stickler syndrome. This syndrome includes the ocular and non-ocular abnormalities present in type I Stickler syndrome with the addition of short trunk and kyphoscoliosis.24,25
Type II Sticker syndrome is characterized by a beaded condensation in the retrolenticular space and is associated with dominant mutations in the COL11A1 gene, which encodes for collagen type XI.20,26–28 Marshall syndrome includes the findings of “ectodermal dysplasia,” hearing defects, a flat nasal bridge, and ocular abnormalities that include myopia, clear vitreous gel, and congenital cataract. It is also associated with autosomal dominant mutations in the COL11A1, indicating that they are overlapping genetic syndromes.29,30
Type III Stickler syndrome and Weissenbacher–Zweymuller syndrome are caused by dominant mutations in COL11A2, but are not associated with an ocular phenotype.31,32 A rare autosomal recessive form Stickler syndrome that does have ocular involvement has also recently been described and is associated with mutations in the COL9A1 gene.33
Wagner disease and erosive vitreoretinopathy
Wagner disease and erosive vitreoretinopathy are related entities associated with autosomal dominant mutations in the gene that encodes for Versican, a large extracellular matrix proteoglycan (chondroitin sulfate proteoglycan type II) that is involved with maintaining the structure of the vitreous body by keeping collagen molecules apart.34 In 1938, Wagner reported a family with low myopia, vitreoretinal degeneration with fluid vitreous, and bone spicule type pigmentary changes.35 This, the first described hereditary vitreoretinal disorder, has become known as Wagner disease. It is associated with impaired dark adaptation and decreased B-wave on electroretinogram. Most patients with Wagner disease have normal vision into the third decade of life, but it is associated with adult onset retinal detachment, chorioretinal atrophy and optic atrophy.34 It is therefore not a common cause of pediatric RRD, but discussion of it is included here as Wagner and Stickler syndrome were historically thought of as being one entity called Wagner–Stickler syndrome. It now understood that the clinical manifestations are quite different, and different gene mutations are responsible for the diseases. In addition to the differing ocular phenotypes between Wagner disease and Stickler syndrome, Wagner disease is not associated with any systemic abnormalities. Erosive vitreoretinopathy includes the clinical findings seen in Wagner disease with the addition of progressive nyctalopia and visual field constriction due to a much more marked chorioretinal atrophy.34
Knobloch syndrome
Knobloch syndrome is defined as a triad of occipital defect, high myopia, and vitreoretinal degeneration. The occipital finding range from scalp defects to encepholocele and ectopic gray matter in the brain. A minority of patients exhibit developmental delay.36–40 Compared to Stickler syndrome, Knobloch syndrome is quite rare. Knobloch syndrome is associated with autosomal recessive mutations in the COL18A1 gene, which codes for type XVIII collagen.41 The antiangiogenic protein, endostatin is also derived from type XVIII collagen.37 A single family has also been reported with Knobloch syndrome and an autosomal recessive mutation in the ADAMTS18 gene, which encodes for a zinc metalloproteinase of unknown function that is expressed highly in the lens and retina.42 In the largest case series involving multiple probands with Knobloch syndrome, all eight affected individuals, aged 4–15, were noted to have high myopia, between −10 and −20 diopters, marked peripheral and macular RPE degeneration, fibrillar vitreous condensation, and smooth irides. Ectopia lentis as well as distinctive lens opacities were also common, but not constant features in children with the syndrome.37 It is hypothesized that the anterior segment abnormalities that are present are a result of a loss of endostatin function.37 Retinal detachment was only reported in one of the children in this case series. However, other reports that included older patients with COL18A1 mutations, report a high incidence of retinal detachment and phthisis. A number of the older relatives of patients in the above-mentioned study were noted to have phthisis.37 The poorly dilating pupil and ectopia lentis that is common in Knobloch syndrome may make diagnosis and management of detachment more difficult in this population. This is also true of Marfan syndrome that is discussed below.
Marfan syndrome
Marfan syndrome is cause by autosomal dominant mutations in the gene that encodes fibrillin-1, an extracellular matrix protein involved in the deposition of elastin. Systemic findings of the disease include increased length of extremities, especially the finger and toes; hyperextendable joints; aortic aneurysm and dissection; and mitral valve prolapse. The most common ocular manifestation is non-traumatic subluxation of the crystalline lens that occurs in 50–80% of affected individuals.43,44 Axial myopia is also common in Marfan syndrome with over 20% of eyes having myopia of greater than 7 diopters.44 High myopia, ectopia lentis, and high rates of surgery for dislocated lenses are predisposing factors for retinal detachment. Rates of bilateral detachment may be as high as 70% and giant retinal tears are common.45,46 In addition to ectopia lentis and poorly dilating pupils, surgery can be more complicated due to thin, friable sclera that can lead to difficulties with wound closure and placement of a scleral buckle.
