

Abstract
Background
Homologous recombination is the key process that generates genetic diversity and drives evolution. SPO11 protein triggers recombination by introducing DNA double stranded breaks at discreet areas of the genome called recombination hotspots. The hotspot locations are largely determined by the DNA binding specificity of the PRDM9 protein in human, mice and most other mammals. In budding yeast Saccharomyces cerevisae, which lacks a Prdm9 gene, meiotic breaks are formed opportunistically in the regions of accessible chromatin, primarily at gene promoters. The genome-wide distribution of hotspots in this organism can be altered by tethering Spo11 protein to Gal4 recognition sequences in the strain expressing Spo11 attached to the DNA binding domain of the Gal4 transcription factor. To establish whether similar re-targeting of meiotic breaks can be achieved in PRDM9-containing organisms we have generated a Gal4BD-Spo11 mouse that expresses SPO11 protein joined to the DNA binding domain of yeast Gal4.
Results
We have mapped the genome-wide distribution of the recombination initiation sites in the Gal4BD-Spo11 mice. More than two hundred of the hotspots in these mice were novel and were likely defined by Gal4BD, as the Gal4 consensus motif was clustered around the centers in these hotspots. Surprisingly, meiotic DNA breaks in the Gal4BD-Spo11 mice were significantly depleted near the ends of chromosomes. The effect is particularly striking at the pseudoautosomal region of the X and Y chromosomes – normally the hottest region in the genome.
Conclusions
Our data suggest that specific, yet-unidentified factors influence the initiation of meiotic recombination at subtelomeric chromosomal regions.
Keywords: Homologous recombination, Recombination hotspots, SPO11, Subtelomeres, Double stranded DNA breaks
Background
Homologous recombination is initiated by the generation of DNA double stranded breaks (DSBs) by the SPO11 protein [1]. Subsequent repair of these breaks culminates in the formation of crossing overs (COs) between homologous chromosomes that, in turn, are required for faithful chromosomal segregation [2,3]. Failure to produce at least one CO per chromosome pair leads to meiotic arrest or to the formation of aneuploid gametes [4-6]. In mammalian males, the X and Y chromosomes face a particular challenge since they share only a very short homologous area called the pseudoautosomal region (PAR). Accordingly, numerical abnormalities of sex chromosomes collectively represent the most common human aneuploidies [6].
The SPO11 protein has two major isoforms that have different expression patterns [7-11]. The beta isoform appears early and maintains a relatively constant level throughout the first meiotic prophase [11]. This form is sufficient for the production of the majority of meiotic DSBs [12]. Expression of the alpha isoform in males predominantly occurs in late prophase, beginning in early pachynema [11], and lack of SPO11α correlates with a reduction in the number of late-forming DSBs in the PAR [12]. Since mice expressing only the SPO11β isoform are also deficient in X/Y synapsis SPO11α is thought to be specifically required for efficient recombination in the PAR [12].
The majority of meiotic DSBs are formed at discreet areas of the genome called recombination hotspots [13-16]. In mice and human the hotspot locations are determined by the sequence specificity of the DNA binding domain of the PRDM9 protein [17-19]. This domain is highly polymorphic with different Prdm9 alleles predicted to recognize dissimilar DNA sequences [18,20,21]. This leads to different hotspot locations in individuals carrying different Prdm9 alleles (reviewed in [22]). The only hotspots that are shared between mouse strains with different Prdm9 alleles, as well as with the Prdm9 knockout mouse are found in the PAR and in the adjacent area [19], suggesting the existence of a Prdm9-independent DSB pathway. The PAR contains a large (~40 Kb) cluster of overlapping hotspots that collectively represent the hottest area of DSB formation in the mouse [23]. Such extensive DSB formation in the PAR is likely important to ensure that PAR undergoes an obligatory CO in every meiosis [24]. Although supporting evidence is not yet available, the DSB targeting to PRDM9-dependent hotspots could be explained, at least in principle, by a physical interaction between the DSB machinery and the PRDM9 protein (see [19] for discussion). However, the mechanisms that target DSBs to the PAR are not understood.
In this study we generate a Gal4BD-Spo11 mouse carrying a hypomorphic Spo11 allele that is deficient in the formation of DSBs in the PAR. This deficiency does not represent a specific defect in the PRDM9-independent DSB pathway, because PRDM9-dependent hotspots are also depleted in the region adjacent to the PAR in these mice. Furthermore, DSB reduction is also apparent at the subtelomeric regions of other chromosomes as well. Our data suggest that specific factors influence early steps of homologous recombination in subtelomeric regions including the PAR, and that the Gal4BD-Spo11 mice have a specific defect that compromises the proper execution of the recombination program in these areas.
Results and discussion
Generation of the Gal4BD-Spo11 knock-in mouse
The yeast Gal4 transcription factor binds to the promoters of several S. cerevisiae GAL genes through its N-terminal DNA-binding domain (reviewed in [25]). This domain recognizes the CGGN11CCG consensus sequence and, when attached to the yeast Spo11 protein, it is able to tether Spo11 to Gal4 recognition sites leading to the formation of Gal4BD-Spo11 specific recombination hotspots [26-28]. To evaluate if such tethering is possible in mice we used gene targeting in embryonic stem cells to introduce the DNA fragment coding for the DNA binding domain of the yeast Gal4 protein upstream of the start codon of the mouse Spo11 gene (Additional file 1: Figure S1). The expression and correct splicing of the resulting gene was confirmed by sequencing PCR fragments generated from cDNA of the Gal4BD-Spo11 homozygous (Spo11Gal/Gal) mice. We confirmed that both major isoforms of SPO11 – alpha and beta – were transcribed (data not shown). Nevertheless, testes of the Spo11Gal/Gal mice were reduced in size and germ cells beyond the spermatocyte stage were absent (Figure 1A). Furthermore, no sperm was detected in the epididymus (Figure 1B) indicating that Spo11Gal/Gal males were infertile. Similarly, Spo11Gal/Gal ovaries were smaller than in wild type and showed a greatly diminished number of follicles (Figure 1C). Young Spo11Gal/Gal females did produce progeny, however no viable litters have been obtained from mice older than 6 months, suggesting a premature cessation of fertility. Females carrying only one copy of the Gal allele and one null copy of Spo11 (Spo11Gal/–) showed a further reduction in ovarian size, no visible follicles and an underdeveloped uterus, indicating that a single Gal4BD-Spo11 allele is insufficient to maintain fertility (Figure 1C and data not shown).
Figure 1.

The Gal4BD-Spo11 allele is insufficient to maintain normal fertility. H&E-stained histological sections are shown. “Gal” indicates the Gal4BD-Spo11 allele, “+” indicates the wild type allele and “–”indicates the null allele of the Spo11 gene. A. Testes. Note the abundance of spermatocytes, but lack of spermatids in the seminiferous tubules of Spo11Gal/Gal mice. B. Epididymus. Note the lack of spermatozoa in the Spo11Gal/Gal mice. C. Ovaries. A few follicles are evident in the Spo11Gal/Gal 3 month old females, but none in the Spo11Gal/- mice.
