

Abstract
Cobalamin malabsorption accompanied by selective proteinuria is an autosomal recessive disorder known as Imerslund-Gräsbeck syndrome in humans and was previously described in dogs due to amnionless (AMN) mutations. The resultant vitamin B12 deficiency causes dyshematopoiesis, lethargy, failure to thrive, and life-threatening metabolic disruption in the juvenile period. We studied 3 kindreds of border collies with cobalamin malabsorption and mapped the disease locus in affected dogs to a 2.9 Mb region of homozygosity on canine chromosome 2. The region included CUBN, the locus encoding cubilin, a peripheral membrane protein that in concert with AMN forms the functional intrinsic factor-cobalamin receptor expressed in ileum and a multi-ligand receptor in renal proximal tubules. Cobalamin malabsorption and proteinuria comprising CUBN ligands were demonstrated by radiolabeled cobalamin uptake studies and SDS-PAGE, respectively. CUBN mRNA and protein expression were reduced ~10 fold and ~20 fold, respectively, in both ileum and kidney of affected dogs. DNA sequencing demonstrated a single base deletion in exon 53 predicting a translational frameshift and early termination codon likely triggering nonsense mediated mRNA decay. The mutant allele segregated with disease in the border collie kindred. The border collie disorder indicates that a CUBN mutation far C-terminal from the intrinsic factor-cobalamin binding site can abrogate receptor expression and cause Imerslund-Gräsbeck syndrome.
Keywords: vitamin B12, amnionless, cubilin, inborn error, methylmalonic aciduria, animal mode
1. Introduction
Imerslund-Gräsbeck syndrome (I-GS; OMIM #261100; http://omim.org/entry/261100) in humans is clinically defined by inherited selective intestinal cobalamin malabsorption accompanied by specific medium- to low-molecular-weight proteinuria [1-3]. The disorder is an autosomal recessive trait, but signs manifest variably in the late infantile to early juvenile periods as fetal stores of the vitamin wane [4]. Many patients exhibit a classical megaloblastic anemia, but neurologic signs or failure to thrive may predominate in others. The selectivity of intestinal nutrient malabsorption coupled with failed renal tubular protein reabsorption is conferred by mutations in cubilin (CUBN) [5] or amnionless (AMN) [6], the subunits of the heteromeric receptor designated cubam [7].
Cubam is a multi-ligand, endocytic receptor expressed in epithelial brush borders of distal small intestine and renal proximal tubules. CUBN is an ~460 kDa glycosylated protein that comprises an N-terminal alpha helix followed by 8 tandem EGF domains and 27 tandem CUB domains. The large protein provide binding sites for various ligands including the intrinsic factor-cobalamin complex (IF-Cbl), vitamin D binding protein, transferrin, albumin, apolipoprotein A1, haptoglobin, and others [8]. CUB domains 5-8 comprise the essential IF-Cbl binding site [9], and the N-terminal alpha helical region mediates association with AMN [10]. AMN is an ~50 kDa glycosylated type I transmembrane protein. The extracellular cysteine-rich domain interacts with CUBN, and the cytoplasmic domain has two endocytic signal motifs that arbitrate nucleation of clathrin-coated pits [11]. Though data [9,12] suggest that a coiled-coil structure of CUBN N-terminal helix regions creates a CUBN trimer bound to AMN, the stoichiometry of subunits in the functional receptor is still unclear. Regardless, formation of a heteromeric complex including both proteins was demonstrated in vitro [7]. Moreover, there is evidence from human, mouse, and canine tissues harboring mutations in either gene [13-16] that both CUBN and AMN must be present with near-normal structure for epithelial brush border expression of the functional receptor complex. The CUBN protein is encoded in 67 exons, but oddly, of 33 CUBN mutations defined in human I-GS patients to date, none has been found more 3′ than exon 28, which encodes the C-terminal half of CUB domain 8 [17].
Dogs closely model human clinical cobalamin deficiency and cubam function. Genetic studies in mice on the receptor complex have been hampered by rodent-specific functions of cubam essential for embryonic development. We previously characterized autosomal recessive I-GS in giant schnauzers and Australian shepherds [14,18]. Affected dogs exhibited signs of cobalamin deficiency including failure-to-thrive, dyshematopoiesis, and methylmalonic aciduria within 8-12 weeks of birth. The disorder was life-threatening if not treated, but parenteral cyanocobalamin administration (~1 mg/month) provided rapid and sustained hematopoietic and metabolic remission. Selective proteinuria persisted life-long despite treatment. In each breed, a distinct AMN mutation abrogated expression of immunodetectable AMN protein and caused CUBN retention in cytoplasmic vesicles of ileal and renal tubule cells [14].
Sporadic cases of a similar but milder disorder have been described in a herding dog breed known as border collies (BC) [19-22]. Owners of affected BC had not sought veterinary attention until the dogs were 8-42 months of age, but by the time of diagnosis the affected dogs exhibited intermittent anorexia, chronic anemia and neutropenia, poor weight gain, and poor muscle mass for several weeks or months. Severe metabolic dysregulation, including hepatic encephalopathy and/or ketoacidosis, occurred in some cases. Other documented abnormalities included low serum cobalamin concentrations, methylmalonic aciduria, and mild proteinuria. Here, we report results of a genome-wide positional gene approach leading to discovery of a single base deletion causing I-GS in BC.