Other inherited syndromes and congenital abnormalities predisposing to retinal detachment
X-linked juvenile retinoschisis
Juvenile X-link retinoschisis is characterized by radial streaks in the macula due to foveal schisis as well as peripheral splitting of the retina that occurs most commonly in the temporal periphery.47,48 It is one of the most common inherited retinal disorders affecting macular function in males, with prevalence between 1 in 5000 to 20,000.49 It is associated with mutations in the RS1 gene that encodes for retinoschisin, a secreted protein that is expressed in and tightly binds to the surface of photoreceptor and bipolar cells. The function for retinoschisin is not entirely clear, but it is thought to play a role in cell-to-cell adhesion. It is present in both the inner and outer plexiform layers.47,48
The foveal schisis is usually associated with moderate vision loss and is present in virtually all cases of the disease,47 with only one recent case report describing an affected patient without this finding.50 The peripheral schisis, which can predispose patients to more severe vision loss, is present in less than half of affected individuals. The inner retinal layer can degenerate leaving blood vessels free within the vitreous cavity. This can lead to vitreous hemorrhage and subsequent amblyopia with severe vision loss. Peripheral schisis cavities can also obscure or extend into the macula. If inner retinal degeneration or breaks occur in conjunction with outer retina breaks, retinal detachment can occur.47 Repair of these retinal detachments requires careful consideration of the detached inner retinal wall. Management options include performing an inner wall retinectomy or dissecting the posterior hyaloid off the inner retinal wall. The latter can be challenging and pre-operative intravitreal autologous plasmin may be beneficial for these cases as it facilitate separation of the posterior hyaloid.51 Recombinant microplasmin may serve a similar role.
Choroidal coloboma
Choroidal colobomas are congenital abnormalities caused by failure of the proper closure of the embryonic fissure. They may be associated with other abnormalities or syndromes or may present sporadically. They occur in the general population at a rate of 0.14%.52,53 Retinal breaks can occur at the borders of the coloboma, and a high rate of retinal detachment has been reported with colobomas of the choroid. In case series of pediatric RRD, choroidal coloboma associated detachments account for up to 2% of the cases of RRD8,14 and up to 8% in children under age 10.14 Their often posterior location usually necessitates vitrectomy for repair.
Non-traumatic inferotemporal retinal dialysis
Non-traumatic inferotemporal retinal dialysis is hypothesized to progress from peripheral cystic degeneration. It tends to occur in emmetropic eyes and can present with retinal detachment, usually in boys in the second decade of life. These detachments are slowly progressive and the retina often becomes thinned, mimicking retinoschisis. Demarcation lines and intraretinal cysts are also often present. PVR is rare in this condition and most patients do well with scleral buckling procedures.53 Several lines of evidence suggest a strong genetic component to non-traumatic retinal dialysis. In his analysis of 24 sibships with atraumatic retinal dialysis, Verdaguer et al. reported eight instances where more than one family member was affected.54 The presence of a single family with three affected children with bilateral retinal detachments suggests a strong genetic component in at least some cases.53 The true prevalence of this condition is not known. It is possible that some cases of retinal detachment caused by retinal dialysis that is attributed to minor trauma reflect a strong genetic predisposition. Alternatively, apparently spontaneous inferior retinal dialysis may also result from an occult history of trauma.
Other causative factors
Atopic dermatitis
An association between atopic dermatitis and pediatric RRD has also been reported, mainly in case series from Japan.3,4,9,55 In these series it is suggested to be the causative factor in 3–9% of cases. It is hypothesized that trauma from rubbing the eyes can lead to retinal detachments in these cases as retinal dialysis is common55 and has been noted in up to 75% of these cases.4 This association is not noted in the Western literature. As atopic dermatitis is not uncommon in this age group, it is not clear whether there is a causal relationship between the two diseases.
Retinal vascular disease
RRD can also occur as late sequelae of tractional retinal detachments and proliferative retinal vascular disease. There is a broad differential that includes persistent fetal vasculature, incontinentia pigmenti, familial exudative vitreoretinopathy (FEVR), regressed retinopathy of prematurity (ROP), uveitis, and hemoglobinopathies. These etiologies occur in variable frequency depending on the patient population. FEVR, for instance, is the etiologic factor in 13-18% of pediatric RRDs in case series from East Asia.3,9,10 Though a detailed discussion of these diseases is beyond the scope of this article, it is important to recognize them, as they often require very different approaches to their management.