The number of meiotic DSBs is reduced in the Spo11Gal/Gal mice
The yeast counterpart of the chimeric GAL4BD-SPO11 gene complements the SPO11Δ mutation, indicating that the fusion Gal4BD-Spo11 protein is proficient at DSB formation [26]. To determine whether DSBs are formed in the Spo11Gal/Gal mice we immunostained chromosomal spreads prepared from spermatocytes (Figure 2) or oocytes (Figure 3) with antibodies to the DMC1 protein, known to bind to the single stranded DNA tails of DSBs [29,30]. Although DMC1 foci were observed, their number was significantly reduced compared to wild type. Precise counting of DSBs is complicated by the dynamic nature of DSB formation and repair. While one can estimate the steady state level of DMC1 foci on each chromosomal spread, the DSBs that have already been repaired and the DSBs that have not yet formed will be under-counted. This problem is particularly pronounced when mutants with delayed or defective DSB repair are compared to wild type. To eliminate the effect of possible timing differences in the formation and/or repair of DSBs between Spo11Gal/Gal and Spo11+/+ mice, we compared the number of DMC1 foci on a Hop2−/− genetic background, i.e., in Spo11Gal/GalHop2−/−versus Spo11+/+Hop2−/− cells. The HOP2 protein is directly involved in the repair of meiotic DSBs [31-33], accordingly, Hop2−/− mice undergo meiotic arrest at the stage when all DSBs are already produced but none are repaired [34]. Therefore, the analysis of the mutants on the Hop2−/− background will reduce counting bias resulting from DSB formation/repair dynamics. We found that the number of DMC1 foci in Spo11Gal/GalHop2−/− spermatocytes was approximately 2 fold lower than that in the Spo11+/+Hop2−/− spermatocytes (Figure 4). Moreover, in Spo11Gal/– spermatocytes, where one Gal and one null allele of Spo11 were present, the number of DMC1 foci was reduced even further – to approximately 1/6th of the wild type level (data not shown). The reduction of the DSB number in Spo11Gal/GalHop2−/− females was more profound than in males, with only 1/4th of the normal number of DMC1 foci being formed (Figure 4).
Figure 2.

The number of meiotic DSBs is reduced in Gal4BD-Spo11 males. Chromosomal spreads from earlier (zygotene) and later (pachytene) stage spermatocytes were immunostained with antibodies to the DMC1 protein (red) to indicate the appearance and repair of DSBs in wild type mice and mice carrying one or two Gal4BD-Spo11 alleles. Chromosome cores were stained with antibodies to the axial element component SCP3 (green), and progression of homologous synapsis was monitored with antibodies to the component of the central element of the synaptonemal complex SCP1 (white).
Figure 3.

The number of meiotic DSBs is reduced in Gal4BD-Spo11 females. Chromosomal spreads from E15.5 ovaries were immunostained with antibodies to the DMC1 (red) and SCP3 (green) proteins.
Figure 4.
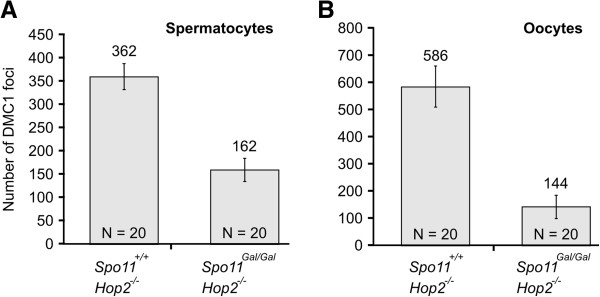
Estimate of the number of meiotic DSBs in Gal4BD-Spo11 mice. A. The number of DMC1 foci in spermatocytes. B. The number of DMC1 foci in oocytes. The number of analyzed cells is indicated within the bars. The foci were counted at late zygotene stage.
To understand what could be the reason for the reduced number of DSBs we examined the mRNA levels of the chimeric GAL4BD-SPO11 protein in testes and ovaries by quantitative PCR. Gal4BD-Spo11 male mice undergo meiotic arrest (Figure 1A and see below) and do not produce spermatids, which normally account for a large fraction of the testis germ cell population. Therefore, the cell type composition in testes of the Gal4BD-Spo11 and wild type mice is vastly different, creating a challenge for a meaningful comparison of gene expression levels. To minimize this difference we again employed Hop2−/− mice, which undergo meiotic arrest at a similar stage [34] as the Gal4BD-Spo11 mice. We found that the mRNA level of the GAL4BD-SPO11 protein in the Gal4BD-Spo11 mice is significantly lower than the mRNA level of the SPO11 protein in wild type or in Hop2−/− mice (Figure 5). More specifically, in Gal4BD-Spo11 males, the level of the beta isoform was reduced 3.3 fold compared to that in Hop2−/− mice, and the level of the alpha isoform was reduced 5.3-fold (Figure 5A). In Gal4BD-Spo11 females the expression of the beta isoform was reduced 4.5 fold compared to wild type mice and the expression of the alpha isoform was reduced 3.8 fold (Figure 5B). These data indicate that the mRNA level of the chimeric GAL4BD-SPO11 protein is profoundly reduced and that the observed reduction in the number of introduced DSBs could be, at least in part, a consequence of this reduction. It is also conceivable that the addition of the Gal4 binding domain to the mouse SPO11 protein might compromise the activity of SPO11, contributing to the reduced DSB count in Gal4BD-Spo11 mice.
Figure 5.
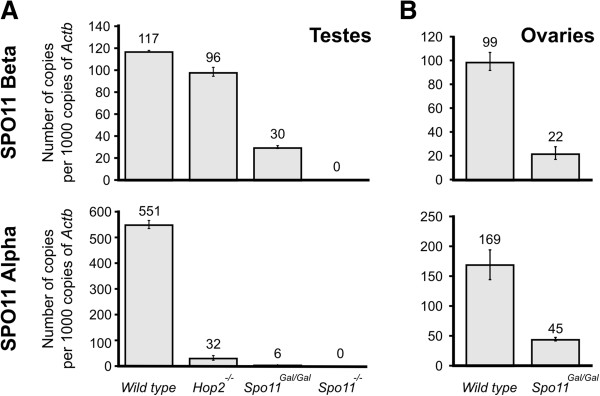
The level of Gal4BD-SPO11 mRNA is reduced compared to wild-type SPO11 mRNA levels. QPCR analysis of SPO11-β (top) and SPO11-α (bottom) mRNA levels in (A) testes and (B) ovaries. Note that in males the alpha isoform of SPO11 is expressed later than the beta isoform [11], resulting in drastic reduction of the alpha isoform mRNA levels in arrested Hop2−/− and Gal4BD-Spo11 spermatocytes.
Homologous chromosome synapsis in SpoGal/Gal mice is incomplete
The formation of meiotic DSBs is followed by rapid phosphorylation of histone H2AX [35]. During break repair homologous chromosomes are brought together and align throughout their entire length within a proteinaceous structure called the synaptonemal complex (SC) (reviewed by [36]). This coincides with the disappearance of the phosphorylated H2AX (γH2AX) and subsequent localization of the MLH1 protein to the sites of crossovers [37]. We analyzed homologous synapsis in Gal4BD-Spo11 spermatocytes by immunostaining the SCP1 protein, a component of the central element of the SC [38] (Figure 2). Although a number of chromosomes in Spo11Gal/Gal spermatocytes appeared properly synapsed, no cells had undergone complete synapsis. A large fraction of chromosomes synapsed only partially or formed branched structures indicative of non-homologous synapsis. γH2AX staining was lost from those chromosomes that had undergone complete synapsis, indicating successful DSB repair (Figure 6). Partially synapsed and branched chromosomes retained substantial γH2AX staining. Synaptic defects were even more prominent in the Spo11Gal/– spermatocytes, indicating that a further reduction in the number of DSBs further compromises homology search and homologous synapsis (Figures 2 and 6). Although some oocytes completed meiosis and resulted in a progeny, the majority showed defects in homologous synapsis. Nevertheless, unlike in spermatocytes (data not shown), MLH1 foci were evident in oocytes even when only a fraction of homologous chromosomes were successfully synapsed (Figure 7).