2. Methods
2.1 Animals
Dogs were handled according to principles and protocols approved by the respective Institutional Animal Use and Care Committees at Michigan State University, University of Pennsylvania, and University of Saskatchewan. Owners submitted samples for DNA isolation and urinalysis from dogs of the BC kindreds and unrelated BC dogs. Additional DNA samples of non-BC dogs were from the laboratory archives. Clinical characterization of cobalamin-deficient dogs included various combinations of compatible clinical signs, routine complete blood counts, serum chemistry panels, serum cobalamin and folate concentrations, urinalysis, and examination of bone marrow aspirates or core biopsies.
In affected dogs, excess methylmalonic acid excretion in urine was documented either qualitatively by a specific reaction with diazotized p-nitroaniline or quantitatively by gas chromatography-mass spectroscopy. Urine proteins were concentrated by centrifugation through a 10 kDa cut-off membrane and separated by one-dimensional SDS-PAGE. Protein identities were confirmed by immunoblotting as previously [23-26]. In two affected dogs, gastrointestinal cobalamin absorption was investigated after parenteral cobalamin replacement therapy by one-stage, dual-label ([58Co] cyanocobalamin and [57Co] cyanocobalamin bound to human gastric juice) radiolabeled cyanocobalamin uptake (Dicopac® Kit, Medi-Physics, Inc., Princeton, NJ) administered according to the manufacturer’s instructions with measurement of excreted radioactivity in 24-hour urine (collected via in-dwelling catheter) and 48-hour fecal collections. Renal, ileal, and pancreatic biopsy samples were obtained from each of the same 2 affected dogs by exploratory laparotomy. Unsaturated cobalamin binding capacity (UCBC) was determined in serum and pancreatic homogenates by [57Co] cyanocobalamin labeling and albumin-coated charcoal separation of free-from bound-radiolabel [18].
2.2.1 Homozygosity mapping and mutation discovery
Genomic DNA was prepared variously from buccal brush, whole blood, or frozen tissue by standard methods [14]. DNA samples from 19 members of the BC kindreds were submitted to GeneSeek® Veterinary Diagnostics (Lincoln, NE) for genome-wide, single nucleotide polymorphism (SNP) genotyping on the Illumina® canine HD (170K) bead chip array derived from the CanFam 2.0 canine reference genome assembly. Genotypes were sorted by chromosome and position in Excel® (Microsoft®, Redmond, WA) and examined for areas of contiguous homozygosity shared by affected dogs. CUBN exons were amplified by PCR [14] from genomic DNA from one affected dog and his dam using primers that flanked exon and splice-site consensus sequences (table S1). CUBN cDNA was amplified in overlapping segments by RT-PCR from kidney total RNA as previously [14] using oligo-dT priming of the RT reaction and PCR primer sequences chosen from GenBank accession no. AF137068.1 (table S1). Amplicons were sequenced and assembled as previously [14] for comparison to the CanFam 3.1 canine reference genome assembly. Functional significance of missense sequences was examined by SIFT analysis [27] and resequencing DNA of clinically normal dogs.
2.2.2 CUBN steady-state expression analysis
CUBN mRNA expression was assessed by real-time quantitative PCR of cDNA templates (RT-qPCR). Normal and control tissues were obtained by surgical biopsy or immediately post-euthanasia, snap frozen in liquid N2, and maintained at −80 C. Total RNA was extracted using the RNeasy® Mini kit with on-column DNase treatment according to manufacturer instructions (Qiagen® Sciences, Inc., Germantown, MD). RNA integrity was assessed on an Agilent 2100 bioanalyzer (Agilent Technologies, Inc., Waldbronn, Germany) using the RNA 6000 Pico microcapillary electrophoresis chip. cDNA was prepared in triplicate from 500 ng of total RNA in 50 μL reactions using Superscript ® III (Life Technologies Corp., Carlsbad, CA) primed with oligo-dT. RT reaction mixes were pre-incubated for 5 min at 65 C, and incubated at 55 C for 50 min after reverse transcriptase was added. RT-qPCR reactions were performed in 3 technical replicates of 15 μL containing 10 ng cDNA, 50 nM each PCR primer, and 1X SYBR® Green PCR Mastermix (Life Technologies Corp., Carlsbad, CA). RT-qPCR was performed on a StepOnePlusTM real-time PCR system (Applied Biosystems, Foster City, CA) preheating the reaction mixtures at 95 C for 10 min followed by 40 cycles of 95 C for 15 s and 60 C for 1 min. No template controls were included in each run. Primer specificity was confirmed by examination of dissociation (melt) curves generated immediately after amplification and by gel electrophoresis of products. Amplification efficiencies of PCR primer pairs were determined from the slopes of Cq determinations in reactions using a 10-fold cDNA dilution series over 6 orders of magnitude. Relative mRNA quantification (ΔCq) was calculated using GAPDH expression as internal control, and statistical comparison between normal and affected dog samples was performed on data calculated as 2−ΔCq. Information on qPCR primers is in table S2.
