

Abstract
Genome-wide association studies (GWAS) have identified several loci associated with many common, multifactorial diseases which have been recently used to market genetic testing directly to the consumers. We here addressed the clinical utility of such GWAS-derived genetic information in predicting type 2 diabetes mellitus (T2DM) and coronary artery disease (CAD) in diabetic patients. In addition, the development of new statistical approaches, novel technologies of genome sequencing and ethical, legal and social aspects related to genetic testing have been also addressed. Available data clearly show that, similarly to what reported for most common diseases, genetic testing offered today by commercial companies cannot be used as predicting tools for T2DM and CAD, both in the general and in the diabetic population. Further studies taking into account the complex interaction between genes as well as between genetic and non genetic factors, including age, obesity and glycemic control which seem to modify genetic effects on the risk of T2DM and CAD, might mitigate such negative conclusions. Also, addressing the role of relatively rare variants by next-generation sequencing may help identify novel and strong genetic markers with an important role in genetic prediction. Finally, statistical tools concentrated on reclassifying patients might be a useful application of genetic information for predicting many common diseases. By now, prediction of such diseases, including those of interest for the clinical diabetologist, have to be pursued by using traditional clinical markers which perform well and are not costly.
Keywords: Common, multifactorial disease, Genome-wide association studies, Genetic counseling, Direct-to consumer testing, Ethical, legal, social issues, Diseases prediction models, Next generation sequencing
Introduction
Multifactorial, common, diseases are caused by the combination of many environmental and genetic and factors with the latter being mostly unknown Since 2006, more than 950 genome-wide association studies (GWAS) have been carried out by analyzing hundreds of thousands of single-nucleotide polymorphisms (SNPs) in very large samples [1; 2] which have identified several loci associated with many common diseases [http://www.genome.gov/gwastudies].
GWAS have addressed only common variants (i.e. minor allele frequency, MAF, > 5%) and have shown they have moderate effect with each allele increasing disease risk only by 10–50% (odds ratios, ORs, ranging 1.10–1.50)[2] ; thus, a large number of loci are needed to significantly influence any single disease.
Based on these results, in the last 2–3 years we have faced the explosion of commercial ventures which are marketing GWAS-based genetic testing directly to the consumers (DTC) for medical purposes.
The possibility of using these data for predicting the development of common diseases is intriguing; in fact, in contrast to clinical markers, genetic markers do not change with time and might help identify high risk subjects several decades before disease onset so that early preventive programs can be implemented.
The aim of the present article is to address the issue of clinical utility of genetic information in predicting T2DM and coronary artery disease (CAD) in diabetic patients. Several collateral, but extremely important, issues including the development of new statistical approaches and novel technologies of genome sequencing which will be largely available in the close future and ethical, legal and social aspects generated by such testing will be also addressed.
Prediction of T2DM
T2DM is a classical example of a common disease in which both genetic and non genetic factors play a pathogenic role [3–5].
In the last 5 years several GWAS have identified more than 40 loci with modest effects (i.e. ORs, ranging 1.10–1.40) which are strongly associated with T2DM. Most of these are likely to modulate insulin secretion while a few are related to obesity and/or insulin resistance [3]. Unfortunately, as for many other common diseases, data from GWAS explain only a small proportion of T2DM genetic background, thus leaving a high proportion of missing heritability yet to be unraveled [3]. This may well be due to some intrinsic characteristics of the genetic background of T2DM which have not been addressed by study designs so far utilized. Firstly, it has been recently reported that heritability for T2DM is higher in patients with disease onset in the range of 35–65 yrs than in those diagnosed at older age [6]. Secondly, both gene-gene [7] and gene-environment (i.e. diet) [8; 9] interaction has been reported to modulate the risk of T2DM. Thus, concentrating our future efforts on specific subset of patients according to their clinical features including age at diabetes onset [6, 10–12] as well as taking into account the role of gene effect “modifiers” (including the protective effect on body weight of whole grain intake [13] is likely to help unravel, at least partly, the yet unknown genetic background of T2DM. Given the yet preliminary status of our understanding of the genetics of T2DM, it is not surprising that using GWAS data for predicting T2DM in the clinical set has been frustrating [12, 14–18] (Table 1). While most of these studies [12, 14–17] described a strongly significant association between a GWAS data-derived genetic risk score and the development of future T2DM (with an increased allelic risk ranging 4–12%), none of them turned out to be of clinical utility in predicting incident events when genetic information was added to models based on classical, non genetic factors.
Table 1.
Genetic prediction of incident T2DM in prospective studies
| Study | Subjects (n) | SNPs (n) | AUC-ROC | ||
|---|---|---|---|---|---|
| Genetic model | Clinical model | Both models | |||
| MPP+Botnia Study (14) | 18,831 | 11 | 0.62 | 0.74 | 0.75* |
| FOS (15) | 2.377 | 18 | 0.58 | 0.9 | 0.901* |
| DESIR (16) | 3,877 | 3 | 0.56 | nr | nr |
| Rotterdam Study (17) | 6,544 | 18 | 0.6 | 0.66 | 0.68* |
| Whitehall II Study (18) | 5,535 | 20 | 0.55 | 0.78 | 0.78 |
| FOS (12) | 3,471 | 40 | 0.6 | 0.903 | 0.906* |
MPP: Malmoe Preventive Program
FOS: Framingham Offspring Study
DESIR: Data from the Epidemiological Study on the Insulin Resistance Syndrome
AUC-ROC: area under the receiver-operating-characteristic curve
nr: not reported, though assessed
= significantly different as compared to clinical model
Prediction of CAD in patients with T2DM
Atherosclerosis too is heavily influenced by genes, with 30 to 50% of its variance being accounted for by heritable factors [19–21]. Thus, genetic markers could also be used to predict the risk of vascular complications in the general population as well as among diabetic subjects. In this latter subgroup, great effort has been dedicated to coronary artery disease (i.e. CAD), a principal cause of increased morbidity and mortality in T2DM [22].
One strategy has been to search for such genetic markers among GWAS-derived loci associated with CAD in the general population [23–28].
In a joint analysis of the Joslin Heart Study (JHS), the Nurses Health Study (NHS), and the Health Professional Follow-up Study (HPFS), five of these loci were found to be associated with CAD also among individuals with T2DM (Table 2) [29].
Table 2.