Evaluation and management
History
When evaluating pediatric patients with retinal detachment, a thorough history, including a birth history is important. A history of prematurity may point to a history of ROP that may be a contributing factor. Congenital TORCH infections have also rarely been implicated as a causative factor.2,9 A thorough medical history may point to an underlying etiologic factor in the cases of detachment related to a hereditary syndrome with systemic manifestations. Likewise, a family history, especially of retinal detachment or of need for ocular surgery at a young age, could point to a specific etiology. Elucidating any history of trauma or ocular surgery, and determining the onset of symptoms are both important to determine the prognosis for visual gain after repair. Leucocoria can be the presenting sign noted by parents. It has been reported as the presenting symptom in up to 15% of cases.2,16 The presence of strabismus in a patient with a retinal detachment may indicate a chronic detachment, and the duration of the ocular misalignment can inform the clinician on the time course of visual loss.
Examination
Examination in the pediatric population poses unique challenges. Diligent attempts should be made to assess visual acuity in each eye. Inability to determine pre-operative acuity has been reported as a poor prognostic factor.18 Ocular alignment should be noted as strabismus may indicate chronicity. Nystagmus may also indicate long-term or congenital vision loss or may help point to a specific underlying disease or syndrome. The refractive error in the unaffected eye can also be useful in the determination of etiologic factors, which has implications for determining the risk of detachment in the fellow eye. The presence of cataract, hypotony, choroidals, and uveitis all are suggestive of a longstanding detachment and are poor prognostic indicators. A thorough examination of the peripheral retina of the fellow eye is vital given the high incidence of bilateral detachments. This can sometimes be accomplished in the clinic in older pediatric patients, but warrants an examination under anesthesia in most pediatric patients. This can usually be done in conjunction with surgery of the fellow eye if surgery is planned. Auxiliary testing such as ultrasound can be helpful when there is insufficient view of the retina due to cataract, ectopia lentis, or poorly dilating pupil. Ultrasound can also help delineate areas of schisis and detachment in combined schisis/RRD. When the view is suboptimal, an ultrasound becomes critical to search for a mass or calcification, as the diagnosis of retinoblastoma must be excluded in a timely fashion. Optical coherence tomography often can be performed even on small children and can help differentiate juvenile X-linked retinoschisis based on the characteristic macular cystoid schisis.
In addition to the ophthalmic exam, evaluation of patient’s appearance is important. Other abnormalities of the face, trunk or extremities or the presence of hearing loss or cognitive deficits may point to a specific genetic syndrome. The authors have found that evaluation by a geneticist can be useful in cases were a genetic syndrome is strongly suspected as a causative factor for the retinal detachment. This evaluation can aid with diagnosis and allow for the identification of other, potentially life threatening, associated conditions, such as aortic aneurysm in Marfan syndrome.
In these cases, examination of family members should be carried out when possible given the high incidence of retinal pathology, including RRD associated with hereditary vitreoretinal syndromes. The importance of the evaluation of family members is illustrated by a family of patients under the care of the authors. The initial presentation was of a 50-year-old man for a dislocated intraocular lens in the left eye and blind painful eye on the right as a result of previous retinal detachment with failed surgical repair. A diagnosis of Stickler syndrome was suspected based on his history and clinical exam. His 8-year-old son was examined and found to have an asymptomatic macula-involving detachment in the right eye that was successfully repaired with scleral buckling (Fig. 1). His 13-year-old daughter was found to have a macula-sparing retinal detachment in the left eye that has been stable on serial examinations (Fig. 2).
Figure 1.
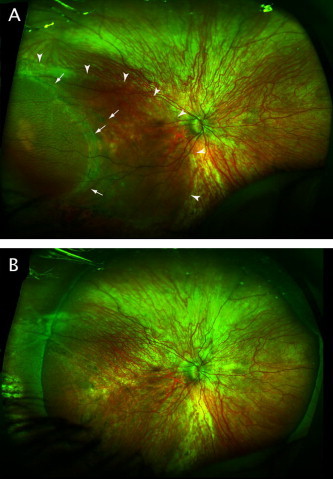
(A) 200° pseudocolor image of right eye of 8-year-old boy. Arrowheads indicate extent of subretinal fluid. Arrows indicate demarcation line and area of schisis. (B) Retina is attached after successful scleral buckling procedure.
Figure 2.
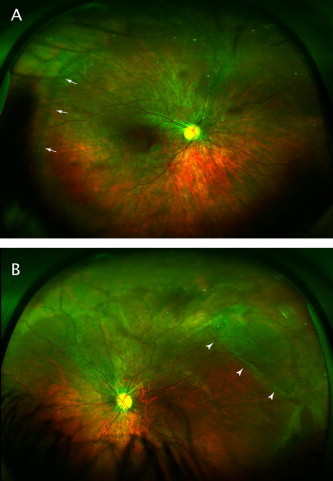
(A) 200° pseudocolor image of right eye of 13-year-old girl. Arrows indicate area of abnormal vitreoretinal interface. (B) Images of left eye. Arrowheads indicate area of stable retinal detachment.