Figure 6.

DSB repair in Gal4BD-Spo11 spermatocytes is incomplete. Chromosomal spreads from earlier (zygotene) and later (pachytene) stage spermatocytes were immunostained with antibodies to γH2AX (red) and SCP3 (green). Note the disappearance of γH2AX staining at homologously synapsed autosomes in later stage wild type and Spo11Gal/Gal spermatocytes consistent with proper repair of DSBs. γH2AX persist in Spo11Gal/- spermatocytes and on asynapsed chromosomes in Spo11Gal/Gal spermatocytes. Sex body (white arrow) is evident in wild type, but not Gal4BD-Spo11 spermatocytes.
Figure 7.

MLH1 foci are formed in a fraction of Spo11Gal/Galoocytes. Chromosomal spreads from E18.5 ovaries were immunostained with antibodies to MLH1 (red) and SCP3 (green). Note incomplete synapsis and consequent reduction in the number of MLH1 foci in Spo11Gal/Gal oocytes.
The X and Y chromosomes synapse and recombine at the short region of homology within the PAR [24]. Unsynapsed parts of X and Y trigger transcriptional silencing of sex chromosomes associated with their assembly into a specialized chromatin domain called the sex or XY body (reviewed by [39-41]). At this stage the X/Y chromatin is decorated by a number of proteins including γH2AX. We found that a sex body was not assembled in the Spo11Gal/Gal spermatocytes, as no area of dense and discrete γH2AX staining was observed (Figure 6). The failure of sex body formation and impairment of transcriptional silencing is common in recombination mutants with compromised DSB repair and homologous synapsis (reviewed by [42]). Transcriptional silencing of sex chromosomes is required for meiotic progression beyond the pachytene stage in males [42,43], potentially explaining the dimorphic phenotype (sterility of males and fertility or subfertility of females) in a number of mouse mutants with mild to moderate synaptic defects; e.g., Sycp3−/−[44,45], H2AX−/−[46], Sycp2−/−[47], Dmc1Mei11/+[48], Brca1Δ11/Δ11p53+/−[49], Spo11β-only[12]. Similar mechanisms are likely to be the reason for the dimorphic phenotype observed in Gal4BD-Spo11 mice.
The mouse GAL4BD-SPO11 fusion protein can target meiotic DSBs to Gal4 consensus binding sites
We have mapped the distribution of DSBs in the Gal4BD-Spo11 mouse using anti-DMC1 chromatin immunoprecipitation (ChIP) followed by high throughput sequencing [19,50]. To increase the sensitivity of the hotspot detection we have also mapped DSBs in the Gal4BD-Spo11 mice on the Hop2 knockout background (SpoGal/GalHop2−/−). This allowed identification of 13,445 hotspots. 98% of the hotspots detected in Spo11Gal/Gal were also present in Spo11Gal/GalHop2−/− mice, reaffirming that the Hop2 mutation does not affect the distribution of meiotic DSBs genome-wide [23]. We have compared the distribution of Gal4BD-Spo11 hotspots to hotspots in wild type mice [19] and to those in Hop2 knockout mice. We found that 97.7% of the top 10,000 Gal4BD-Spo11 hotspots correspond to wild type hotspots. When all 13,445 Gal4BD-Spo11 hotspots are considered the overlap between Gal4BD-Spo11 and wild type hotspots is 94.7%. Therefore, the overall distribution of recombination hotspots in Gal4BD-Spo11 mice is not affected. The relative strength of the hotspots shared between wild type and Gal4BD-Spo11 mice is also highly correlated (Additional file 2: Figure S2).
There are 81,710 Gal4 consensus CGGN11CCG sequences present in the mouse genome. Approximately 2% of DSB hotspots overlapped these sites in either wild type or Hop2−/− mice (Table 1). However, when the Spo11Gal/GalHop2−/− mice were examined, the number of consensus-bearing hotspots increased to ~4%. These extra hotspots are weak, and although some of them are also apparent in the Spo11Gal/Gal mice upon visual examination, they were beyond the detection threshold in this sample (Table 1). Out of 546 Gal4BD-Spo11 hotspots with a Gal4 consensus site 292 were also present in wild type mice (Figure 8). A prominent peak in the distribution of wild type hotspot motif [19] was found at the center of these hotspots, whereas the distribution of the Gal4 consensus appeared random. The remaining 254 Gal4BD-Spo11 hotspots with a Gal4 consensus were Gal4BD-Spo11-specific. We found that the Gal4 recognition sequence rather than the hotspot consensus motif is enriched at the centers of these hotspots. Furthermore, only 3% of these Gal4BD-Spo11-specific hotspots overlapped PRDM9-dependent histone H3 lysine 4 trimethylation marks (H3K4me3). In aggregate, these data indicate that a small fraction of DSBs in the Gal4BD-Spo11 mice are likely targeted through a PRDM9-independent mechanism by tethering of the GAL4BD-SPO11 protein to Gal4 binding sites. In addition to the H3K4me3 introduced by the PRDM9 protein, gene promoters and enhancers are also decorated with H3K4me3 [51-53]. We have previously found that such sites represent preferred DSB formation loci in mice lacking Prdm9[19]. We now show that 84% of Gal4BD-Spo11-specific hotspots overlap transcription start sites. Importantly, the vast majority (96%) of Gal4BD-Spo11-specific hotspots overlap H3K4me3, indicating that H3K4me3 marks or/and events preceding H3K4 trimethylation are still essential when DSB targeting is GAL4-mediated.
Table 1.
Gal4BD-SPO11 directs a population of hotspots to the sites with Gal4 consensus motif
| Sample | Sequenced ssDNA tags, ×106 | Identified hotspots | Hotspots with Gal4 consensus | Hotspots present in the wild type mice | ||
|---|---|---|---|---|---|---|
| Wild type |
49.4 |
18,313 |
416 |
(2.3%) |
18,313 |
(100%) |
|
Hop2−/− |
3.4 |
8,221 |
204 |
(2.5%) |
8,036 |
(97.7%) |
|
Spo11Gal/Gal |
17.5 |
10,313 |
224 |
(2.2%) |
10,086 |
(97.8%) |
| Spo11Gal/GalHop2−/− | 46.3 | 13,445 | 546 | (4.1%) | 12,730 | (94.7%) |
The genotype of mouse strains is listed in the left column. Sequenced ssDNA tags indicate the number of ssDNA-derived fragments (e.g., those derived from ssDNA tails of DSBs) that have been sequenced for this analysis. The number of hotspots identified in each mouse strain is also indicated (column 3) and the overlap of these hotspots with the hotspots found in the wild type mice (“wild type” sample) is specified (column 5). The number of hotspots that contain Gal4 consensus motif (column 4, left) and the percentage of Gal4-containing hotspots in the total number of hotspots identified in the corresponding samples (column 4, right) are shown to demonstrate the increase in the Gal4-containing hotspots in the Spo11Gal/GalHop2−/− mice (see text for more explanation).