CUBN protein expression in tissue homogenates was assessed by capture on rat gastric intrinsic factor-cobalamin-agarose beads, as described previously [14], followed by western blotting. CUBN was detected on membranes incubated sequentially with previously characterized rabbit polyclonal anti-dog CUBN serum [28], goat anti-rabbit IgG-horse radish peroxidase conjugate (cat. # 141506, Kirkegaard & Perry Laboratories, Inc., Gaithersburg, MD), and chemiluminescence reagents (Western Lightning ECL Plus, PerkinElmer, Inc., Waltham, MA). Relative quantification of western blot signals was performed with the Image Lab™ software of the Bio-Rad Chemi Doc™ XRS+ Molecular Imager (Bio-Rad Laboratories, Inc., Hercules, CA).
2.2.3 CUBN mutation genotyping
The CUBN exon 53 mutation site was amplified from genomic DNA using the primers 5′-AACCTGTGCTTGGGATTCTG-3′ and 5′-CGCAGAGAATGTTTATTTTACCTAAAGACT-3′. The -3 nucleotide (bold) of the reverse primer introduced an HpyCH4 III restriction enzyme cleavage site on the normal allele that is not in the native sequence and which is lost on the mutant allele. Amplification products were digested with HpyCH4 III, and fragments were separated on 4% agarose gels.
3. Results
3.1 Clinical phenotype
Ten cobalamin deficient BC dogs were ascertained in distantly related familial clusters (Fig 1) and 2 unrelated cases. Diagnosis was delayed in the propositus of each cluster and in sporadic cases until 9 to 24 months of age at which time expressed complaints were inappetence, failure to gain weight, lethargy, and malaise that was exacerbated after the dogs ate. In 2 dogs of the CA kindred, the disorder progressed within 2 and 8 weeks of initial presentation, respectively, to fatal metabolic crises characterized by non-regenerative anemia with circulating erythroblasts, anisocytosis, schistocytosis, neutropenia, hyperammonemia, metabolic acidosis, ketonuria without glucosuria, and methylmalonic aciduria. Bone marrow examinations revealed erythropoietic maturation arrest at the metarubricyte/rubricyte stage and giant metamyelocytes.
Figure 1. Pedigrees of 3 border collie families segregating canine I-GS.

Squares indicate males, circles indicate females, and filled symbols indicate affected dogs. Offspring of a mating are arranged on a horizontal line extending between vertical lines that descend from the parents. Clinical findings of the dogs labeled D and T in the CA kindred are described in detail in Results. The filled diamond indicates an affected dog of unreported sex. The double line connecting two males in the UK2 kindred indicates that they share common great-grandparents but of unknown relationship to the other BC.
In repeat matings, pups were screened from young age for similar signs and methylmalonic aciduria. In one litter, two male pups (D and T of the CA kindred in Fig 1) examined at 14 weeks of age exhibited poor growth compared to littermates, non-regenerative anemia, neutropenia with hypersegmentation, methylmalonic aciduria (6,700 and 8,800 mmol MMA/mol creatinine, respectively; reference range < 4 mmol MMA/mol creatinine). In each dog, serum cobalamin concentration was undetectable (<20 pM; reference range 166-487 pM). A 3-week course of oral cyanocobalamin supplementation in one dog had no effect, whereas subsequent parenteral administration of the vitamin (1 mg SQ) to both dogs caused a reticulocyte response within 4 days and corrected all clinical and laboratory abnormalities within a few weeks. These two dogs were maintained on parenteral cyanocobalamin supplementation (~100 μg monthly) for 14 months, during which time they were in complete clinical remission. At 17 months of age, both affected dogs underwent a dual radiolabeled intestinal cobalamin absorption test. Cobalamin labeled with [57Co] was bound to human gastric juice as a source of intrinsic factor. In both dogs, <0.4% of the administered dose of unbound cobalamin and <0.1% of the gastric juice-bound cobalamin were recovered in 24-hour urine collections. Nearly all of each administered isotope was recovered in feces. No histologic abnormalities were observed in pancreatic, ileal, or renal biopsies of either dog. The unsaturated cobalamin binding capacity (UCBC) of pancreatic tissue extracts of the 2 affected dogs was not different from that of normal controls (3.6±1.3 nmol/g tissue, n=6), and intrinsic factor represented 52-63% of the total UCBC in pancreas as determined by cobinamide blocking. The UCBC of an affected dog serum sample (1.74 nM) was similar to controls (1.6±0.1 nM, n=3), and nearly all serum-bound cobalamin eluted from a gel filtration column at Ve/Vo = 2.08, as previously determined for canine transcobalamin (TCII) [18].
Other affected BC were studied in less detail but were minimally ascertained by development of similar clinical signs with documentation of methylmalonic aciduria and/or serum cobalamin deficiency and rapid clinical response to parenteral cyanocobalamin administration. Additional studies demonstrated selective proteinuria in the affected dogs that persisted lifelong despite cobalamin replenishment. Total urine protein excretion was not in excess of what is recognized as normal in dogs (7-12 mg protein/kg body weight/day; protein/creatinine <0.4), but greater than normal excretion of cubam ligands including albumin, haptoglobin, apo A1, vitamin D-binding protein and transferrin was observed (Fig 2). The pattern of CUBN ligand excretion was similar to what has been demonstrated in I-GS affected giant schnauzers [23-26] and Australian shepherds with documented mutations in AMN [14], as well as in a human CUBN deficient patient [13].
Figure 2. Selective proteinuria in BC with I-GS.