Surgical approach and outcomes
The repair of pediatric RRD involves the same surgical principles that guide repair of adult detachment: relief of traction on the causative retinal defect or defects, reapproximation of the neurosensory retina to the retinal pigment epithelium (RPE), and creation of a retina to RPE adhesion. However, there are several key differences in the anatomy and function of the pediatric eye that demand careful consideration and change the relative advantages of different techniques.
In younger children, a detailed examination under anesthesia is often the first step in repairing a retinal detachment. Visualizing the peripheral retina of a young child can be difficult for the clinician and traumatizing for the child. Once the clinician has determined that an examination under anesthesia is necessary and surgery likely, further examination in the clinic is not necessary and is often counterproductive. Once in the operating room, both eyes are examined in detail, with careful attention to the location of all retinal breaks and the extent of the subretinal fluid. A suitable site for external drainage can be identified at this point. A certain degree of flexibility on the part of operating team and patient’s family is necessary as the specific surgical approach is often determined in the operating room minutes before the surgery begins. More than once, the authors have scheduled a child for repair of a chronic macula-involving retinal detachment in one eye, only to discover, during the examination under anesthesia, a peripheral macula-sparring retinal detachment in the other eye.
The majority of pediatric RRDs should be approached initially with a scleral buckle. There are at least three reasons to avoid vitrectomy if at all possible. First, in contrast to the classic adult posterior vitreous detachment, a child’s posterior hyaloid often remains quite adherent to the retinal surface and difficult to elevate anteriorly beyond the causative pathology. Vitreous that is left near the break often contracts, leading to recurrent detachment. Second, pediatric vitreous is often more formed than adult vitreous. In cases with broad peripheral pathology, the surgeon can take advantage of this. Once the traction has been relieved with a scleral buckle, the vitreous itself can help to tamponade an inferior break, allowing resorption of subretinal fluid. Finally, vitrectomy is associated with a much higher rate of cataract formation compared to scleral buckling. Because of amblyopia risk and loss of accommodation, pediatric cataracts are considerably more morbid than adult cataracts. Thus, approaches that minimize the cataract risk are preferable. Prior to the routine use of vitrectomy, scleral buckling alone was successful for the repair of 70-80% of pediatric RRDs (Table 1),5,7,8,14 though the more complex cases that are now addressed with vitrectomy were likely excluded from these studies as they would have been deemed inoperable at the time.
In adults, vitrectomy and scleral buckle have similar success rates. Typically, the surgeon aims to address all three of the above-mentioned goals. In order to achieve the reapproximation of the retina to RPE, a vitrectomy with intraoperative heavy fluid tamponade or a posterior drainage retinotomy is often employed. This last step is not always necessary. Subretinal fluid is expected to resorb over time if the traction is relieved and the break is closed. Particularly for children, the desire to leave the retina completely flat at the end of the case must be weighed carefully against the additional morbidity of a pediatric vitrectomy. Even cases with extensive PVR can be successfully addressed with a sclera buckle. In the case series by Akabane et al., the authors report successful repair of 7/7 retinal detachments attempted with scleral buckling alone in cases with at least PVR grade C.3
In pediatric cases that involve vitrectomy, large retinectomies are quite common, as the strength of the pediatric vitreoretinal interface often precludes a dissection that relieves all the surface traction. Lensectomy at the time of the repair is common as well (Table 1). Because of the challenges of postoperative positioning and the difficulty of monitoring postoperative intraocular pressure, silicon oil is often preferred to long-acting gas for tamponade (Table 1).