Associations with CAD of previously reported SNPs in the general population and in T2DM.
| General Population | T2DM† | |||||
|---|---|---|---|---|---|---|
| Locus | Closest Gene(s) | Risk allele | OR (95%CI) | Ref. | OR (95%CI) | P value |
| 9p21 | CDKN2A/CDKN2B | G | 1.29 (1.25–1.34) | (23; 24; 65; 66) | 1.21 (1.08–1.35) | 0.0007 |
| 6p24 | PHACTR1 | C | 1.12 (1.08–1.17) | (65) | 1.25 (1.10–1.41) | 0.0002 |
| 1p21 | CELSR2-PSRC1-SORT1 | T | 1.19 (1.13–1.26) | (23; 65) | 1.17 (1.02–1.34) | 0.03 |
| 12q24 | HNF1A | T | 1.08 (1.05–1.11) | (25) | 1.17 (1.04–1.32) | 0.0048 |
| 1p32 | PCSK9 | T | 1.15 (1.10–1.21) | (65) | 1.26 (1.09–1.47) | 0.0013 |
Data are from the study by Qi et al. (29)
While the effect of each genetic variant was as modest as in the general population, considered jointly these five loci had an influence on CAD risk among diabetic subjects similar in size to that of major cardiovascular risk factors. Carriers of more than eight risk alleles at any of these five loci (about 20% of the diabetic population) had a two-fold increase in CAD risk as compared to carriers of less than five risk alleles (about 30% of the diabetic population). This effect, however, did not translate into a large improvement in risk discrimination over that provided by clinical predictors of CAD. The area under the ROC curve (AUC-ROC; see below for methodological details) was 0.59 for the five loci considered together as compared to 0.70 for the clinical model. Addition of the five loci to the clinical predictors produced only a small, though statistical significant, increase in AUC-ROC (from 0.70 to 0.715; p=0.001). The improvement in risk prediction seemed somewhat more substantial when expressed as Net Reclassification Improvement (NRI; see below), with about 15% of diabetic individuals being correctly reclassified into the correct risk group by the addition of genetic information in the predictive model.
Another approach to identify genetic predictors of CAD in diabetes has been based on the “common soil hypothesis”, which postulates that insulin resistance and T2DM share some of their genetic background with coronary artery disease [30]. On this basis, one recent study has evaluated whether loci that have been found to date to be associated with T2DM may also predict CAD events [31]. A genetic risk score based on the 38 T2DM loci was associated with a modest increase in the risk of incident CAD (OR per score tertile=1.08, p=0.02). However, this effect lost its significance after adjustment for diabetes status (OR per score tertile=1.05, p=0.09), indicating that most of the predisposing effect on CAD was mediated by the increased risk of T2DM. The AUC-ROC was unaffected by the addition of this genetic score to a clinical model. These negative results may have been due to the fact that the T2DM genes identified to date are mostly involved in insulin secretion rather than insulin action. In this regard, genes involved in insulin signaling, such as ENPP1, IRS1, and TRIB3 have yielded better results. In three cohorts of subjects at high cardiovascular risk, about half of whom with diabetes, a risk score based on three non-synonymous variants placed in these genes and previously reported to be associated with insulin-resistance [32–34] predicted cardiovascular events with an hazard ratio per risk allele equal to 1.18 (p=0.0009). The risk score improved the survival AUC-ROC from 0.678 to 0.688 in the overall population and from 0.759 to 0.789 among obese subjects, and determined the reclassification in the correct risk group of 18% of study subjects, which went up to 59% in the obese subgroup (V. Trischitta, personal communication). Along the same line a recent study reported that variability at the FTO obesity locus is associated with cardiovascular events in people with abnormal glucose homeostasis [35]. These studies, though preliminary, stresses how important may be to look at the effect of genetic variability occurring in specific pathways known to modulate disease risk.
Despite these somewhat encouraging results, the efforts to improve the prediction of cardiovascular disease in diabetic patients by means of genetic markers have not met the expectations. Very similar negative results have been obtained also in the general population [36]. At least among diabetic patients such negative conclusions should be mitigated by the possibility that as yet unidentified genetic factors that predispose to CAD specifically in the presence of T2DM do exist. Identification of these genes through GWAS specifically targeted to the diabetic population may provide more powerful tools to predict CAD risk in these subjects.
New statistical tools for predicting complex diseases
Because of the specific scope of this review article, the present section is mainly focused on the novel measures of classification/prediction ability which allows taking into account the interplay between genetic and clinical risk factors. In contrast, because of space constrain, important aspects related to how to address gene-environment interactions [37–41] are not discussed here.
A clinical model performs well when subjects who will experience say a cardiovascular event receive a higher probability of such event than subjects who will not. To this purpose, the simple statistical significance (i.e. based on p-values) which may be obtained in presence of large sample studies regardless of clinical relevance, is not appropriate [42].
Alternative established approaches such as the geometrical area under a receiver-operating-characteristic curve (AUC-ROC) which contrasts the false positive and true positive rates by the different cut-points for predictive probability of the event) The perfect classification, therefore, corresponds to a AUC-ROC equal to one, while the minimum value they can assume is 0.50 (corresponding to classification by chance, say, as if determined by the toss of a coin). Unfortunately, AUC-ROC are scarcely sensitive to the inclusion of new risk predictors (i.e. genetic information) within a well-established clinical model [42].
Another common criterion to define an accurate predictive tool is the ability of measuring the agreement between observed and predicted proportion of events in the whole sample, known as calibration [42]. Indeed, high discriminatory capacity of individuals at high or low risk (i.e. an AUC-ROC close to 1) does not necessarily correspond to high ability to estimate disease probability in the whole sample (i.e. good calibration).
Greater attention has been recently given to novel indices which are focused on what a new marker can actually add to clinically-established prediction models (i.e. the ability in reclassifying what had been incorrectly classified before) [42, 43] namely Integrated Discrimination Improvement (IDI) and Net Reclassification Improvement (NRI). IDI is defined as a difference in the means of the model-based event probabilities, that is a subtraction of the nonevents from the events. IDI has been proved to be closely related to the R-square.
Suppose we have mean predictive values p for the old model (i.e., the clinically established model; Old) and the new model (i.e. the model which includes, say, the genetic factor whose added value we want to assess; New) separately for subject with (D) and without (ND) disease, that is respectively: pOld,D, pNew,D pOld,ND pNew,ND, absolute IDI corresponds to (pNew,D-pOld,D)-(pNew,ND-pOld,ND), while relative IDI is (pNew,D-pOld,D)/(pNew,ND-pOld,ND)-1.
NRI assesses the correct movement (upward for diseased and downward for non-diseased) in pre-defined categories of the predicted values based on reclassification tables built separately for subjects with and without disease. A worked example is as follows. In a sample of 177 obese patients with T2DM (extracted from the Gargano Heart Study, prospective design [44]) a clinical model, including baseline patients’ age, gender, smoking status, BMI and presence of hypertension, was used to predict incident major cardiovascular events. As a didactic example of NRI as compared to ROC curves, we here assess the improvement in model performance introduced by the inclusion of ENPP1 K121Q genotype, a functional aminoacid change which is associated with insulin resistance [32]. For simplicity of presentation, all performance measures are assessed for a binary outcome, ignoring follow-up time. The ROC curves for the model without and with K121Q genotype are presented in Figure 1. The corresponding AUCR-OCs were 0.82 and 0.86, with the difference being not statistically significant (p-value=0.228). For NRI, three risk categories of potential clinical utility (i.e. low, medium, high risk) were created and used for both the old model and the new model. Table 3 cross-classifies subjects who experienced (i.e. patients, upper panel) or not (lower panel) cardiovascular events. For patients, the upper off-diagonal elements (light grey background) represent those who were reclassified correctly (i.e. into a higher category of risk by the new model); in contrast, the lower off-diagonal elements (dark grey background) are patients who were reclassified incorrectly. A symmetrical approach is used for the non-diseased subjects with the new model performing better if it reclassifies non-diseased subjects into lower category of risk. NRI is obtained by the sum of these differences between correct and incorrect reclassification. In the upper panel, classification on the new model improved for 4 and became worse for 1 patient, with the net gain in reclassification proportion of 13% (3/23). In the lower panel (i.e. non-diseased individuals), 50 individuals were reclassified correctly down and 23 were reclassified incorrectly up with the net gain in reclassification proportion of 17.5% (27/154). Overall, the NRI was estimated as 30.5% (i.e. 13+17.5) and was highly significant (p-value=0.006), that is the new model including ENPP1 K121Q genotype correctly re-classifies in the correct risk group 30.5% individuals of the whole sample. This didactic example serves the important function to show how new statistical tools, focused on what a new marker can add to clinically-established models, may provide novel and possibly alternative information as compared to classical tools. In conclusion, although strengths and weaknesses of their application are still matter of ongoing research [45, 46], IDI and NRI now do belong to the standards for transparent reporting of genetic risk prediction studies [47].