Shown are lanes from a silver-stained 15% SDS-PAGE gel loaded with urine proteins of an affected (lane 1) and a clinically normal BC (lane 2). Proteins in each lane were concentrated from urine samples containing 200 μg of creatinine. The affected dog was in metabolic remission at the time of urine collection due to previous parenteral cyanocobalamin administration. Identities of the labeled protein bands were confirmed by immunoblotting as previously reported [23-26].
3.2 Genetic investigations
Segregation analysis of I-GS in the BC kindreds was consistent with simple autosomal recessive inheritance. The number of affected pups born in such matings was not different from what is expected under that hypothesis (χ2=0.35, p>0.55, df=1), and both male and female affected dogs were observed, always born in matings of clinically normal parents. Due to incomplete breeding records, we could not determine an ancestor that was common to all obligate carriers of all kindreds. However, affected dogs of this study appeared to be distantly related despite that ascertainment of the 3 kindreds occurred over 17 years and 2 continents.
3.2.1 CUBN mutation discovery
Two strong functional-candidate genes for I-GS are those that encode the known constituents of cubam, CUBN and AMN. However, a sizable proportion of human I-GS patients do not have mutations in exons of either CUBN or AMN, and there are a handful of weaker candidates based on emerging evidence of their function in cobalamin absorption [17]. In order to narrow the search for the disease gene we sought to establish a positional-candidate interval by genome-wide, high density, homozygosity mapping. SNP genotypes (table S3) were determined in 17 members of the UK1 kindred and two affected dogs of the CA kindred for which we had samples (D and T in Fig 1). SNP genotyping demonstrated only one region of contiguous homozygosity of length compatible with linkage disequilibrium commonly observed in dogs of a single breed. In the nearly 2.9 Mbp interval of homozygosity on chromosome 2 (chr2:21,068,544-23,940,272; CanFam 2 assembly coordinates) none of the informative alleles shared by the 4 affected dogs were homozygous in any of the clinically normal dogs, but 29 SNPs within the interval (chr2:21,386,833-23,524,746) were heterozygous in both tested parents and some normal littermates in the UK1 kindred. The interval of homozygosity was centered over the CUBN locus (chr2:22,619,020-22,876,740), thus conferring positional as well as functional candidacy to that gene.
We amplified overlapping segments of CUBN cDNA by RT-PCR from kidney cortex RNA of an affected dog in the CA kindred (D in Fig 1) and a healthy control dog. We also amplified the 67 CUBN exons with flanking splice sites from genomic DNA of an affected dog of the UK1 kindred and its healthy dam. The cDNA and genomic exon sequences of the two affected dogs were identical. In comparison to the canine reference sequence assembly (CanFam 3.1), there were no variations observed in splice sites, and there was no evidence of missplicing in the affected dog cDNA sequence. The protein coding sequences had 5 silent variations from the reference sequence and 2 variations that predicted well-tolerated amino acid changes by SIFT analysis (V52A and S3121T). The latter were also both homozygous in clinically normal dogs of other breeds that we sequenced. Most importantly, however, the affected dogs exhibited a homozygous deletion of one cytosine in exon 53 in both genomic and cDNA that predicted a translation reading frameshift and early termination 2 codons later (c.8392delC; p. Q2798Rfs*3; Fig 3). The deletion was heterozygous in the affected dog’s dam.
Figure 3. Frameshift mutation near the CUBN exon/intron boundary 53.
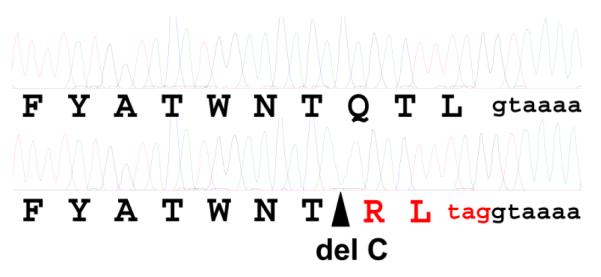
Shown are electophoretograms obtained by Sanger sequencing of exon 53 with flanking intronic sequence amplified by PCR from a normal (above) and an I-GS affected BC (below). The site of a single cytosine deletion in the affected BC sequence is indicated by the triangle. The deduced amino acid sequence is shown in single letter code below each electrophoretogram. The translation sequence and premature stop codon predicted by the frameshift are shown in red. The nucleotides indicated in lower case black are the splice donor sequence of intron 53.
3.2.2 CUBN steady-state expression analysis
To quantify the effect of the observed single base deletion on CUBN mRNA expression, we amplified ileal and renal cortex RNA by real-time RT-qPCR from the two affected dogs of the CA kindred for which we had biopsy material and from clinically normal, unrelated dogs of other breeds. In ileum, CUBN mRNA expression of the two affected dogs was 5-12% of normal control dogs (n=3), depending on which of the CUBN primer pairs we used. The coefficient of variation in repeated assays using the CUBN exon 59-60 primer pair was 0.04 (9.12 ± 0.34%, mean ± SD). In kidney, affected dog CUBN expression was 11% of normal (CUBN exon 40-41 primer pair).