Figure 8.

A subset of Gal4BD-Spo11 hotspots is targeted to the Gal4 consensus.Gal4BD-Spo11 hotspots containing a match to the Gal4 DNA binding consensus were sub-divided into those found in the wild type strain (C57Bl/6) and those absent in the wild type. The coverage of the Gal4 consensus and of the previously identified C57Bl/6 hotspot consensus motif were calculated around the hotspot centers.
Gal4BD-Spo11 mice lack a DSB hotspot cluster in the PAR
We have previously demonstrated that the PAR carries a large cluster of overlapping DSB hotspots [23] that are most likely required to ensure an obligate DSB (and CO) in the PAR. Although individual hotspots cannot be resolved within the hotspot cluster it is clear that a large fraction, if not all, PAR hotspots are fundamentally different from those in the rest of the genome, because their formation does not depend on PRDM9 [19]. The formation of DSBs in the PAR has been reported to occur in two rounds: one (early) set is introduced at the same time as the majority of the autosomal breaks and the second one is introduced at a relatively late stage, presumably, by the alpha isoform of the SPO11 protein [12]. We found that Gal4BD-Spo11 mice are severely deficient in DSB formation in the PAR (Figure 9). The contribution of the DSBs in the PAR hotspot cluster to the total number of DSBs introduced genome-wide in Hop2−/− mice is estimated as 0.33%, but drops over 30-fold (to 0.01%) in the Spo11Gal/GalHop2−/− (data not shown). Although this defect may be explained to some extent by the extremely low expression of the SPO11 alpha isoform in the mutant, the complete lack of SPO11 alpha has been reported to result in only a 3-fold reduction of the DSBs in the PAR [12]. Reduced expression of the beta isoform in the Gal4BD-Spo11 mice could be another reason for the DSB reduction. However, the overall number of DSBs in the GAL4BD-Spo11 mice (presumably, introduced by the beta isoform) is reduced only 2.2-fold. It is therefore likely that, in addition to reduced protein level, a specific defect of the GAL4BD-SPO11 protein contributes to the drastic reduction of the DSBs in the PAR.
Figure 9.
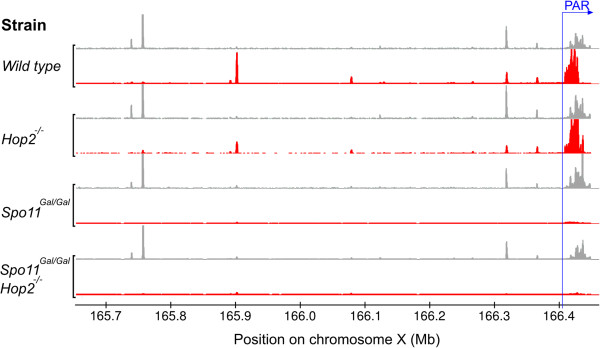
Gal4BD-Spo11 mice lack DSB hotspot cluster in the PAR. Red tracks represent the DSB coverage (ssDNA fragments). Grey tracks represent the H3K4me3 coverage (tags).
Gal4BD-Spo11 mice exhibit a deficiency of DSBs at chromosomal ends
The PAR is located at the end of the X and Y chromosomes. We therefore examined whether DSB formation in Gal4BD-Spo11 mice is also affected at the ends of autosomes. Indeed, we found that the strength of DSB hotspots near chromosome ends is significantly reduced in Spo11Gal/Gal and Spo11Gal/GalHop2−/−mice (Figure 10), and as a consequence, many subtelomeric hotspots that are present in wild-type mice are not detected. This phenomenon was not observed in Hop2 knockout mice indicating that the effect is not related to the meiotic arrest in Gal4BD-Spo11 spermatocytes.
Figure 10.
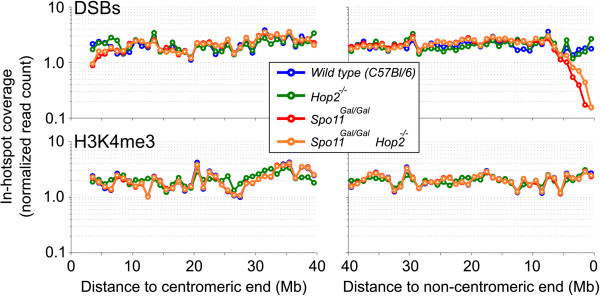
DSBs are depleted in the autosomal subtelomeric regions of Gal4BD-Spo11 mice. The coverage of DSBs (top panels) and H3K4me3 (bottom panels) in the regions corresponding to the wild type hotspots was calculated in 1 Mb windows across all autosomes. Values were normalized as a percentage of total in-hotspot coverage. DSB depletion is evident at the non-centromeric (right) chromosomal ends in Gal4BD-Spo11 mice. PRDM9-dependent H3K4me3 coverage in these regions does not change from the chromosomal average. Note that the sequence of the first 3 Mb of centromeric ends (left) of mouse chromosomes is not available.
We propose that specific suppression of homologous recombination at subtelomeres of the Gal4BD-Spo11 mice indicates the potential difference in the DSBs formed in these areas compared to DSBs in the rest of the genome. For example, it is conceivable that DSB formation close to chromosome ends is influenced by telomere attachment to the nuclear periphery [54] or by a specific chromosome organization and chromatin structure in the telomere-adjacent regions. Specific interactions may occur in these areas between the DSB machinery and proteins enriched at telomeres and/or subtelomeres. In fact, one such protein has recently been implicated in the regulation of transcription [55]. Incidentally, SPO11 itself was reported to bind to the telomeres of mouse embryonic stem cells [56]. It is therefore possible that the GAL4BD-SPO11 protein alone or as a component of the DSB formation complex is deficient in overcoming the inhibitory effects of the specific chromatin environment of subtelomeric regions. Such a deficiency can result from (i) the inability of GAL4BD-SPO11 to interact with a particular protein or protein complex, (ii) attenuated GAL4BD-SPO11 DNA binding in the context of such a chromatin environment or even (iii) a defect in GAL4BD-SPO11 removal from DNA in these regions that would prevent processing of meiotic DSB ends and loading RAD51 and DMC1. It is important to note that PRDM9-dependent H3K4me3 is introduced normally in hotspot-depleted regions of Gal4BD-Spo11 subtelomeres (Figure 10), therefore PRDM9 accessibility to these areas is not an issue.
Conclusions
Although the pivotal role of PRDM9 in defining the positions of individual recombination hotspots is established, a large number of potential PRDM9 binding sites in the genome are not being utilized. It is not clear what makes such sites refractory to the DSB machinery, but higher order chromatin structure and chromosome organization within the nucleus are likely to play a role. These features might provide slowly evolving physical constraints, which, according to a two-stage model of recombination initiation [57], ensure relatively similar recombination frequencies over large chromosomal domains between individuals in spite of Prdm9-dependent variability at individual hotspots within such domains. The Gal4BD-Spo11 mutant shows a domain-specific defect in initiation of homologous recombination without visible changes in the rest of the genome. This model will be instrumental in dissecting specific interactions occurring between subtelomeric chromatin and the DSB machinery and in providing molecular insights into the megabase-scale control of initiation of homologous recombination.
Methods
Mouse strains
All animal procedures have been approved by the USUHS Animal Care and Use Committee or were performed according to NIH Guide for the Care and Use of Laboratory Animals.