Retinal tears and other significant peripheral retinal pathology are often present in the fellow eye at the time of diagnosis (Table 1).2,7,8,11,15 Typically, this is treated with either cryotherapy or laser retinopexy at the time of diagnosis. Children have an increased likelihood of bilateral retinal detachments. For this reason, many surgeons consider 360° prophylactic retinopexy for fellow eyes deemed to be at high risk for detachment.22,56 In the case of Stickler syndrome, there is some retrospective evidence to suggest this is beneficial.21,22 Interestingly, the retinal cryopexy in this study was applied anteriorly, just posterior to the ora serrata. Stickler eyes have abnormal vitreous bases. When retinal tears develop, they are often quite posterior. This treatment is thought to decrease the risk for development and progression of giant retinal tears. As would be expected, the eyes in this study that developed retinal detachment following prophylaxis did so as a result of posterior tears.22
Compared to the success rates of retinal detachment repair in the adult population, repair of RRD in children has a modestly lower final anatomic success rate of 70–80% in most series (Table 1). However, the reattachment rate with one surgery is considerably lower at 50–80% (Table 1). Visual outcomes also tend to be significantly worse in children with most series showing only 30–40% of patients reaching a final acuity of 20/200–20/400 (Table 1). This is likely due in part to the increased proportion of chronic detachments as well as amblyopia. Anatomic success rates and visual outcomes are lowest among the younger age groups. Initial and final anatomic success rates for children less than eleven years old are only 50% and 60%.17 These patients are also more likely to undergo vitrectomy and lensectomy.17 When different etiologies of retinal detachment are considered, the poorest outcomes tend to be in patients with congenital abnormalities, trauma, and prior surgery.2,4,10,15 with only 22–44% success rate for the latter in some series.2,4 Other risk factors for poor outcome are macular involvement, the presence of PVR, the presence of giant retinal tear, and inability to determine pre-operative acuity.2,11,15,17,18 Four quadrant detachments also have worse outcome compared to three quadrant detachments.14
Especially in younger children the importance of refraction and amblyopia therapy cannot be over emphasized. Eyes with longstanding detachment are already affected by amblyopia at presentation. The visual potential of an eye with a successfully attached retina can be further compromised due to amblyopia induced by refractive changes or strabismus following scleral buckle, as a result of aphakia, or as a result of long-term silicone oil tamponade. Eyes, including those with silicone oil tamponade, should be refracted, and glasses or contact lenses should be prescribed promptly. Penalization of the fellow eye should be recommended when appropriate. Strong consideration should also be given to the removal of cataracts when they become visually significant. A visit to the pediatric ophthalmologist is also another opportunity to emphasize the importance of monocular precautions.
The informed consent process is a critical part of the care of any patient with a retinal detachment. In the case of pediatric retinal detachments, particular challenges must be addressed. As evident in Table 1, the anatomic and visual success rate is relatively low. Often it takes a lengthy conversation for the family to comprehend the extent of the care required and to develop realistic expectations. Before surgery, they must understand the likelihood of multiple surgeries, the importance of follow-up, and the amount of visual recovery that can be realistically expected. When faced with this prognosis, it is not unreasonable for a family to conclude that the benefit of pursuing surgical repair is not worth the risks and hardship involved. This is particularly true of monocular cases with low-risk fellow eyes. Conversely, it is important not to discount the role of relatively low levels of vision to a child’s well-being.57–59 Forty percent of the time, the vision recovered from retinal detachment repair is better than the fellow eye.2 As always, families will rely on an informed and thoughtful surgeon to help them decide how best to care for their child.