We determined CUBN protein expression in affected and normal dog ileum and kidney by IF-Cbl affinity capture and western blotting (Fig 4). CUBN expression in ileum of affected dogs (lanes 2 and 3) was 16 and 21 fold less than the mean of that observed in two normal controls (lanes 4 and 5). CUBN expression in kidney of an affected dog (lanes 6) was 24 fold less than the mean of that observed in two normal controls (lanes 7 and 8). Overexposure of the blot revealed normal sized CUBN proteins of ~460 and ~190 kDa in affected dog ileum (lane 1) but none of a size consistent with truncation in exon 53 and loss of ~90 kDa of amino acid sequence. CUBN was undetectable on western blots of 50 μg protein/lane of affected dog tissue homogenates without affinity capture (not shown).
Figure 4. Deficient CUBN expression in ileum and kidney of BC with I-GS.
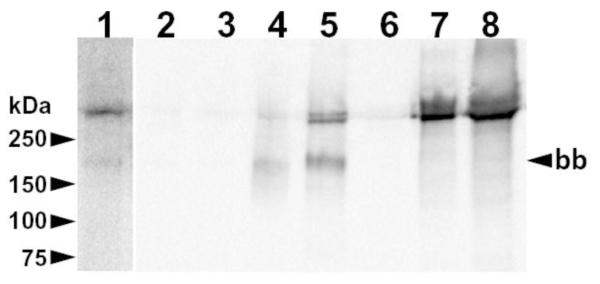
Shown is a blot of proteins accumulated from homogenates of ileal mucosa (lanes 1-5) and kidney cortex (lanes 6-8) of I-GS affected dogs (lanes 1-3 and 6) and normal controls (lanes 4, 5 and 7, 8). Lane 1 is an overexposure of lane 2 on the same blot. Twenty-five mg of total protein in ileal homogenates and 2 mg in kidney homogenates were incubated with 20 μL of IF-Cbl-agarose beads. Bound proteins were separated by non-reducing SDS-PAGE (4-20% gradient gel) and electroblotted. The membrane was incubated sequentially with rabbit polycolonal anti-canine CUBN serum [28] (1:20,000 dilution), goat anti-rabbit IgG-HRP conjugate, and chemiluminescent detection reagents. Migration of molecular weight markers is indicated on the left. The ileal brush-border size (~190 kDa) of CUBN that occurs after exposure of full-length CUBN to luminal proteases is indicated on the right (bb).
3.2.3 CUBN mutation genotyping
To examine segregation of the exon 53 deletion in BC we designed a convenient allele-specific genotyping assay for the mutation based on PCR amplification of genomic DNA followed by restriction enzyme digestion of the amplicon (Fig 5). The reverse primer introduced a change of the amplicon sequence that created an HpyCH4III cleavage site in the normal but not in the mutant allele. We genotyped the affected dogs and 27 other members of the 3 ascertained kindreds, 2 additional sporadic I-GS cases in BC of unknown relatedness, 95 unrelated and clinically normal BC, 3 I-GS cases in other breeds and 37 clinically normal dogs of other breeds. All affected BC were homozygous for the deletion, and the 3 obligate carriers tested were heterozygous. Thirteen clinically normal littermates of affected dogs plus one grandsire were also heterozygous, and all other dogs tested in the kindreds were homozygous for the normal allele. Therefore, the deletion mutation segregated completely with the disease allele in the ascertained BC kindreds. No healthy or I-GS affected dog of a different breed exhibited the BC mutation allele, except for one I-GS dog of mixed BC heritage that was homozygous for the BC mutation. We observed 6 carriers among the 95 BC (6.3%) that were unrelated to the kindreds investigated here or to each other within 3 generations.
Figure 5. CUBN exon 53 cytosine deletion genotyping in BC.
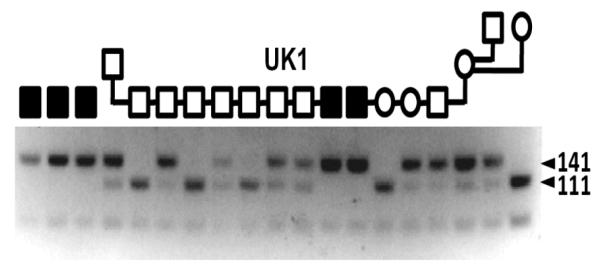
Shown are results of an allele-specific, HpyCH4III restriction digest of 212 bp PCR products amplified from genomic DNA. The cytosine deletion mutation abrogates a cut site, creating a 141 bp fragment upon digestion of the initial PCR amplicon. The wildtype sequence is cut to create a 111 bp fragment. The amplicon has a second HpyCH4III recognition site distant from the mutation site that creates a 71 bp fragment regardless of genotype. The symbols (see Fig 1 legend) above indicate the disease phenotype of several dogs of the UK1 kindred, D and T of the CA kindred and one sporadic case (S) of an I-GS BC from Michigan.