Gal4BD-Spo11 mice were generated on a C57Bl/6 background. The targeting construct was based on the pLoxpNEO vector [58] (a gift from Dr. Chu-Xia Deng, NIH). The Spo11 gene was obtained from the BAC clone RP23-52C7 (CHORI). A 4.3 Kb HincII/SmaI fragment of the 5′ region of the mouse Spo11 gene including the first exon was subcloned, and the DNA corresponding to the Gal DNA binding domain (from pGBKT7 plasmid, Clontech) was inserted upstream of the start codon of the Spo11 gene. The resulting fragment was then cloned into the pLoxpNEO vector upstream of the NEO gene cassette (Additional file 1: Figure S1A). A 3.8 Kb XmaI/XmaI fragment containing exon II of the Spo11 gene was cloned between the NEO and TK genes. Gene targeting and blastocyst injections were performed by Xenogen Biosciences Corporation (Caliper Life Sciences). Confirmation of targeting was done by Southern blot analysis (Additional file 1: Figure S1B and S1C). To excise the NEO targeting cassette Gal4BD-Spo11 heterozygous mice were crossed with a CRE-expressing mice (Jackson Laboratories stock # 003724) and the excision was confirmed by PCR. Expression of the correctly spliced Gal4BD-Spo11α and Gal4BD-Spo11β isoforms have been confirmed by RT PCR followed by sequencing. Gal4BD-Spo11 mice were genotyped by PCR using the following primers: Gal/dir: CTCAGAGCGGCTCCGCATCC; Gal/rev: GGCGCCACGAGGAACCTTCC. Spo11−/− mice (strain deltaSpo.BC/B6) have been produced by the targeted deletion of exons 2–6 of the Spo11 gene resulting in the absence of the SPO11 protein (Romanienko and Camerini-Otero, unpublished). The phenotype of this knockout strain is identical to the phenotype of the previously described Spo11−/− strain Spo11tm1Rdco[8]. Hop2 knockout mice have been previously described [34]. C57Bl/6 was used as a wild type strain and all mutant strains were on a C57Bl/6 background. Adult (2–6 month old) mice were used for the analyses.
Histology
Testes or ovaries of adult animals were dissected in PBS solution and placed in 10% formalin. Tissue sections and hematoxylin/eosin staining was performed by American Histo Labs Inc.
Quantitative PCR
Total RNA was extracted using RNeasy plus mini kit (Qiagen) and the cDNA was prepared using the Trancriptor First Strand cDNA Synthesis Kit (Roche). Quantitative PCR was performed using a Maxima SYBR Green/Rox Kit (Fermentas) according to the manufacturer’s instructions for a 7500 Real-Time PCR system. Gene copy number was calculated with ABI SDS Software. PCR amplification of the coding region of β actin gene was used for normalization. The following primer pairs have been used: Spo11 beta: CTCTAGTTCTGAGGTTCTTACAGCT, GGACAATACTTTCAGAATCAGAGCG; Spo11 alpha: GCGTGGCCTCTAGGTTTGATGATT, TCATCGATGGCGCTGTCCAC; ActB: CCAACTGGGACGACATGGAG, CCAACTGGGACGACATGGAG.
Antibodies
Rabbit anti-DMC1 antibodies to a full-length protein were generated by New England Peptide and affinity purified. Mouse anti-SCP3 antibodies were previously described [34]. Rabbit anti-SCP1 and anti-SCP3 antibodies were a gift from C. Hoog. The following commercial antibodies have been used: Anti-H3K4me3, Milipore (07–473); anti-γH2AX, Trevigen (4411-PC-100); mouse anti-MLH1, BD Biosciences (551092); rabbit anti-RAD51 and goat anti-DMC1, Santa Cruz (H-92 sc-8349 and C-20 sc-8973, respectively). Secondary antibodies were from Jackson IR Laboratories.
Meiotic chromosome spreads
Spermatocytes: Seminiferous tubules were chopped in RPMI 1640 high-glucose media (GIBCO-BRL). The cells were released from the tubules by pipetting and filtered through a 40 μm cell strainer (Falcon). The cells were pelleted and washed with RPMI. The resulting pellet was resuspended in 0.5% NaCl, added to the glass slides, and allowed to adhere for 10–15 min. The slides were fixed in 2% paraformaldehyde with 0.03% SDS for 3 min, 2% paraformaldehyde for 3 min, washed 3 times in 0.4% Photo-Flo 200 (Kodak) for 1 min, and air dried.
Oocytes: Ovaries were dissected in PBS at E15.5 for DMC1 staining and at E18.5 for MLH1 staining. Ovaries were placed in 20 μl of 100 mM sucrose, disrupted with tweezers and pipetted up and down until cell suspension was formed. The cells were added to 100 μl of 1% paraformaldehyde, 0.1% Triton X-100 solution that was spread over the slide. Slides were kept 2-4 h in humidified chamber at room temperature, then air dried. After four 1 min washes in 0.4% Kodak Photo Flo the slides were air dried again.
Immunofluorescence
The slides were incubated with blocking solution (1% donkey serum, 0.3% BSA, 0.005% Triton X-100 in PBS) for 20 minutes at 37°C in a humidity chamber. Primary antibodies were diluted in blocking buffer and incubated under the same conditions for 1–2 hr. After two 5 min washes in 0.4% Photo-Flo/PBS solution, slides were blocked for an additional 5 min and incubated with secondary antibodies for 20 min at room temperature. The slides were washed twice with 0.4% Photo-Flo in PBS, rinsed twice with 0.4% Photo-Flo, and allowed to air dry.
Chromatin immunoprecipitation and high throughput sequencing
Chromatin immunoprecipitation was done as previously described [19,23] with small modifications. Testes were fixed for 10 min in 1% formaldehyde. After quenching the tissue was homogenised, filtered through 40 μm cell strainer, and washed in the following buffers: 1) PBS (twice); 2) 0.25% Triton X-100, 10 mM EDTA, 0.5 mM EGTA, 10 mM Tris pH8; 3) 0.2 M NaCl, 1 mM EDTA, 0.5 mM EGTA, 10 mM Tris pH8. Cells were lysed in 1.5 ml of the lysis buffer (1% SDS, 10 mM EDTA, 50 mM TrisCl pH8 with complete protein inhibitor cocktail (Roche) and the chromatin was sheared to ~1000 bp by sonication. The sample was dialyzed against ChIP buffer (0.01% SDS, 1.1% Triton X-100, 1.2 mM EDTA, 16.7 mM TrisHCl, 167 mM NaCl). Chromatin was incubated with appropriate antibodies overnight at 4°C followed by 2 h incubation with Protein G beads. Beads were washed in the following buffers: 1) 0.1% SDS, 1% Triton X-100, 2 mM EDTA, 20 mM TrisHCl, 150 mM NaCl; 2) 0.1% SDS, 1% Triton X-100, 2 mM EDTA, 20 mM TrisCl pH8, 500 mM NaCl; 3) 0.25 M LiCl, 1% Igepal, 1 mM EDTA, 10 mM TrisCl, pH8, 1% Deoxycholic acid; 4) TE (twice). The chromatin was eluted by 1% SDS, 0.1 M NaHCO3 pH9 at 65°C and crosslinking was reversed at 65°C overnight. DNA was deproteinized for 2 h at 45°C and DNA was purified with a MinElute Reaction Clean up kit (QIAGEN).