Figure 1.
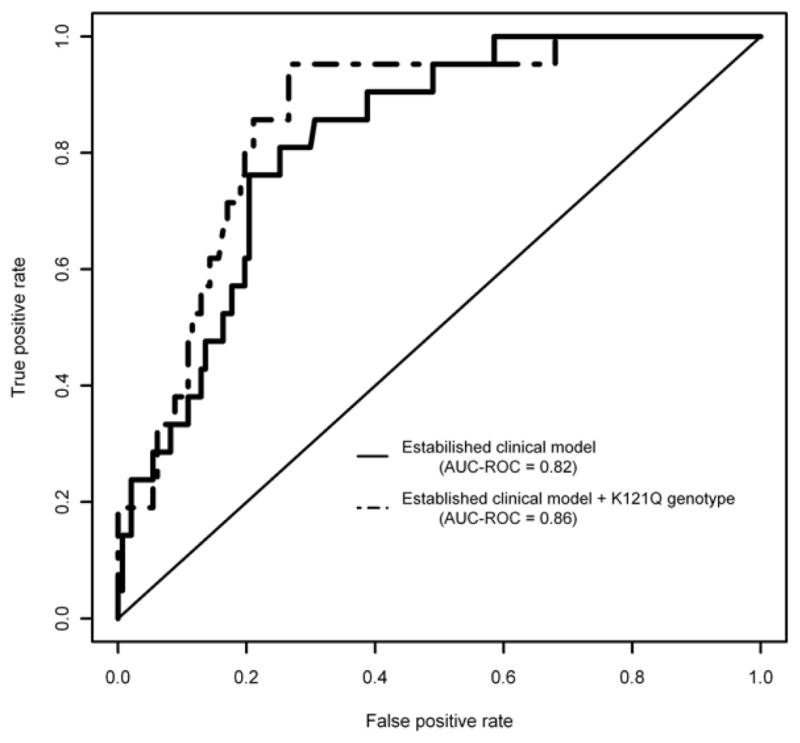
AUC-ROC curves for cardiovascular events probabilities estimated by a clinical model, including baseline patient’s age, gender, smoking status, BMI and hypertension and by the same model plus ENPP1 genotype.
Table 3.
Reclassification among people who experienced or not incident cardiovascular event during follow-up.
| Old model (without genetic information) | New model (with genetic information) | |||
|---|---|---|---|---|
| Frequency | Low risk | Medium risk | High risk | Total |
| Participants with event | ||||
| Low risk | 2 | 0 | 0 | 2 |
| Medium risk | 1 | 0 | 4 | 5 |
| High risk | 0 | 0 | 16 | 16 |
| Total | 3 | 0 | 20 | 23 |
| Participants without event | ||||
| Low risk | 55 | 8 | 0 | 63 |
| Medium risk | 38 | 2 | 15 | 55 |
| High risk | 3 | 9 | 24 | 36 |
| Total | 96 | 19 | 39 | 154 |
Light grey background cells represent patients who were reclassified correctly, while dark grey background cells represent patients who were reclassified incorrectly (see also the text for further details).
New technologies of genome sequencing
Beside the “common disease-common variants hypothesis”[48], the “common disease-multiple rare variants hypothesis”, in which few rare alleles are determining a large effect [2, 49], is also believed to underlie the genetic background of common diseases.
To capture such rare variants, it will be necessary to sequence entire genomes, instead of genotyping a catalogue of variants. Until recently, this was a daunting task.
The automated sequencing (also known as “automated Sanger sequencing”) had dominated the research in the genetic field for almost two decades, leading to a number of monumental accomplishments, including the completion of the only “finished-grade” human genome sequence [50]. However, the limitations of Sanger sequencing, due to the small size of fragments to be run beyond the specific sequencing primer and to the long time required to sequence discrete chromosomal regions, have called for new technologies able to massively sequence large numbers of human genomes in a reasonable time, referred to as Next- Generation Sequencing (NGS).
NGS technologies, which include a number of methods that are grouped broadly as template preparation, sequencing, imaging and, finally, data analysis, have the potential to discover the entire spectrum of sequence variations. The unique combination of specific protocols distinguishes one technology from another and determines the type of data produced from each platform [51].
Today’s commercially available platforms are typically able to generate tens or hundreds of Giga bases (i.e 1Gb=109 bases) of short reads sequence. As a result, the average NGS experiment generates terabytes (i.e 1Tbyte=~1012 bytes) of raw data, making data analysis and management particularly laborious. Given the vast amount of data produced by NGS, developing a massive data storage and management solution and creating informatics tools to effectively analyze data is essential to the successful application of this technology [51].
The ability to sequence the whole genome of several individuals, as for the “1000 Genomes Project” [52], in which whole genomes from 1,000 individuals of several ethnicities have been entirely sequenced have already revolutionized our knowledge in terms of numbers of genes or genomic regions actually known involved in the pathogenesis of several human diseases including both Mendelian and complex disease [53,54]. A major advance offered by NGS is the ability to produce an enormous volume of data at reasonable cost (~7,500 US$ for a whole genome; ~ 2,500 US$ for an exome).
NGS technologies are not restricted to the only DNA sequencing. A variety of additional application are actually available for the analysis of chromatin immunoprecipitation coupled to DNA microarray (ChIPchip) or sequencing (ChIP-seq), for RNA sequencing and quantification (RNAseq), for profiling of epigenetic marks, including analysis of methylation (methyl–seq and DNase–seq) [51].
Overall, there is no doubts that, thanks to NGS, the rapidly evolving field of genomics is dramatically changing our understanding of many diseases, by documenting the large pathogenic contribution of human genomic variations.
Ethical, legal and social issues
So far, we have provided clear evidences that genetic testing is not clinically useful for predicting T2DM and CAD in diabetic patients. Very similar conclusions can be drawn for most common diseases [55].
Nonetheless, as a likely consequence of the several Web-mediated commercial ventures, DTC genetic testing is becoming more and more familiar to patients and clinicians.
A critical appraisal of such commercial tests, based on predictive genomic profiling offered by seven companies, pointed out that there was “insufficient scientific evidence to conclude that genomic profiles are useful in measuring genetic risks for common diseases or in developing personalized diet and lifestyle recommendations for disease prevention” [55].