4. Discussion
Beginning with Nobel Prize-winning investigations by George Whipple [29], observations in dogs have provided important insight into nutritional support of metabolism for nearly a century. Dogs continue to be a rich source of genetic variation that informs our understanding of molecular mechanisms mediating cobalamin absorption and renal function. Selective intestinal cobalamin malabsorption with selective proteinuria is a unique inborn error of metabolism known as Imerslund-Gräsbeck syndrome. To date, I-GS indicates mutations of either CUBN or AMN and loss of cubam function in both ileum and renal tubules. The nosology is somewhat confused because certain reports speak of I-GS without proteinuria. In the original description of Norwegian cases by Olga Imerslund [1], affected patients in 2 of her 6 families did not exhibit proteinuria, but molecular diagnosis of these has not been reported. Furthermore, one of the first described CUBN mutations was a missense mutation that abrogates IF-Cbl binding (e.g. the so-called FM1 mutation [5,30]), but not the binding of other ligands. A study of urinary protein excretion in I-GS patients demonstrated high variability even among FM1-homozygous individuals, suggesting there is incomplete penetrance of proteinuria in some patients with CUBN mutations [31,32]. Nevertheless, when selective proteinuria is recognized, it is a useful clinical diagnostic feature because it excludes other causes of selective cobalamin malabsorption such as gastric intrinsic factor (GIF) deficiency. Since the reagents for radiolabeled cobalamin absorption testing in the clinical setting became unavailable [4,33,34], several patients harboring GIF mutations have been misdiagnosed as I-GS [17,35]. At present, the most efficient means to discriminate among known types of inherited selective cobalamin malabsorption without proteinuria is by molecular diagnosis, undertaking screens of GIF, CUBN, and AMN for causative mutations [17]. Other genes (e.g. ABCD4 [36,37] and LMBRD1 [38]) have been implicated in the intracellular cobalamin processing disorders, cblJ and cblF, respectively, that include a component of selective cobalamin malabsorption without proteinuria. While the onset of signs in these disorders is typically in the infantile period, rare patients may not come to medical attention for several years. A newly described transporter of free cobalamin (ABCC1) across the enterocyte basolateral membrane [39] has yet to be implicated in cobalamin deficiency [40].
Urine samples of affected dogs ascertained in this investigation contained 1500-2000 fold elevated MMA concentrations that were concordant with their undetectable serum cobalamin concentrations. The ensuing dyshematopoiesis, ketoacidosis, and hyperammonemia are potentially lethal disruptions that occur due to secondary inhibition of key metabolic pathways when cobalamin deficiency is severe and prolonged [41]. Affected BC dogs also excreted a constellation of CUBN ligands in urine that are filtered at the glomerulus. Remission of all clinical and laboratory abnormalities, other than specific proteinuria, after parenteral cobalamin supplementation suggested I-GS. Diagnosis of selective cobalamin malabsorption was confirmed by unequivocal results of a radiolabeled cobalamin absorption study similar to the traditional Schilling test. Thus, the disease phenotype of affected BC is florid cobalamin deficiency caused by selective intestinal malabsorption that is accompanied by selective proteinuria. As in humans, I-GS proteinuria in dogs is a failure of receptor function that is not associated with hypertension or diabetes, nor is it a precursor of progressive renal failure.
The BC I-GS mutation demonstrated here is a single base deletion in exon 53 (c.8392delC; p.Q2798Rfs*3) that predicts a translational frameshift and early stop after 2 codons of mistranslation. This mutation has the effect of reducing stable CUBN mRNA expression, most probably due to induction of nonsense-mediated decay [42], and is a hypomorphic allele. Interestingly, the remaining mRNA is measurable with PCR primers placed variously along the length of the message, including the exon 59-60 primers that are well 3′ of the mutation site. There was no evidence of a truncated protein product, but it appeared there is remnant translation of full-length CUBN products in ileum and kidney. An early stop codon may be ignored on a minority of transcripts if the translating ribosome occasionally introduces a compensating frameshift. Such a phenomenon confers partial rescue of carbonic anhydrase deficiency in some human patients [43] and could be operating here. Moreover, appearance of the 190 kDa form of CUBN in ileum of the affected dogs suggests that at least some of the protein reaches the brush-border and is exposed to luminal enzymes [28]. Regardless, CUBN expression is insufficient in homozygous dogs to mediate enough cobalamin absorption to maintain metabolic homeostasis.
A similar exon 53 mutation (c.8355delA; p.S2785fsX19) that predicted truncation at the same codon as the BC mutation was recently described as causing fluctuating proteinuria in a pair of Egyptian siblings of consanguineous parents [44]. The report focused on kidney function and did not include results of an adequate assessment of the patients’ cobalamin status. In all cases of proteinuria due to CUBN or AMN mutations, a full diagnosis should be pursued aggressively because it is intestinal malabsorption and cobalamin deficiency that is pathogenic, not the proteinuria. CUBN mutations anywhere in the coding or splicing sequences that destabilize the message or the protein are likely to adversely affect cubam function in both ileum and kidney. Our results indicate that the reported Egyptian patients probably suffer from limited cobalamin absorption and need lifelong cobalamin supplementation.
The affected BC in this study and other reports [19-22] came to medical attention at variable ages as late as 42 months. This is in sharp contrast to the I-GS giant schnauzers and Australian shepherds harboring AMN mutations which were inappetent and exhibiting failure-to-thrive as early as 8 weeks of age [14,18]. This may be due to a low-level remnant of cubam expression and cobalamin absorption in affected BC that is somewhat protective for several months. However, the fact that affected BC could be recognized and exhibited methylmalonic aciduria as early as 14 weeks of age when new litters were examined with a high index of suspicion suggests that the later recognition of disease in BC may not reflect a difference of cubam function or a mutation-specific effect. It may be that as working farm dogs that are typically quite lean, the affected BC simply did not seem abnormal to their owners until the signs of disease became severe or were long-standing. The late recognition of disease may also have contributed to a wide dispersion and 6 % carrier rate of the disease allele in BC.