Sequencing library construction was done according to the SSDS protocol that was previously described [19,50]. Sequencing was performed on an Illumina HiSeq 2000 using the standard paired-end cluster generation kit and sequencing reagents.
Samples (Table 2)
Table 2.
The list of samples used in this study
| Mouse strain | Replicate | Antibody | Method | Accession # |
|---|---|---|---|---|
|
Spo11Gal/Gal |
1 |
Dmc1 |
SSDS |
GSM1179920 |
|
Spo11Gal/Gal |
2 |
Dmc1 |
SSDS |
GSM1179921 |
|
Spo11Gal/- |
1 |
Dmc1 |
SSDS |
GSM1179919 |
|
Spo11Gal/GalHop2−/− |
1 |
Dmc1 |
SSDS |
GSM1179922 |
|
Hop2−/− |
1 |
Dmc1 |
SSDS |
GSM1179917 |
|
Hop2−/− |
2 |
Dmc1 |
SSDS |
GSM1179918 |
|
Wild type (C57Bl/6) |
* |
Dmc1 |
SSDS |
GSM869781*, GSM869782* |
|
Spo11Gal/Gal |
1 |
H3K4me3 |
ChIP-Seq |
GSM1179925 |
|
Spo11Gal/GalHop2−/− |
1 |
H3K4me3 |
ChIP-Seq |
GSM1179926 |
| Hop2−/− | 1 | H3K4me3 | ChIP-Seq | GSM1179924 |
* Published data [19].
Hotspot identification and peak calling
We have previously shown that specific usage of ssDNA increases DSB detection sensitivity [50]. For all SSDS samples, the computational pipeline described in the aforementioned work was used to align paired-end reads to the mouse reference genome (mm9) and to identify ssDNAs. We subsequently discarded ssDNA fragments where either the first or second end read had a quality score < 30. We also retained only a maximum of 20 duplicate ssDNA fragments at any locus. Data for replicates were pooled and DSB hotspots were identified using the peak calling method described in [19]. H3K4me3 reads were aligned to the mouse mm9 genome using CASAVA 1.8. H3K4me3 peaks were called using MACS 1.3.7 and the parameters described in [19].
The effect of using different overlap windows between hotspots has been described in [19]. Overlaps between hotspots were limited to the central 400 nt. Overlaps between hotspots and H3K4me3 were limited to the central 1 Kb. Overlaps between hotspots and Gal4 binding consensus were limited to the central 2 Kb.
Motif search
GAL4 binding sites were identified using an exhaustive genomic search for the consensus CGGN11CCG. No mismatches were permitted. Sites matching the putative C57Bl/6 PRDM9 binding site were identified with MAST, using standard parameters.
Availability of supporting data
All sequencing data for this study are publicly available and have been deposited in National Center for Biotechnology Information Gene Expression Omnibus under accession number GSE48493.
Abbreviations
DSB: Double stranded DNA breaks; CO: Crossing overs; PAR: Pseudoautosomal region.
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
FS and YP carried out most of the experiments, KB performed the computational analysis, US made the targeting vector. All authors participated in the design of the study and editing of the manuscript. RDCO and GVP supervised the study. All authors read and approved the final manuscript.
Supplementary Material
The generation of the Gal4BD-Spo11 knock-in mouse. A. Schematic of the targeting vector. Nucleotide coordinates correspond to nucleotide #1 being located at the position -7,000 upstream of the first exon of the Spo11 gene. B. Schematic of the Southern blot strategy. Genomic DNA was cut with either NdeI+HpaI or ApaLI and hybridized with 5’ or 3’ probes, respectively. C. Southern blots for 5’ (left) and 3’ (right) homology arms. The genotype of mice (lanes 1-3) and ES clones (lanes 4-6) is indicated.
The strength of Gal4BD-Spo11 hotspots correlates with the strength of hotspots in wild type mice. The number of ssDNA fragments in wild type (C57Bl/6) hotspots was calculated for each dataset. Density scatter plots are shown for all hotspots. Log(strength) is shown on the y-axes. The Spearman Correlation Coefficient is also shown between all samples (inset). Over 94% of Gal4BD-Spo11 Hop2-/- hotspots corresponded to with type hotspots.
Contributor Information
Fatima Smagulova, Email: fatima.smagulova@univ-rennes1.fr.
Kevin Brick, Email: brickkm@niddk.nih.gov.
Yongmei Pu, Email: yongmei.pu@usuhs.edu.
Uttara Sengupta, Email: uttara.sengupta@gmail.com.
R Daniel Camerini-Otero, Email: rdcamerini@niddk.nih.gov.
Galina V Petukhova, Email: Galina.Petukhova@usuhs.edu.
Acknowledgements
The authors would like to thank Pavel Khil for helpful discussions. This research was supported by the NIH grant 1R01GM084104-01A1 from NIGMS (G.V.P.); grants FS71HU, R071HU and CS71HU from USUHS (G.V.P.) and by the NIDDK Intramural Research Program (RDCO).
References
- Neale MJ, Keeney S. Clarifying the mechanics of DNA strand exchange in meiotic recombination. Nature. 2006;442:153–158. doi: 10.1038/nature04885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yanowitz J. Meiosis: making a break for it. Curr Opin Cell Biol. 2010;22:744–751. doi: 10.1016/j.ceb.2010.08.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Youds JL, Boulton SJ. The choice in meiosis - defining the factors that influence crossover or non-crossover formation. J Cell Sci. 2011;124:501–513. doi: 10.1242/jcs.074427. [DOI] [PubMed] [Google Scholar]
- Handel MA, Schimenti JC. Genetics of mammalian meiosis: regulation, dynamics and impact on fertility. Nat Rev Genet. 2010;11:124–136. doi: 10.1038/nrg2723. [DOI] [PubMed] [Google Scholar]
- Hassold T, Hall H, Hunt P. The origin of human aneuploidy: where we have been, where we are going. Hum Mol Genet. 2007;16 Spec No. 2:R203–R208. doi: 10.1093/hmg/ddm243. [DOI] [PubMed] [Google Scholar]
- Hall H, Hunt P, Hassold T. Meiosis and sex chromosome aneuploidy: how meiotic errors cause aneuploidy; how aneuploidy causes meiotic errors. Curr Opin Genet Dev. 2006;16:323–329. doi: 10.1016/j.gde.2006.04.011. [DOI] [PubMed] [Google Scholar]
- Neale MJ, Pan J, Keeney S. Endonucleolytic processing of covalent protein-linked DNA double-strand breaks. Nature. 2005;436:1053–1057. doi: 10.1038/nature03872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romanienko PJ, Camerini-Otero RD. The mouse Spo11 gene is required for meiotic chromosome synapsis. Mol Cell. 2000;6:975–987. doi: 10.1016/s1097-2765(00)00097-6. [DOI] [PubMed] [Google Scholar]
- Keeney S, Baudat F, Angeles M, Zhou ZH, Copeland NG, Jenkins NA, Manova K, Jasin M. A mouse homolog of the Saccharomyces cerevisiae meiotic recombination DNA transesterase Spo11p. Genomics. 1999;61:170–182. doi: 10.1006/geno.1999.5956. [DOI] [PubMed] [Google Scholar]
- Romanienko PJ, Camerini-Otero RD. Cloning, characterization, and localization of mouse and human SPO11. Genomics. 1999;61:156–169. doi: 10.1006/geno.1999.5955. [DOI] [PubMed] [Google Scholar]