Conclusions
Compared to adult RRD, pediatric RRDs have distinct etiologies and differ in their presentation and characteristics. Repair of pediatric RRDs pose unique challenges to the surgeon due to their characteristics as well as the anatomy and physiology of the pediatric eye. While visual outcomes are poor compared to the adult population, the high rate of bilateral detachments as well as the importance of even low levels of vision in these patients mean that the repair of these detachments can make a significant impact on children’s lives.
Footnotes
Peer review under responsibility of King Saud University.

References
- 1.Haimann M.H., Burton T.C., Brown C.K. Epidemiology of retinal detachment. Arch Ophthalmol. 1982;100(2):289–292. doi: 10.1001/archopht.1982.01030030291012. [DOI] [PubMed] [Google Scholar]
- 2.Fivgas G.D., Capone A., Jr. Pediatric rhegmatogenous retinal detachment. Retina. 2001;21(2):101–106. doi: 10.1097/00006982-200104000-00001. [DOI] [PubMed] [Google Scholar]
- 3.Akabane N. Surgical outcomes in juvenile retinal detachment. Jpn J Ophthalmol. 2001;45(4):409–411. doi: 10.1016/s0021-5155(01)00361-6. [DOI] [PubMed] [Google Scholar]
- 4.Chang P.Y. Clinical characteristics and surgical outcomes of pediatric rhegmatogenous retinal detachment in Taiwan. Am J Ophthalmol. 2005;139(6):1067–1072. doi: 10.1016/j.ajo.2005.01.027. [DOI] [PubMed] [Google Scholar]
- 5.Hudson J.R. Retinal detachments in children. Trans Ophthalmol Soc UK. 1965;85:79–91. [PubMed] [Google Scholar]
- 6.Rumelt S. Paediatric vs adult retinal detachment. Eye. 2007;21(12):1473–1478. doi: 10.1038/sj.eye.6702511. [DOI] [PubMed] [Google Scholar]
- 7.Scharf J., Zonis S. Juvenile retinal detachment. J Pediatr Ophthalmol. 1977;14(5):302–304. [PubMed] [Google Scholar]
- 8.Winslow R.L., Tasman W. Juvenile rhegmatogenous retinal detachment. Ophthalmology. 1978;85(6):607–618. doi: 10.1016/s0161-6420(78)35641-4. [DOI] [PubMed] [Google Scholar]
- 9.Yokoyama T. Characteristics and surgical outcomes of paediatric retinal detachment. Eye. 2004;18(9):889–892. doi: 10.1038/sj.eye.6701341. [DOI] [PubMed] [Google Scholar]
- 10.Chen S.N., Jiunn-Feng H., Te-Cheng Y. Pediatric rhegmatogenous retinal detachment in Taiwan. Retina. 2006;26(4):410–414. doi: 10.1097/01.iae.0000238546.51756.cd. [DOI] [PubMed] [Google Scholar]
- 11.Gonzales C.R. Pediatric rhegmatogenous retinal detachment: clinical features and surgical outcomes. Retina. 2008;28(6):847–852. doi: 10.1097/IAE.0b013e3181679f79. [DOI] [PubMed] [Google Scholar]
- 12.Haring G., Wiechens B. Long-term results after scleral buckling surgery in uncomplicated juvenile retinal detachment without proliferative vitreoretinopathy. Retina. 1998;18(6):501–505. doi: 10.1097/00006982-199806000-00001. [DOI] [PubMed] [Google Scholar]
- 13.Kocaoglan H. The efficacy of conventional rhegmatogenous retinal detachment surgery in the pediatric population. J Pediatr Ophthalmol Strabismus. 2003;40(1):4–5. doi: 10.3928/0191-3913-20030101-03. [DOI] [PubMed] [Google Scholar]
- 14.Okinami S. Juvenile retinal detachment. Ophthalmologica. 1987;194(2–3):95–102. doi: 10.1159/000309743. [DOI] [PubMed] [Google Scholar]
- 15.Soheilian M. Clinical features and surgical outcomes of pediatric rhegmatogenous retinal detachment. Retina. 2009;29(4):545–551. doi: 10.1097/IAE.0b013e318194fd1a. [DOI] [PubMed] [Google Scholar]
- 16.Wadhwa N. Rhegmatogenous retinal detachments in children in India: clinical characteristics, risk factors, and surgical outcomes. J AAPOS. 2008;12(6):551–554. doi: 10.1016/j.jaapos.2008.05.002. [DOI] [PubMed] [Google Scholar]
- 17.Wang N.K. Pediatric rhegmatogenous retinal detachment in East Asians. Ophthalmology. 2005;112(11):1890–1895. doi: 10.1016/j.ophtha.2005.06.019. [DOI] [PubMed] [Google Scholar]
- 18.Weinberg D.V. Rhegmatogenous retinal detachments in children: risk factors and surgical outcomes. Ophthalmology. 2003;110(9):1708–1713. doi: 10.1016/S0161-6420(03)00569-4. [DOI] [PubMed] [Google Scholar]
- 19.Stickler G.B. Hereditary progressive arthro-ophthalmopathy. Mayo Clin Proc. 1965;40:433–455. [PubMed] [Google Scholar]