More recently, the United States Government Accountability Office (GAO) has evaluated the results of DTC genetic testing on five donors, including 10 tests, purchased from four companies. Fifteen common diseases that were tested, including CAD and T2DM. Two DNAs samples from each of five donors were sent to the four companies, one using factual information about the donor, and one using fictitious information, such as incorrect age and race or ethnicity. The GAO’s fictitious consumers received test results that were misleading and contradictory across the four companies and of little or no practical use. For example, one donor was told that he was below-average, average, and above average risk for prostate cancer and hypertension. In addition, DNA-based disease predictions conflicted with the actual medical conditions. For example, a patient who had a peacemaker implanted 13 years before to treat an irregular heartbeat, was defined at low risk for developing this condition. Finally, three companies failed to provide an expert advice. These observations suggested that although these tests show promise for the future, the consumers should not rely on any of these results at the present time [www.gao.gov/new.items/d10847t.pdf]. The Italian Network of Public Health Genomics has collected available knowledge on the potential use of genomics for preventing complex diseases and highlighted a number of principles that should direct the access to genomic information and services [http://istituti.unicatt.it/igiene_1830.html]. This document concluded that the predictive value of most markers identified so far is insufficient to meet the standards for clinical use.
Specific concerns on DTC genetic testing have been raised also by the European Society of Human Genetics [56] and by the Italian Committee for Bioethics and the National Committee for Bio-security, Biotechnology and Life Sciences (http://www.governo.it/biotecnologie/documenti/Test_genetici2.pdf) has reached similar conclusion on DTC. It is also of note that awareness of genetic risks may have negative social consequences, in terms of stigma, employment, insurance or other. Also personal consequences have to be taken into proper account. While is has been reported that genetic testing for common diseases would make patients much more motivated to adhere to medications [57], there are no data about the psychological harm that such tests may cause in both users and their relatives with increased asking for costly medical advices and laboratory testing. So, appropriate studies are definitively needed to assess both benefits and harms of such type of information [58].
Finally, is the general practitioner (GP) or the specialist who should help the direct consumer to manage such genetic information? Should the specialist be a non-genetic clinicians (i.e. diabetologist and/or cardiologist in our case) or a clinical geneticist? Are these clinicians trained to independently do their job or do they need to collaborate? In this respect, it is likely that genomic knowledge needs to be increased for all health professionals [59] with core competences being different for primary (GP), secondary (non genetic specialist) and tertiary (genetic specialist) care [60].
In conclusion, advances in the comprehension of the genetic background of common diseases have generated a number of ethical, legal and social implications which tend to be overlooked in the rush to have these genetic tests spread in the market.
Improved public and professional knowledge about genetics of common diseases definitively needs to be paralleled by adequate assessments of such implications if translation of genetic testing into clinical care with a most favorable benefits-to-harms ratio is our ultimate goal for the near future.
Perspective and conclusion
Available data clearly indicate that genetic testing offered by commercial companies cannot be used for predicting most common, multifactorial diseases, including T2DM and cardiovascular disease [55, 61]. This is mainly due to the fact that genetic data thus far obtained refer mostly to common variants with modest effect size in a scenario where a large proportion of disease risk is under the control of environmental factors. Further discovery of yet unidentified predisposing genetic factors might importantly change the present scenario. These new discoveries are likely to take place by the use of novel study designs and tools which are here briefly listed.
Firstly, when creating prediction models in which genetic factors are considered, it should be taken into account the complex gene-gene interaction that might occur in modulating common diseases and which has been, in fact, suggested for T2DM [7].
Secondly, the interaction between genetic and non genetic factors should be also taken into account. In fact, preliminary evidences point to age [10, 12], body mass index [7, 11, 44, 62, 63] and glycemic control [64] as potential modifiers of genetic effect on the risk of T2DM [7, 10–12, 62] and cardiovascular disease [44, 64].
Third, new statistical tools concentrated in reclassifying patients subgroup of patients might also play a role in rendering GWAS-derived genetic testing clinically useful for predicting many common, multifactorial diseases.
Finally, all the GWAS conducted to date have been focused on relatively common variants with minor allele frequency generally greater than 5%. The study of less frequent variants, such as those included in the recently released ExomeChip or identified by ad hoc re-sequencing efforts using NGS (see above), may lead to the identification of stronger genetic effects, which may have a larger aggregate impact on risk discrimination of both T2DM and cardiovascular disease than that offered by common variants.
By now, prediction of such diseases, including those of interest for the clinical diabetologist, have to be pursued by using traditional clinical markers which perform well and are not costly.
Acknowledgments
This study was supported in part by: Italian Ministry of Health, Ricerca Corrente 2011 and 2012 to SP, BD, FP and VT; Fondazione Roma, “Sostegno alla ricerca scientifica biomedica 2008” to VT; National Institute of Health, USA, grant HL073168 to AD.
Footnotes
Additional Information:
- The National Human Genome Research Institute (NHGRI) catalog of published genome-wide association studies http://www.genome.gov/gwastudies
- United States Government Accountability office. “Direct to consumer genetic test. Misleading test results are further complicated by deceptivefmarketing and other questionable practices”, GAO-10-847T, July 22, 2010 www.gao.gov/new.items/d10847t.pdf
- Network Italiano di Genomica in Sanità Pubblica: “La genomica in sanità pubblica” http://istituti.unicatt.it/igiene_1830.html
- Comitato Nazionale per la Bioetica, Comitato Nazionale per la Biosicurezza, le Biotecnologie e le Scienze della Vita: “Test genetici di suscettibilità e medicina personalizzata”, Presidenza del Consiglio dei Ministri, July 15 2010 http://www.governo.it/biotecnologie/documenti/Test_genetici2.pdf.