- Bellani MA, Boateng KA, McLeod D, Camerini-Otero RD. The expression profile of the major mouse SPO11 isoforms indicates that SPO11beta introduces double strand breaks and suggests that SPO11alpha has an additional role in prophase in both spermatocytes and oocytes. Mol Cell Biol. 2010;30:4391–4403. doi: 10.1128/MCB.00002-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kauppi L, Barchi M, Baudat F, Romanienko PJ, Keeney S, Jasin M. Distinct properties of the XY pseudoautosomal region crucial for male meiosis. Science. 2011;331:916–920. doi: 10.1126/science.1195774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arnheim N, Calabrese P, Tiemann-Boege I. Mammalian meiotic recombination hot spots. Annu Rev Genet. 2007;41:369–399. doi: 10.1146/annurev.genet.41.110306.130301. [DOI] [PubMed] [Google Scholar]
- Paigen K, Petkov P. Mammalian recombination hot spots: properties, control and evolution. Nat Rev Genet. 2010;11:221–233. doi: 10.1038/nrg2712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buard J, de Massy B. Playing hide and seek with mammalian meiotic crossover hotspots. Trends Genet. 2007;23:301–309. doi: 10.1016/j.tig.2007.03.014. [DOI] [PubMed] [Google Scholar]
- Lichten M. Meiotic Chromatin: the Substrate for Recombination Initiation. Genome Dyn Stab. 2008;3:165–193. doi: 10.1007/7050_2008_040. [DOI] [Google Scholar]
- Grey C, Barthes P, Chauveau-Le Friec G, Langa F, Baudat F, de Massy B. Mouse PRDM9 DNA-Binding Specificity Determines Sites of Histone H3 Lysine 4 Trimethylation for Initiation of Meiotic Recombination. PLoS Biol. 2011;9:e1001176. doi: 10.1371/journal.pbio.1001176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baudat F, Buard J, Grey C, Fledel-Alon A, Ober C, Przeworski M, Coop G, de Massy B. PRDM9 is a major determinant of meiotic recombination hotspots in humans and mice. Science. 2010;327:836–840. doi: 10.1126/science.1183439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brick K, Smagulova F, Khil P, Camerini-Otero RD, Petukhova GV. Genetic recombination is directed away from functional genomic elements in mice. Nature. 2012;485:642–645. doi: 10.1038/nature11089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Myers S, Bowden R, Tumian A, Bontrop RE, Freeman C, MacFie TS, McVean G, Donnelly P. Drive against hotspot motifs in primates implicates the PRDM9 gene in meiotic recombination. Science. 2010;327:876–879. doi: 10.1126/science.1182363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parvanov ED, Petkov PM, Paigen K. Prdm9 controls activation of mammalian recombination hotspots. Science. 2010;327:835. doi: 10.1126/science.1181495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Segurel L, Leffler EM, Przeworski M. The case of the fickle fingers: how the PRDM9 zinc finger protein specifies meiotic recombination hotspots in humans. PLoS Biol. 2011;9:e1001211. doi: 10.1371/journal.pbio.1001211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smagulova F, Gregoretti IV, Brick K, Khil P, Camerini-Otero RD, Petukhova GV. Genome-wide analysis reveals novel molecular features of mouse recombination hotspots. Nature. 2011;472:375–378. doi: 10.1038/nature09869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burgoyne PS. Genetic homology and crossing over in the X and Y chromosomes of Mammals. Hum Genet. 1982;61:85–90. doi: 10.1007/BF00274192. [DOI] [PubMed] [Google Scholar]
- Johnston M. A model fungal gene regulatory mechanism: the GAL genes of Saccharomyces cerevisiae. Microbiol Rev. 1987;51:458–476. doi: 10.1128/mr.51.4.458-476.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pecina A, Smith KN, Mezard C, Murakami H, Ohta K, Nicolas A. Targeted stimulation of meiotic recombination. Cell. 2002;111:173–184. doi: 10.1016/S0092-8674(02)01002-4. [DOI] [PubMed] [Google Scholar]
- Robine N, Uematsu N, Amiot F, Gidrol X, Barillot E, Nicolas A, Borde V. Genome-wide redistribution of meiotic double-strand breaks in Saccharomyces cerevisiae. Mol Cell Biol. 2007;27:1868–1880. doi: 10.1128/MCB.02063-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukuda T, Kugou K, Sasanuma H, Shibata T, Ohta K. Targeted induction of meiotic double-strand breaks reveals chromosomal domain-dependent regulation of Spo11 and interactions among potential sites of meiotic recombination. Nucleic Acids Res. 2008;36:984–997. doi: 10.1093/nar/gkm1082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bishop DK. RecA homologs Dmc1 and Rad51 interact to form multiple nuclear complexes prior to meiotic chromosome synapsis. Cell. 1994;79:1081–1092. doi: 10.1016/0092-8674(94)90038-8. [DOI] [PubMed] [Google Scholar]
- San Filippo J, Sung P, Klein H. Mechanism of eukaryotic homologous recombination. Annu Rev Biochem. 2008;77:229–257. doi: 10.1146/annurev.biochem.77.061306.125255. [DOI] [PubMed] [Google Scholar]
- Chen YK, Leng CH, Olivares H, Lee MH, Chang YC, Kung WM, Ti SC, Lo YH, Wang AH, Chang CS. et al. Heterodimeric complexes of Hop2 and Mnd1 function with Dmc1 to promote meiotic homolog juxtaposition and strand assimilation. Proc Natl Acad Sci USA. 2004;101:10572–10577. doi: 10.1073/pnas.0404195101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Enomoto R, Kinebuchi T, Sato M, Yagi H, Shibata T, Kurumizaka H, Yokoyama S. Positive role of the mammalian TBPIP/HOP2 protein in DMC1-mediated homologous pairing. J Biol Chem. 2004;279:35263–35272. doi: 10.1074/jbc.M402481200. [DOI] [PubMed] [Google Scholar]
- Petukhova GV, Pezza RJ, Vanevski F, Ploquin M, Masson JY, Camerini-Otero RD. The Hop2 and Mnd1 proteins act in concert with Rad51 and Dmc1 in meiotic recombination. Nat Struct Mol Biol. 2005;12:449–453. doi: 10.1038/nsmb923. [DOI] [PubMed] [Google Scholar]
- Petukhova GV, Romanienko PJ, Camerini-Otero RD. The Hop2 protein has a direct role in promoting interhomolog interactions during mouse meiosis. Dev Cell. 2003;5:927–936. doi: 10.1016/S1534-5807(03)00369-1. [DOI] [PubMed] [Google Scholar]
- Mahadevaiah SK, Turner JM, Baudat F, Rogakou EP, de Boer P, Blanco-Rodriguez J, Jasin M, Keeney S, Bonner WM, Burgoyne PS. Recombinational DNA double-strand breaks in mice precede synapsis. Nat Genet. 2001;27:271–276. doi: 10.1038/85830. [DOI] [PubMed] [Google Scholar]
- Yang F, Wang PJ. The Mammalian synaptonemal complex: a scaffold and beyond. Genome Dyn. 2009;5:69–80. doi: 10.1159/000166620. [DOI] [PubMed] [Google Scholar]