- 20.Snead M.P., Yates J.R. Clinical and molecular genetics of stickler syndrome. J Med Genet. 1999;36(5):353–359. [PMC free article] [PubMed] [Google Scholar]
- 21.Carroll C, et al. The clinical effectiveness and safety of prophylactic retinal interventions to reduce the risk of retinal detachment and subsequent vision loss in adults and children with Stickler syndrome: a systematic review. Health Technol Assess 2011;15 (16):iii–xiv, 1–62. [DOI] [PMC free article] [PubMed]
- 22.Ang A. Retinal detachment and prophylaxis in type 1 Stickler syndrome. Ophthalmology. 2008;115(1):164–168. doi: 10.1016/j.ophtha.2007.03.059. [DOI] [PubMed] [Google Scholar]
- 23.Richards A.J. A novel mutation of COL2A1 resulting in dominantly inherited rhegmatogenous retinal detachment. Invest Ophthalmol Vis Sci. 2005;46(2):663–668. doi: 10.1167/iovs.04-1017. [DOI] [PubMed] [Google Scholar]
- 24.Spranger J. Kniest dysplasia is caused by dominant collagen II (COL2A1) mutations: parental somatic mosaicism manifesting as Stickler phenotype and mild spondyloepiphyseal dysplasia. Pediatr Radiol. 1994;24(6):431–435. doi: 10.1007/BF02011911. [DOI] [PubMed] [Google Scholar]
- 25.Winterpacht A. Alternative splicing as the result of a type II procollagen gene (COL2A1) mutation in a patient with Kniest dysplasia. Hum Mol Genet. 1994;3(10):1891–1893. doi: 10.1093/hmg/3.10.1891. [DOI] [PubMed] [Google Scholar]
- 26.Richards A.J. A family with Stickler syndrome type 2 has a mutation in the COL11A1 gene resulting in the substitution of glycine 97 by valine in alpha 1 (XI) collagen. Hum Mol Genet. 1996;5(9):1339–1343. doi: 10.1093/hmg/5.9.1339. [DOI] [PubMed] [Google Scholar]
- 27.Snead M.P. Hereditary vitreopathy. Eye. 1996;10(Pt 6):653–663. doi: 10.1038/eye.1996.158. [DOI] [PubMed] [Google Scholar]
- 28.Snead M.P. Stickler syndrome type 2 and linkage to the COL11A1 gene. Ann N Y Acad Sci. 1996;785:331–332. doi: 10.1111/j.1749-6632.1996.tb56300.x. [DOI] [PubMed] [Google Scholar]
- 29.Annunen S. Splicing mutations of 54-bp exons in the COL11A1 gene cause Marshall syndrome, but other mutations cause overlapping Marshall/Stickler phenotypes. Am J Hum Genet. 1999;65(4):974–983. doi: 10.1086/302585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Marshall D. Ectodermal dysplasia; report of kindred with ocular abnormalities and hearing defect. Am J Ophthalmol 1958;45 (4, Part 2):143–56. [PubMed]
- 31.Pihlajamaa T. Heterozygous glycine substitution in the COL11A2 gene in the original patient with the Weissenbacher–Zweymuller syndrome demonstrates its identity with heterozygous OSMED (nonocular Stickler syndrome) Am J Med Genet. 1998;80(2):115–120. doi: 10.1002/(sici)1096-8628(19981102)80:2<115::aid-ajmg5>3.0.co;2-o. [DOI] [PubMed] [Google Scholar]
- 32.Vikkula M. Autosomal dominant and recessive osteochondrodysplasias associated with the COL11A2 locus. Cell. 1995;80(3):431–437. doi: 10.1016/0092-8674(95)90493-x. [DOI] [PubMed] [Google Scholar]
- 33.Van Camp G. A new autosomal recessive form of Stickler syndrome is caused by a mutation in the COL9A1 gene. Am J Hum Genet. 2006;79(3):449–457. doi: 10.1086/506478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Mukhopadhyay A. Erosive vitreoretinopathy and Wagner disease are caused by intronic mutations in CSPG2/Versican that result in an imbalance of splice variants. Invest Ophthalmol Vis Sci. 2006;47(8):3565–3572. doi: 10.1167/iovs.06-0141. [DOI] [PubMed] [Google Scholar]
- 35.Wagner H. Ein Bisher Unbekanntes Erbleiden des Auges (Degeneratio Hyaloideo Hereditaria), Beobachtet im Kanton Zurich. Klin Monatsbl Augenheilkd. 1938;100:840. [Google Scholar]
- 36.Czeizel A.E. The second report of Knobloch syndrome. Am J Med Genet. 1992;42(6):777–779. doi: 10.1002/ajmg.1320420605. [DOI] [PubMed] [Google Scholar]
- 37.Khan A.O. The distinct ophthalmic phenotype of Knobloch syndrome in children. Br J Ophthalmol. 2012;96(6):890–895. doi: 10.1136/bjophthalmol-2011-301396. [DOI] [PubMed] [Google Scholar]
- 38.Knobloch W.H., Layer J.M. Retinal Detachment and encephalocele. J Pediatr Ophthalmol. 1971;8:181–184. [Google Scholar]
- 39.Passos-Bueno M.R. Knobloch syndrome in a large Brazilian consanguineous family: confirmation of autosomal recessive inheritance. Am J Med Genet. 1994;52(2):170–173. doi: 10.1002/ajmg.1320520209. [DOI] [PubMed] [Google Scholar]