5. Conclusions
This work demonstrates that selective cobalamin malabsorption with proteinuria in BC is an autosomal recessive trait caused by a single-base deletion in CUBN and documents the first naturally-occurring animal model of CUBN deficiency. Cobalamin malabsorption and urinary excretion of CUBN ligands were demonstrated in affected dogs by radiolabeled cobalamin uptake studies and protein electrophoresis, respectively, indicating cubam dysfunction in ileum and renal tubules. The deletion (c.8392delC) causes a frameshift and early stop codon leading to ~10 fold reduction of steady-state CUBN mRNA and ~20 fold reduction of protein in ileum and kidney. A survey of healthy, unrelated BC suggests that ~6% of the breed are carriers of the disease-causing mutation. Thus, a mutation far C-terminal from the intrinsic factor-cobalamin binding site can abrogate receptor expression and cause Imerslund-Gräsbeck syndrome. It is relevant to search for this type of mutation in future genetic screens of patients with Imerslund-Gräsbeck syndrome.
Supplementary Material
Highlights.
Imerslund-Gräsbeck syndrome is a potentially lethal, inherited defect of receptor-mediated endocytosis in humans characterized by cobalamin malabsorption, proteinuria, and the metabolic consequences of cobalamin deficiency. This manuscript describes experimental characterization of a unique orthologous canine model of cobalamin malabsorption caused by a CUBN mutation and demonstrates effects on mRNA and protein expression in both ileum and kidney. This is the most distal CUBN mutation described to date.
Acknowledgments
The authors thank Stephan M. Tanner for helpful discussion and access to a preprint version of reference 17. We also thank dog owners, breeders, and veterinarians of study dogs, James Mickelson, Nora Berghoff, and Paula Henthorn for DNA samples from clinically normal BC. This work was supported in part by NIH grant OD010939 and revenues of the MSU Laboratory of Comparative Medical Genetics. The sponsors had no part in study design or producing this report.
Abbreviations
- AMN
amnionless
- BC
border collie
- CUB
an ~110 residue protein domain structure first defined in Complement proteins C1r/C1s, sea Urchin EGF-domain-containing protein (Uegf), and Bone morphogenic protein 1 (Bmp1)
- cubam
the functional receptor complex composed of AMN and CUBN subunits
- CUBN
cubilin
- GIF
gastric intrinsic factor
- I-GS
Imerslund-Gräsbeck syndrome
- MMA
methylmalonic acid
- qPCR
quantitative polymerase chain reaction
- RT
reverse transcriptase
- SNP
single nucleotide polymorphism
- UCBC
unsaturated cobalamin binding capacity; the single-letter amino acid code is used throughout
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Contributions: J.C.F., C.A.O., S.L.M, C.A.F., and U.G. performed clinical analyses and collected patient samples; C.A.O. and S.L.M. designed and performed radiolabeled cobalamin uptake studies and tissue biopsies; S.L.H., and J.C.F. performed molecular genetics experiments and western blots; J.C.F., P.J.V., and U.G. designed the genetics studies, analyzed results, wrote the manuscript, and made figures.
Conflict of Interest Disclosure: The authors declare no competing financial interests.
References
- [1].Imerslund O. Idiopathic chronic megaloblastic anemia in children. Acta Paedr. Scand. 1960;49(Suppl.119):1–115. [PubMed] [Google Scholar]
- [2].Imerslund O, Bjørnstad P. Familial vitamin B12 malabsorption. Acta Haemat. 1963;30:1–7. doi: 10.1159/000208104. [DOI] [PubMed] [Google Scholar]
- [3].Gräsbeck R, Gordin R, Kantero I, Kuhlback B. Selective vitamin B12 malabsorption and proteinuria in young people. Acta Med. Scand. 1960;167:289–296. doi: 10.1111/j.0954-6820.1960.tb03549.x. [DOI] [PubMed] [Google Scholar]
- [4].Stabler SP. Vitamin B12 deficiency. N. Engl. J. Med. 2013;386:149–160. doi: 10.1056/NEJMcp1113996. [DOI] [PubMed] [Google Scholar]
- [5].Aminoff M, Carter JE, Chadwick RB, Johnson C, Gräsbeck R, Abdelaal MA, Broch H, Jenner LB, Verroust PJ, Moestrup SK, de la Chapelle A, Krahe R. Mutations in CUBN, encoding the intrinsic factor-vitamin B12 receptor, CUBN, cause hereditary megaloblastic anaemia 1. Nat. Genet. 1999;21:309–313. doi: 10.1038/6831. [DOI] [PubMed] [Google Scholar]
- [6].Tanner SM, Aminoff M, Wright FA, Liyanarachchi S, Kuronen M, Saarinen A, Massika O, Mandel H, Broch H, de la Chapelle A. Amnionless, essential for mouse gastrulation, is mutated in recessive hereditary megaloblastic anemia. Nat. Genet. 2003;33:426–429. doi: 10.1038/ng1098. [DOI] [PubMed] [Google Scholar]