- Baker SM, Plug AW, Prolla TA, Bronner CE, Harris AC, Yao X, Christie DM, Monell C, Arnheim N, Bradley A. et al. Involvement of mouse Mlh1 in DNA mismatch repair and meiotic crossing over. Nat Genet. 1996;13:336–342. doi: 10.1038/ng0796-336. [DOI] [PubMed] [Google Scholar]
- Dobson MJ, Pearlman RE, Karaiskakis A, Spyropoulos B, Moens PB. Synaptonemal complex proteins: occurrence, epitope mapping and chromosome disjunction. J Cell Sci. 1994;107(Pt 10):2749–2760. doi: 10.1242/jcs.107.10.2749. [DOI] [PubMed] [Google Scholar]
- Cloutier JM, Turner JM. Meiotic sex chromosome inactivation. Curr Biol. 2010;20:R962–R963. doi: 10.1016/j.cub.2010.09.041. [DOI] [PubMed] [Google Scholar]
- Ichijima Y, Sin HS, Namekawa SH. Sex chromosome inactivation in germ cells: emerging roles of DNA damage response pathways. Cell Mol Life Sci. 2012;69(15):2559–2572. doi: 10.1007/s00018-012-0941-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inagaki A, Schoenmakers S, Baarends WM. DNA double strand break repair, chromosome synapsis and transcriptional silencing in meiosis. Epigenetics. 2010;5:255–266. doi: 10.4161/epi.5.4.11518. [DOI] [PubMed] [Google Scholar]
- Burgoyne PS, Mahadevaiah SK, Turner JM. The consequences of asynapsis for mammalian meiosis. Nat Rev Genet. 2009;10:207–216. doi: 10.1038/nrg2505. [DOI] [PubMed] [Google Scholar]
- Royo H, Polikiewicz G, Mahadevaiah SK, Prosser H, Mitchell M, Bradley A, de Rooij DG, Burgoyne PS, Turner JM. Evidence that meiotic sex chromosome inactivation is essential for male fertility. Curr Biol. 2010;20:2117–2123. doi: 10.1016/j.cub.2010.11.010. [DOI] [PubMed] [Google Scholar]
- Yuan L, Liu JG, Hoja MR, Wilbertz J, Nordqvist K, Hoog C. Female germ cell aneuploidy and embryo death in mice lacking the meiosis-specific protein SCP3. Science. 2002;296:1115–1118. doi: 10.1126/science.1070594. [DOI] [PubMed] [Google Scholar]
- Yuan L, Liu JG, Zhao J, Brundell E, Daneholt B, Hoog C. The murine SCP3 gene is required for synaptonemal complex assembly, chromosome synapsis, and male fertility. Mol Cell. 2000;5:73–83. doi: 10.1016/S1097-2765(00)80404-9. [DOI] [PubMed] [Google Scholar]
- Celeste A, Petersen S, Romanienko PJ, Fernandez-Capetillo O, Chen HT, Sedelnikova OA, Reina-San-Martin B, Coppola V, Meffre E, Difilippantonio MJ. et al. Genomic instability in mice lacking histone H2AX. Science. 2002;296:922–927. doi: 10.1126/science.1069398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang F, De La Fuente R, Leu NA, Baumann C, McLaughlin KJ, Wang PJ. Mouse SYCP2 is required for synaptonemal complex assembly and chromosomal synapsis during male meiosis. J Cell Biol. 2006;173:497–507. doi: 10.1083/jcb.200603063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bannister LA, Pezza RJ, Donaldson JR, de Rooij DG, Schimenti KJ, Camerini-Otero RD, Schimenti JC. A dominant, recombination-defective allele of Dmc1 causing male-specific sterility. PLoS Biol. 2007;5:e105. doi: 10.1371/journal.pbio.0050105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu X, Aprelikova O, Moens P, Deng CX, Furth PA. Impaired meiotic DNA-damage repair and lack of crossing-over during spermatogenesis in BRCA1 full-length isoform deficient mice. Development. 2003;130:2001–2012. doi: 10.1242/dev.00410. [DOI] [PubMed] [Google Scholar]
- Khil PP, Smagulova F, Brick KM, Camerini-Otero RD, Petukhova GV. Sensitive mapping of recombination hotspots using sequencing-based detection of ssDNA. Genome Res. 2012;22:957–965. doi: 10.1101/gr.130583.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guenther MG, Levine SS, Boyer LA, Jaenisch R, Young RA. A chromatin landmark and transcription initiation at most promoters in human cells. Cell. 2007;130:77–88. doi: 10.1016/j.cell.2007.05.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pekowska A, Benoukraf T, Zacarias-Cabeza J, Belhocine M, Koch F, Holota H, Imbert J, Andrau JC, Ferrier P, Spicuglia S. H3K4 tri-methylation provides an epigenetic signature of active enhancers. EMBO J. 2011;30:4198–4210. doi: 10.1038/emboj.2011.295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ernst J, Kheradpour P, Mikkelsen TS, Shoresh N, Ward LD, Epstein CB, Zhang X, Wang L, Issner R, Coyne M. et al. Mapping and analysis of chromatin state dynamics in nine human cell types. Nature. 2011;473:43–49. doi: 10.1038/nature09906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alsheimer M. The dance floor of meiosis: evolutionary conservation of nuclear envelope attachment and dynamics of meiotic telomeres. Genome Dyn. 2009;5:81–93. doi: 10.1159/000166621. [DOI] [PubMed] [Google Scholar]
- Martinez P, Thanasoula M, Carlos AR, Gomez-Lopez G, Tejera AM, Schoeftner S, Dominguez O, Pisano DG, Tarsounas M, Blasco MA. Mammalian Rap1 controls telomere function and gene expression through binding to telomeric and extratelomeric sites. Nat Cell Biol. 2010;12:768–780. doi: 10.1038/ncb2081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zalzman M, Falco G, Sharova LV, Nishiyama A, Thomas M, Lee SL, Stagg CA, Hoang HG, Yang HT, Indig FE. et al. Zscan4 regulates telomere elongation and genomic stability in ES cells. Nature. 2010;464:858–863. doi: 10.1038/nature08882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Myers S, Bottolo L, Freeman C, McVean G, Donnelly P. A fine-scale map of recombination rates and hotspots across the human genome. Science. 2005;310:321–324. doi: 10.1126/science.1117196. [DOI] [PubMed] [Google Scholar]
- Yang X, Li C, Xu X, Deng C. The tumor suppressor SMAD4/DPC4 is essential for epiblast proliferation and mesoderm induction in mice. Proc Natl Acad Sci USA. 1998;95:3667–3672. doi: 10.1073/pnas.95.7.3667. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
The generation of the Gal4BD-Spo11 knock-in mouse. A. Schematic of the targeting vector. Nucleotide coordinates correspond to nucleotide #1 being located at the position -7,000 upstream of the first exon of the Spo11 gene. B. Schematic of the Southern blot strategy. Genomic DNA was cut with either NdeI+HpaI or ApaLI and hybridized with 5’ or 3’ probes, respectively. C. Southern blots for 5’ (left) and 3’ (right) homology arms. The genotype of mice (lanes 1-3) and ES clones (lanes 4-6) is indicated.
The strength of Gal4BD-Spo11 hotspots correlates with the strength of hotspots in wild type mice. The number of ssDNA fragments in wild type (C57Bl/6) hotspots was calculated for each dataset. Density scatter plots are shown for all hotspots. Log(strength) is shown on the y-axes. The Spearman Correlation Coefficient is also shown between all samples (inset). Over 94% of Gal4BD-Spo11 Hop2-/- hotspots corresponded to with type hotspots.