- 40.Seaver L.H. Congenital scalp defects and vitreoretinal degeneration: redefining the Knobloch syndrome. Am J Med Genet. 1993;46(2):203–208. doi: 10.1002/ajmg.1320460221. [DOI] [PubMed] [Google Scholar]
- 41.Sertie A.L. Collagen XVIII, containing an endogenous inhibitor of angiogenesis and tumor growth, plays a critical role in the maintenance of retinal structure and in neural tube closure (Knobloch syndrome) Hum Mol Genet. 2000;9(13):2051–2058. doi: 10.1093/hmg/9.13.2051. [DOI] [PubMed] [Google Scholar]
- 42.Aldahmesh M.A. Identification of ADAMTS18 as a gene mutated in Knobloch syndrome. J Med Genet. 2011;48(9):597–601. doi: 10.1136/jmedgenet-2011-100306. [DOI] [PubMed] [Google Scholar]
- 43.Cross H.E., Jensen A.D. Ocular manifestations in the Marfan syndrome and homocystinuria. Am J Ophthalmol. 1973;75(3):405–420. doi: 10.1016/0002-9394(73)91149-5. [DOI] [PubMed] [Google Scholar]
- 44.Maumenee I.H. The eye in the Marfan syndrome. Trans Am Ophthalmol Soc. 1981;79:684–733. [PMC free article] [PubMed] [Google Scholar]
- 45.Abboud E.B. Retinal detachment surgery in Marfan’s syndrome. Retina. 1998;18(5):405–409. doi: 10.1097/00006982-199805000-00003. [DOI] [PubMed] [Google Scholar]
- 46.Sharma T. Retinal detachment in Marfan syndrome: clinical characteristics and surgical outcome. Retina. 2002;22(4):423–428. doi: 10.1097/00006982-200208000-00005. [DOI] [PubMed] [Google Scholar]
- 47.Molday R.S., Kellner U., Weber B.H. X-linked juvenile retinoschisis: clinical diagnosis, genetic analysis, and molecular mechanisms. Prog Retin Eye Res. 2012;31(3):195–212. doi: 10.1016/j.preteyeres.2011.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Tantri A. X-linked retinoschisis: a clinical and molecular genetic review. Surv Ophthalmol. 2004;49(2):214–230. doi: 10.1016/j.survophthal.2003.12.007. [DOI] [PubMed] [Google Scholar]
- 49.George N.D., Yates J.R., Moore A.T. Clinical features in affected males with X-linked retinoschisis. Arch Ophthalmol. 1996;114(3):274–280. doi: 10.1001/archopht.1996.01100130270007. [DOI] [PubMed] [Google Scholar]
- 50.Eadie J.A., Luo C.K., Trese M.T. Novel clinical manifestation of congenital X-linked retinoschisis. Arch Ophthalmol. 2012;130(2):255–257. doi: 10.1001/archopthalmol.2011.1352. [DOI] [PubMed] [Google Scholar]
- 51.Wu W.C. Plasmin enzyme-assisted vitreoretinal surgery in congenital X-linked retinoschisis: surgical techniques based on a new classification system. Retina. 2007;27(8):1079–1085. doi: 10.1097/IAE.0b013e31806196d0. [DOI] [PubMed] [Google Scholar]
- 52.Jesberg D.O., Schepens C.L. Retinal detachment associated with coloboma of the choroid. Arch Ophthalmol. 1961;65:163–173. doi: 10.1001/archopht.1961.01840020165003. [DOI] [PubMed] [Google Scholar]
- 53.Soliman M.M., Macky T.A. Pediatric rhegmatogenous retinal detachment. Int Ophthalmol Clin. 2011;51(1):147–171. doi: 10.1097/IIO.0b013e31820099c5. [DOI] [PubMed] [Google Scholar]
- 54.Verdaguer T.J., Rojas B., Lechuga M. Genetical studies in nontraumatic retinal dialysis. Mod Probl Ophthalmol. 1975;15:34–39. [PubMed] [Google Scholar]
- 55.Oka C. Retinal detachment with atopic dermatitis similar to traumatic retinal detachment. Ophthalmology. 1994;101(6):1050–1054. doi: 10.1016/s0161-6420(94)31219-x. [DOI] [PubMed] [Google Scholar]
- 56.Ang GS, Townend J, Lois N. Interventions for prevention of giant retinal tear in the fellow eye. Cochrane Database Syst Rev 2012;2:CD006909. [DOI] [PMC free article] [PubMed]
- 57.Seaber J.H. Long-term visual results of children after initially successful vitrectomy for stage V retinopathy of prematurity. Ophthalmology. 1995;102(2):199–204. doi: 10.1016/s0161-6420(95)31035-4. [DOI] [PubMed] [Google Scholar]
- 58.de Juan E., Jr. The treatment of pediatric retinal detachment. Arch Ophthalmol. 1993;111(5):599. doi: 10.1001/archopht.1993.01090050033021. [DOI] [PubMed] [Google Scholar]
- 59.Hartnett M.E. Long-term vision results measured with Teller Acuity Cards and a new Light Perception/Projection Scale after management of late stages of retinopathy of prematurity. Arch Ophthalmol. 2003;121(7):991–996. doi: 10.1001/archopht.121.7.991. [DOI] [PubMed] [Google Scholar]