- 1000 Genomes: http://www.1000genomes.org
Conflict of interest
The authors declare no conflict of interest.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errorsmaybe discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Maher B. Personal genomes: The case of the missing heritability. Nature. 2008;456:18–21. doi: 10.1038/456018a. [DOI] [PubMed] [Google Scholar]
- 2.Manolio TA, Collins FS, Cox NJ, Goldstein DB, Hindorff LA, Hunter DJ, McCarthy MI, Ramos EM, Cardon LR, Chakravarti A, Cho JH, Guttmacher AE, Kong A, Kruglyak L, Mardis E, Rotimi CN, Slatkin M, Valle D, Whittemore AS, Boehnke M, Clark AG, Eichler EE, Gibson G, Haines JL, Mackay TF, McCarroll SA, Visscher PM. Finding the missing heritability of complex diseases. Nature. 2009;461:747–753. doi: 10.1038/nature08494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.McCarthy MI. Genomics, type 2 diabetes, and obesity. N Engl J Med. 363:2339–2350. doi: 10.1056/NEJMra0906948. [DOI] [PubMed] [Google Scholar]
- 4.Newman B, Selby JV, King MC, Slemenda C, Fabsitz R, Friedman GD. Concordance for type 2 (non-insulin-dependent) diabetes mellitus in male twins. Diabetologia. 1987;30:763–768. doi: 10.1007/BF00275741. [DOI] [PubMed] [Google Scholar]
- 5.Weijnen CF, Rich SS, Meigs JB, Krolewski AS, Warram JH. Risk of diabetes in siblings of index cases with Type 2 diabetes: implications for genetic studies. Diabet Med. 2002;19:41–50. doi: 10.1046/j.1464-5491.2002.00624.x. [DOI] [PubMed] [Google Scholar]
- 6.Almgren P, Lehtovirta M, Isomaa B, Sarelin L, Taskinen MR, Lyssenko V, Tuomi T, Groop L. Heritability and familiality of type 2 diabetes and related quantitative traits in the Botnia Study. Diabetologia. 2011;54:2811–2819. doi: 10.1007/s00125-011-2267-5. [DOI] [PubMed] [Google Scholar]
- 7.Cauchi S, Meyre D, Durand E, Proenca C, Marre M, Hadjadj S, Choquet H, De Graeve F, Gaget S, Allegaert F, Delplanque J, Permutt MA, Wasson J, Blech I, Charpentier G, Balkau B, Vergnaud AC, Czernichow S, Patsch W, Chikri M, Glaser B, Sladek R, Froguel P. Post genome-wide association studies of novel genes associated with type 2 diabetes show gene-gene interaction and high predictive value. PLoS One. 2008;3:e2031. doi: 10.1371/journal.pone.0002031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Pisabarro RE, Sanguinetti C, Stoll M, Prendez D. High incidence of type 2 diabetes in peroxisome proliferator-activated receptor gamma2 Pro12Ala carriers exposed to a high chronic intake of trans fatty acids and saturated fatty acids. Diabetes Care. 2004;27:2251–2252. doi: 10.2337/diacare.27.9.2251. [DOI] [PubMed] [Google Scholar]
- 9.Fisher E, Boeing H, Fritsche A, Doering F, Joost HG, Schulze MB. Whole-grain consumption and transcription factor-7-like 2 ( TCF7L2) rs7903146: gene-diet interaction in modulating type 2 diabetes risk. Br J Nutr. 2009;101:478–481. doi: 10.1017/S0007114508020369. [DOI] [PubMed] [Google Scholar]
- 10.Morini E, Prudente S, Succurro E, Chandalia M, Zhang YY, Mammarella S, Pellegrini F, Powers C, Proto V, Dallapiccola B, Cama A, Sesti G, Abate N, Doria A, Trischitta V. IRS1 G972R polymorphism and type 2 diabetes: a paradigm for the difficult ascertainment of the contribution to disease susceptibility of ‘low-frequency-low-risk’ variants. Diabetologia. 2009;52:1852–1857. doi: 10.1007/s00125-009-1426-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Prudente S, Morini E, Trischitta V. Insulin signaling regulating genes: effect on T2DM and cardiovascular risk. Nat Rev Endocrinol. 2009;5:682–693. doi: 10.1038/nrendo.2009.215. [DOI] [PubMed] [Google Scholar]
- 12.de Miguel-Yanes JM, Shrader P, Pencina MJ, Fox CS, Manning AK, Grant RW, Dupuis J, Florez JC, D’Agostino RB, Cupples LA, Meigs JB. Genetic risk reclassification for type 2 diabetes by age below or above 50 years using 40 type 2 diabetes risk single nucleotide polymorphisms. Diabetes Care. 2011;34:121–125. doi: 10.2337/dc10-1265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Giacco R, Della Pepa G, Luongo D, Riccardi G. Whole grain intake in relation to body weight: From epidemiological evidence to clinical trials. Nutr Metab Cardiovasc Dis. 2011;21:901–908. doi: 10.1016/j.numecd.2011.07.003. [DOI] [PubMed] [Google Scholar]
- 14.Lyssenko V, Jonsson A, Almgren P, Pulizzi N, Isomaa B, Tuomi T, Berglund G, Altshuler D, Nilsson P, Groop L. Clinical risk factors, DNA variants, and the development of type 2 diabetes. N Engl J Med. 2008;359:2220–2232. doi: 10.1056/NEJMoa0801869. [DOI] [PubMed] [Google Scholar]
- 15.Meigs JB, Shrader P, Sullivan LM, McAteer JB, Fox CS, Dupuis J, Manning AK, Florez JC, Wilson PW, D’Agostino RB, Cupples LA. Genotype score in addition to common risk factors for prediction of type 2 diabetes. N Engl J Med. 2008;359:2208–2219. doi: 10.1056/NEJMoa0804742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Vaxillaire M, Veslot J, Dina C, Proenca C, Cauchi S, Charpentier G, Tichet J, Fumeron F, Marre M, Meyre D, Balkau B, Froguel P. Impact of common type 2 diabetes risk polymorphisms in the DESIR prospective study. Diabetes. 2008;57:244–254. doi: 10.2337/db07-0615. [DOI] [PubMed] [Google Scholar]
- 17.van Hoek M, Dehghan A, Witteman JC, van Duijn CM, Uitterlinden AG, Oostra BA, Hofman A, Sijbrands EJ, Janssens AC. Predicting type 2 diabetes based on polymorphisms from genome-wide association studies: a population-based study. Diabetes. 2008;57:3122–3128. doi: 10.2337/db08-0425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Talmud PJ, Hingorani AD, Cooper JA, Marmot MG, Brunner EJ, Kumari M, Kivimaki M, Humphries SE. Utility of genetic and non-genetic risk factors in prediction of type 2 diabetes: Whitehall II prospective cohort study. BMJ. 2010;340:b4838. doi: 10.1136/bmj.b4838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lange LA, Bowden DW, Langefeld CD, Wagenknecht LE, Carr JJ, Rich SS, Riley WA, Freedman BI. Heritability of carotid artery intima-medial thickness in type 2 diabetes. Stroke. 2002;33:1876–1881. doi: 10.1161/01.str.0000019909.71547.aa. [DOI] [PubMed] [Google Scholar]
- 20.Marenberg ME, Risch N, Berkman LF, Floderus B, de Faire U. Genetic susceptibility to death from coronary heart disease in a study of twins. N Engl J Med. 1994;330:1041–1046. doi: 10.1056/NEJM199404143301503. [DOI] [PubMed] [Google Scholar]
- 21.Wagenknecht LE, Bowden DW, Carr JJ, Langefeld CD, Freedman BI, Rich SS. Familial aggregation of coronary artery calcium in families with type 2 diabetes. Diabetes. 2001;50:861–866. doi: 10.2337/diabetes.50.4.861. [DOI] [PubMed] [Google Scholar]
- 22.Rivellese AA, Piatti PM. Consensus on: Screening and therapy of coronary heart disease in diabetic patients. Nutr Metab Cardiovasc Dis. 2011;21:757–764. doi: 10.1016/j.numecd.2011.07.005. [DOI] [PubMed] [Google Scholar]
- 23.Samani NJ, Erdmann J, Hall AS, Hengstenberg C, Mangino M, Mayer B, Dixon RJ, Meitinger T, Braund P, Wichmann HE, Barrett JH, Konig IR, Stevens SE, Szymczak S, Tregouet DA, Iles MM, Pahlke F, Pollard H, Lieb W, Cambien F, Fischer M, Ouwehand W, Blankenberg S, Balmforth AJ, Baessler A, Ball SG, Strom TM, Braenne I, Gieger C, Deloukas P, Tobin MD, Ziegler A, Thompson JR, Schunkert H. Genomewide association analysis of coronary artery disease. N Engl J Med. 2007;357:443–453. doi: 10.1056/NEJMoa072366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.McPherson R, Pertsemlidis A, Kavaslar N, Stewart A, Roberts R, Cox DR, Hinds DA, Pennacchio LA, Tybjaerg-Hansen A, Folsom AR, Boerwinkle E, Hobbs HH, Cohen JC. A common allele on chromosome 9 associated with coronary heart disease. Science. 2007;316:1488–1491. doi: 10.1126/science.1142447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Erdmann J, Grosshennig A, Braund PS, Konig IR, Hengstenberg C, Hall AS, Linsel-Nitschke P, Kathiresan S, Wright B, Tregouet DA, Cambien F, Bruse P, Aherrahrou Z, Wagner AK, Stark K, Schwartz SM, Salomaa V, Elosua R, Melander O, Voight BF, O’Donnell CJ, Peltonen L, Siscovick DS, Altshuler D, Merlini PA, Peyvandi F, Bernardinelli L, Ardissino D, Schillert A, Blankenberg S, Zeller T, Wild P, Schwarz DF, Tiret L, Perret C, Schreiber S, El Mokhtari NE, Schafer A, Marz W, Renner W, Bugert P, Kluter H, Schrezenmeir J, Rubin D, Ball SG, Balmforth AJ, Wichmann HE, Meitinger T, Fischer M, Meisinger C, Baumert J, Peters A, Ouwehand WH, Deloukas P, Thompson JR, Ziegler A, Samani NJ, Schunkert H. New susceptibility locus for coronary artery disease on chromosome 3q22. 3. Nat Genet. 2009;41:280–282. doi: 10.1038/ng.307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Tregouet DA, Konig IR, Erdmann J, Munteanu A, Braund PS, Hall AS, Grosshennig A, Linsel-Nitschke P, Perret C, DeSuremain M, Meitinger T, Wright BJ, Preuss M, Balmforth AJ, Ball SG, Meisinger C, Germain C, Evans A, Arveiler D, Luc G, Ruidavets JB, Morrison C, van der Harst P, Schreiber S, Neureuther K, Schafer A, Bugert P, El Mokhtari NE, Schrezenmeir J, Stark K, Rubin D, Wichmann HE, Hengstenberg C, Ouwehand W, Ziegler A, Tiret L, Thompson JR, Cambien F, Schunkert H, Samani NJ. Genome-wide haplotype association study identifies the SLC22A3-LPAL2-LPA gene cluster as a risk locus for coronary artery disease. Nat Genet. 2009;41:283–285. doi: 10.1038/ng.314. [DOI] [PubMed] [Google Scholar]
- 27.Schunkert H, Konig IR, Kathiresan S, Reilly MP, Assimes TL, Holm H, Preuss M, Stewart AF, Barbalic M, Gieger C, Absher D, Aherrahrou Z, Allayee H, Altshuler D, Anand SS, Andersen K, Anderson JL, Ardissino D, Ball SG, Balmforth AJ, Barnes TA, Becker DM, Becker LC, Berger K, Bis JC, Boekholdt SM, Boerwinkle E, Braund PS, Brown MJ, Burnett MS, Buysschaert I, Carlquist JF, Chen L, Cichon S, Codd V, Davies RW, Dedoussis G, Dehghan A, Demissie S, Devaney JM, Diemert P, Do R, Doering A, Eifert S, Mokhtari NE, Ellis SG, Elosua R, Engert JC, Epstein SE, de Faire U, Fischer M, Folsom AR, Freyer J, Gigante B, Girelli D, Gretarsdottir S, Gudnason V, Gulcher JR, Halperin E, Hammond N, Hazen SL, Hofman A, Horne BD, Illig T, Iribarren C, Jones GT, Jukema JW, Kaiser MA, Kaplan LM, Kastelein JJ, Khaw KT, Knowles JW, Kolovou G, Kong A, Laaksonen R, Lambrechts D, Leander K, Lettre G, Li M, Lieb W, Loley C, Lotery AJ, Mannucci PM, Maouche S, Martinelli N, McKeown PP, Meisinger C, Meitinger T, Melander O, Merlini PA, Mooser V, Morgan T, Muhleisen TW, Muhlestein JB, Munzel T, Musunuru K, Nahrstaedt J, Nelson CP, Nothen MM, Olivieri O, Patel RS, Patterson CC, Peters A, Peyvandi F, Qu L, Quyyumi AA, Rader DJ, Rallidis LS, Rice C, Rosendaal FR, Rubin D, Salomaa V, Sampietro ML, Sandhu MS, Schadt E, Schafer A, Schillert A, Schreiber S, Schrezenmeir J, Schwartz SM, Siscovick DS, Sivananthan M, Sivapalaratnam S, Smith A, Smith TB, Snoep JD, Soranzo N, Spertus JA, Stark K, Stirrups K, Stoll M, Tang WH, Tennstedt S, Thorgeirsson G, Thorleifsson G, Tomaszewski M, Uitterlinden AG, van Rij AM, Voight BF, Wareham NJ, Wells GA, Wichmann HE, Wild PS, Willenborg C, Witteman JC, Wright BJ, Ye S, Zeller T, Ziegler A, Cambien F, Goodall AH, Cupples LA, Quertermous T, Marz W, Hengstenberg C, Blankenberg S, Ouwehand WH, Hall AS, Deloukas P, Thompson JR, Stefansson K, Roberts R, Thorsteinsdottir U, O’Donnell CJ, McPherson R, Erdmann J, Samani NJ. Large-scale association analysis identifies 13 new susceptibility loci for coronary artery disease. Nat Genet. 2011;43:333–338. doi: 10.1038/ng.784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Coronary Artery Disease (C4D) Genetics Consortium. A genome-wide association study in Europeans and South Asians identifies five new loci for coronary artery disease. Nat Genet. 2011;43:339–344. doi: 10.1038/ng.782. [DOI] [PubMed] [Google Scholar]
- 29.Qi L, Parast L, Cai T, Powers C, Gervino EV, Hauser TH, Hu FB, Doria A. Genetic susceptibility to coronary heart disease in type 2 diabetes 3 independent studies. J Am Coll Cardiol. 2011;58:2675–2682. doi: 10.1016/j.jacc.2011.08.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Stern MP. Diabetes and cardiovascular disease. The “common soil” hypothesis. Diabetes. 1995;44:369–374. doi: 10.2337/diab.44.4.369. [DOI] [PubMed] [Google Scholar]
- 31.Pfister R, Barnes D, Luben RN, Khaw KT, Wareham NJ, Langenberg C. Individual and cumulative effect of type 2 diabetes genetic susceptibility variants on risk of coronary heart disease. Diabetologia. 2011;54:2283–2287. doi: 10.1007/s00125-011-2206-5. [DOI] [PubMed] [Google Scholar]
- 32.Pizzuti A, Frittitta L, Argiolas A, Baratta R, Goldfine ID, Bozzali M, Ercolino T, Scarlato G, Iacoviello L, Vigneri R, Tassi V, Trischitta V. A polymorphism (K121Q) of the human glycoprotein PC-1 gene coding region is strongly associated with insulin resistance. Diabetes. 1999;48:1881–1884. doi: 10.2337/diabetes.48.9.1881. [DOI] [PubMed] [Google Scholar]
- 33.Hribal ML, Federici M, Porzio O, Lauro D, Borboni P, Accili D, Lauro R, Sesti G. The Gly-- >Arg972 amino acid polymorphism in insulin receptor substrate-1 affects glucose metabolism in skeletal muscle cells. J Clin Endocrinol Metab. 2000;85:2004–2013. doi: 10.1210/jcem.85.5.6608. [DOI] [PubMed] [Google Scholar]
- 34.Prudente S, Hribal ML, Flex E, Turchi F, Morini E, De Cosmo S, Bacci S, Tassi V, Cardellini M, Lauro R, Sesti G, Dallapiccola B, Trischitta V. The functional Q84R polymorphism of mammalian Tribbles homolog TRB3 is associated with insulin resistance and related cardiovascular risk in Caucasians from Italy. Diabetes. 2005;54:2807–2811. doi: 10.2337/diabetes.54.9.2807. [DOI] [PubMed] [Google Scholar]
- 35.Lappalainen T, Kolehmainen M, Schwab US, Tolppanen AM, Stancakova A, Lindstrom J, Eriksson JG, Keinanen-Kiukaanniemi S, Aunola S, Ilanne-Parikka P, Herder C, Koenig W, Gylling H, Kolb H, Tuomilehto J, Kuusisto J, Uusitupa M. Association of the FTO gene variant (rs9939609) with cardiovascular disease in men with abnormal glucose metabolism--the Finnish Diabetes Prevention Study. Nutr Metab Cardiovasc Dis. 2011;21:691–698. doi: 10.1016/j.numecd.2010.01.006. [DOI] [PubMed] [Google Scholar]
- 36.Ripatti S, Tikkanen E, Orho-Melander M, Havulinna AS, Silander K, Sharma A, Guiducci C, Perola M, Jula A, Sinisalo J, Lokki ML, Nieminen MS, Melander O, Salomaa V, Peltonen L, Kathiresan S. A multilocus genetic risk score for coronary heart disease: case-control and prospective cohort analyses. Lancet. 2010;376:1393–1400. doi: 10.1016/S0140-6736(10)61267-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Thomas D. Gene--environment-wide association studies: emerging approaches. Nat Rev Genet. 2010;11:259–272. doi: 10.1038/nrg2764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Mukherjee B, Ahn J, Gruber SB, Chatterjee N. Testing gene-environment interaction in large-scale case-control association studies: possible choices and comparisons. Am J Epidemiol. 2012;175:177–190. doi: 10.1093/aje/kwr367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Cornelis MC, Tchetgen EJ, Liang L, Qi L, Chatterjee N, Hu FB, Kraft P. Gene-environment interactions in genome-wide association studies: a comparative study of tests applied to empirical studies of type 2 diabetes. Am J Epidemiol. 2012;175:191–202. doi: 10.1093/aje/kwr368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Thomas DC, Lewinger JP, Murcray CE, Gauderman WJ. Invited commentary: GE-Whiz! Ratcheting gene-environment studies up to the whole genome and the whole exposome. Am J Epidemiol. 2012;175:203–207. doi: 10.1093/aje/kwr365. discussion 208–209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Mukherjee BAJ, Gruber SB, Chatterjee N. Response to Invited Commentary: Mukherjee et al. Respond to “GE-Whiz! Ratcheting Up Gene-Environment Studies”. Am J Epidemiol. 2012;175:208–209. [Google Scholar]
- 42.Cook NR. Use and misuse of the receiver operating characteristic curve in risk prediction. Circulation. 2007;115:928–935. doi: 10.1161/CIRCULATIONAHA.106.672402. [DOI] [PubMed] [Google Scholar]
- 43.Pencina MJ, D’Agostino RB, Sr, D’Agostino RB, Jr, Vasan RS. Evaluating the added predictive ability of a new marker: from area under the ROC curve to reclassification and beyond. Stat Med. 2008;27:157–172. doi: 10.1002/sim.2929. discussion 207–112. [DOI] [PubMed] [Google Scholar]
- 44.Bacci S, Rizza S, Prudente S, Spoto B, Powers C, Facciorusso A, Pacilli A, Lauro D, Testa A, Zhang YY, Di Stolfo G, Mallamaci F, Tripepi G, Xu R, Mangiacotti D, Aucella F, Lauro R, Gervino EV, Hauser TH, Copetti M, De Cosmo S, Pellegrini F, Zoccali C, Federici M, Doria A, Trischitta V. The ENPP1 Q121 variant predicts major cardiovascular events in high-risk individuals: evidence for interaction with obesity in diabetic patients. Diabetes. 2011;60:1000–1007. doi: 10.2337/db10-1300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Pencina MJ, D’Agostino RB, Sr, Steyerberg EW. Extensions of net reclassification improvement calculations to measure usefulness of new biomarkers. Stat Med. 2011;30:11–21. doi: 10.1002/sim.4085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Cook NR, Paynter NP. Comments on ‘Extensions of net reclassification improvement calculations to measure usefulness of new biomarkers. In: Pencina MJ, D’Agostino RB Sr, Steyerberg EW, editors. Stat Med. Vol. 31. 2012. pp. 93–95. [DOI] [PubMed] [Google Scholar]
- 47.Janssens AC, Ioannidis JP, van Duijn CM, Little J, Khoury MJ. Strengthening the reporting of genetic risk prediction studies: the GRIPS statement. Eur J Clin Invest. 2011;41:1004–1009. doi: 10.1111/j.1365-2362.2011.02494.x. [DOI] [PubMed] [Google Scholar]
- 48.Reich DE, Lander ES. On the allelic spectrum of human disease. Trends Genet. 2001;17:502–510. doi: 10.1016/s0168-9525(01)02410-6. [DOI] [PubMed] [Google Scholar]
- 49.Schork NJ, Murray SS, Frazer KA, Topol EJ. Common vs. rare allele hypotheses for complex diseases. Curr Opin Genet Dev. 2009;19:212–219. doi: 10.1016/j.gde.2009.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.International Human Genome Sequencing Consortium. Finishing the euchromatic sequence of the human genome. Nature. 2004;431:931–945. doi: 10.1038/nature03001. [DOI] [PubMed] [Google Scholar]
- 51.Metzker ML. Sequencing technologies - the next generation. Nat Rev Genet. 2010;11:31–46. doi: 10.1038/nrg2626. [DOI] [PubMed] [Google Scholar]
- 52.1000 genomes project Consortium. A map of human genome variation from population-scale sequencing. Nature. 2010;467:1061–1073. doi: 10.1038/nature09534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Bick D, Dimmock D. Whole exome and whole genome sequencing. Curr Opin Pediatr. 2011;23:594–600. doi: 10.1097/MOP.0b013e32834b20ec. [DOI] [PubMed] [Google Scholar]
- 54.Cirulli ET, Goldstein DB. Uncovering the roles of rare variants in common disease through whole-genome sequencing. Nat Rev Genet. 2010;11:415–425. doi: 10.1038/nrg2779. [DOI] [PubMed] [Google Scholar]
- 55.Janssens AC, Gwinn M, Bradley LA, Oostra BA, van Duijn CM, Khoury MJ. A critical appraisal of the scientific basis of commercial genomic profiles used to assess health risks and personalize health interventions. Am J Hum Genet. 2008;82:593–599. doi: 10.1016/j.ajhg.2007.12.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.European Society of Human Genetics. Statement of the ESHG on direct-to-consumer genetic testing for health-related purposes. Eur J Hum Genet. 2010;18:1271–1273. doi: 10.1038/ejhg.2010.129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Grant RW, Hivert M, Pandiscio JC, Florez JC, Nathan DM, Meigs JB. The clinical application of genetic testing in type 2 diabetes: a patient and physician survey. Diabetologia. 2009;52:2299–2305. doi: 10.1007/s00125-009-1512-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Ransohoff DF, Khoury MJ. Personal genomics: information can be harmful. Eur J Clin Invest. 2010;40:64–68. doi: 10.1111/j.1365-2362.2009.02232.x. [DOI] [PubMed] [Google Scholar]
- 59.Ormond KE, Wheeler MT, Hudgins L, Klein TE, Butte AJ, Altman RB, Ashley EA, Greely HT. Challenges in the clinical application of whole-genome sequencing. Lancet. 2010;375:1749–1751. doi: 10.1016/S0140-6736(10)60599-5. [DOI] [PubMed] [Google Scholar]
- 60.Skirton H, Lewis C, Kent A, Coviello DA. Genetic education and the challenge of genomic medicine: development of core competences to support preparation of health professionals in Europe. Eur J Hum Genet. 2010;18:972–977. doi: 10.1038/ejhg.2010.64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Janssens AC, van Duijn CM. Genome-based prediction of common diseases: advances and prospects. Hum Mol Genet. 2008;17:R166–173. doi: 10.1093/hmg/ddn250. [DOI] [PubMed] [Google Scholar]
- 62.Cornelis MC, Qi L, Zhang C, Kraft P, Manson J, Cai T, Hunter DJ, Hu FB. Joint effects of common genetic variants on the risk for type 2 diabetes in U.S. men and women of European ancestry. Ann Intern Med. 2009;150:541–550. doi: 10.7326/0003-4819-150-8-200904210-00008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Timpson NJ, Lindgren CM, Weedon MN, Randall J, Ouwehand WH, Strachan DP, Rayner NW, Walker M, Hitman GA, Doney AS, Palmer CN, Morris AD, Hattersley AT, Zeggini E, Frayling TM, McCarthy MI. Adiposity-related heterogeneity in patterns of type 2 diabetes susceptibility observed in genome-wide association data. Diabetes. 2009;58:505–510. doi: 10.2337/db08-0906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Doria A, Wojcik J, Xu R, Gervino EV, Hauser TH, Johnstone MT, Nolan D, Hu FB, Warram JH. Interaction between poor glycemic control and 9p21 locus on risk of coronary artery disease in type 2 diabetes. JAMA. 2008;300:2389–2397. doi: 10.1001/jama.2008.649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Kathiresan S, Voight BF, Purcell S, Musunuru K, Ardissino D, Mannucci PM, Anand S, Engert JC, Samani NJ, Schunkert H, Erdmann J, Reilly MP, Rader DJ, Morgan T, Spertus JA, Stoll M, Girelli D, McKeown PP, Patterson CC, Siscovick DS, O’Donnell CJ, Elosua R, Peltonen L, Salomaa V, Schwartz SM, Melander O, Altshuler D, Ardissino D, Merlini PA, Berzuini C, Bernardinelli L, Peyvandi F, Tubaro M, Celli P, Ferrario M, Fetiveau R, Marziliano N, Casari G, Galli M, Ribichini F, Rossi M, Bernardi F, Zonzin P, Piazza A, Mannucci PM, Schwartz SM, Siscovick DS, Yee J, Friedlander Y, Elosua R, Marrugat J, Lucas G, Subirana I, Sala J, Ramos R, Kathiresan S, Meigs JB, Williams G, Nathan DM, MacRae CA, O’Donnell CJ, Salomaa V, Havulinna AS, Peltonen L, Melander O, Berglund G, Voight BF, Kathiresan S, Hirschhorn JN, Asselta R, Duga S, Spreafico M, Musunuru K, Daly MJ, Purcell S, Voight BF, Purcell S, Nemesh J, Korn JM, McCarroll SA, Schwartz SM, Yee J, Kathiresan S, Lucas G, Subirana I, Elosua R, Surti A, Guiducci C, Gianniny L, Mirel D, Parkin M, Burtt N, Gabriel SB, Samani NJ, Thompson JR, Braund PS, Wright BJ, Balmforth AJ, Ball SG, Hall AS, Schunkert H, Erdmann J, Linsel-Nitschke P, Lieb W, Ziegler A, Konig I, Hengstenberg C, Fischer M, Stark K, Grosshennig A, Preuss M, Wichmann HE, Schreiber S, Schunkert H, Samani NJ, Erdmann J, Ouwehand W, Hengstenberg C, Deloukas P, Scholz M, Cambien F, Reilly MP, Li M, Chen Z, Wilensky R, Matthai W, Qasim A, Hakonarson HH, Devaney J, Burnett MS, Pichard AD, Kent KM, Satler L, Lindsay JM, Waksman R, Knouff CW, Waterworth DM, Walker MC, Mooser V, Epstein SE, Rader DJ, Scheffold T, Berger K, Stoll M, Huge A, Girelli D, Martinelli N, Olivieri O, Corrocher R, Morgan T, Spertus JA, McKeown P, Patterson CC, Schunkert H, Erdmann E, Linsel-Nitschke P, Lieb W, Ziegler A, Konig IR, Hengstenberg C, Fischer M, Stark K, Grosshennig A, Preuss M, Wichmann HE, Schreiber S, Holm H, Thorleifsson G, Thorsteinsdottir U, Stefansson K, Engert JC, Do R, Xie C, Anand S, Kathiresan S, Ardissino D, Mannucci PM, Siscovick D, O’Donnell CJ, Samani NJ, Melander O, Elosua R, Peltonen L, Salomaa V, Schwartz SM, Altshuler D. Genome-wide association of early-onset myocardial infarction with single nucleotide polymorphisms and copy number variants. Nat Genet. 2009;41:334–341. doi: 10.1038/ng.327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Helgadottir A, Thorleifsson G, Manolescu A, Gretarsdottir S, Blondal T, Jonasdottir A, Jonasdottir A, Sigurdsson A, Baker A, Palsson A, Masson G, Gudbjartsson DF, Magnusson KP, Andersen K, Levey AI, Backman VM, Matthiasdottir S, Jonsdottir T, Palsson S, Einarsdottir H, Gunnarsdottir S, Gylfason A, Vaccarino V, Hooper WC, Reilly MP, Granger CB, Austin H, Rader DJ, Shah SH, Quyyumi AA, Gulcher JR, Thorgeirsson G, Thorsteinsdottir U, Kong A, Stefansson K. A common variant on chromosome 9p21 affects the risk of myocardial infarction. Science. 2007;316:1491–1493. doi: 10.1126/science.1142842. [DOI] [PubMed] [Google Scholar]