- [7].Fyfe JC, Madsen M, Højrup P, Christensen EI, Tanner SM, de la Chapelle A, He Q, Moestrup SK. The functional cobalamin (vitamin B12)-intrinsic factor receptor is a novel complex of CUBN and amnionless. Blood. 2004;103:1573–1579. doi: 10.1182/blood-2003-08-2852. [DOI] [PubMed] [Google Scholar]
- [8].Nielsen MJ, Rasmussen MR, Andersen CBF, Nexø E, Moestrup SK. Vitamin B12 transport from food to the body’s cells - a sophisticated, multistep pathway. Nat. Rev. Gastroenterol. Hepatol. 2012;9:345–354. doi: 10.1038/nrgastro.2012.76. [DOI] [PubMed] [Google Scholar]
- [9].Andersen CBF, Madsen M, Storm T, Moestrup SK, Andersen GR. Structural basis for receptor recognition of vitamin-B12-intrinsic factor complexes. Nature. 2010;464:445–449. doi: 10.1038/nature08874. [DOI] [PubMed] [Google Scholar]
- [10].Kristiansen M, Kozyraki R, Jacobsen C, Nexø E, Verroust PJ, Moestrup SK. Molecular dissection of the intrinsic factor-vitamin B12 receptor, CUBN, discloses regions important for membrane association and ligand binding. J. Biol. Chem. 1999;274:20540–20544. doi: 10.1074/jbc.274.29.20540. [DOI] [PubMed] [Google Scholar]
- [11].Pedersen GA, Chakraborty S, Steinhauser AL, Traub LM, Madsen M. AMN directs endocytosis of the intrinsic factor-vitamin B(12) receptor cubam by engaging ARH or Dab2. Traffic. 2010;11:706–720. doi: 10.1111/j.1600-0854.2010.01042.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Lindblom A, Quadt N, Marsh T, Aeschlimann D, Mörgelin M, Mann K, Maurer P, Paulsson M. The intrinsic factor-vitamin B12 receptor, CUBN, is assembled into trimers via a coiled-coil alpha-helix. J. Biol. Chem. 1999;274:6374–6380. doi: 10.1074/jbc.274.10.6374. [DOI] [PubMed] [Google Scholar]
- [13].Storm T, Emma F, Verroust PJ, Hertz JM, Nielsen R, Christensen EI. A patient with CUBN deficiency. N. Engl. J. Med. 2011;364:89–91. doi: 10.1056/NEJMc1009804. [DOI] [PubMed] [Google Scholar]
- [14].He Q, Madsen M, Kilkenney A, Gregory B, Christensen EI, Vorum H, Højrup P, Schäffer AA, Kirkness EF, Tanner SM, de la Chapelle A, Giger U, Moestrup SK, Fyfe JC. Amnionless function is required for CUBN brush-border expression and intrinsic factor-cobalamin (vitamin B12) absorption in vivo. Blood. 2005;106:1447–1453. doi: 10.1182/blood-2005-03-1197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Amsellem S, Gburek J, Hamard G, Nielsen R, Willnow TE, Devuyst O, Nexo E, Verroust PJ, Christensen EI, Kozyraki R. CUBN is essential for albumin reabsorption in the renal proximal tubule. J. Am. Soc. Nephrol. 2010;21:1859–1867. doi: 10.1681/ASN.2010050492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Fyfe JC, Ramanujam KS, Ramaswamy K, Patterson DF, Seetharam B. Defective brush-border expression of intrinsic factor-cobalamin receptor in canine inherited intestinal cobalamin malabsorption. J. Biol. Chem. 1991;266:4489–4494. [PubMed] [Google Scholar]
- [17].Tanner SM, Sturm AC, Baack EC, Liyanarachchi S, de la Chapelle A. Inherited cobalamin malabsorption. Mutations in three genes reveal functional and ethnic patterns. Orphanet. J. Rare Dis. 2012;7:56. doi: 10.1186/1750-1172-7-56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Fyfe JC, Giger U, Hall CA, Jezyk PF, Klumpp SA, Levine JS, Patterson DF. Inherited selective intestinal cobalamin malabsorption and cobalamin deficiency in dogs. Pediatr. Res. 1991;29:24–31. doi: 10.1203/00006450-199101000-00006. [DOI] [PubMed] [Google Scholar]
- [19].Giger U, Outerbridge CA, Myers SL. Hereditary cobalamin deficiency in border collie dogs. J. Vet. Intern. Med. 1996;10:169. [Google Scholar]
- [20].Morgan LW, McConnell J. Cobalamin deficiency associated with erythroblastic anemia and methylmalonic aciduria in a border collie. J. Am. Anim. Hosp. Assoc. 1999;35:392–395. doi: 10.5326/15473317-35-5-392. [DOI] [PubMed] [Google Scholar]
- [21].Battersby IA, Giger U, Hal EJ. Hyperammonaemic encephalopathy secondary to selective cobalamin deficiency in a juvenile border collie. J. Small Anim. Prac. 2005;46:339–344. doi: 10.1111/j.1748-5827.2005.tb00330.x. [DOI] [PubMed] [Google Scholar]
- [22].Lutz S, Sewell AC, Reusch CE, Kook PH. Clinical and laboratory findings in border collies with presumed hereditary juvenile cobalamin deficiency. J. Am. Anim. Hosp. Assoc. 2013;49:197–203. doi: 10.5326/JAAHA-MS-5867. [DOI] [PubMed] [Google Scholar]
- [23].Nykjaer A, Fyfe JC, Kozyraki R, Leheste JR, Jacobsen C, Nielsen MS, Verroust PJ, Aminoff M, de la Chapelle A, Moestrup SK, Ray R, Gliemann J, Willnow TE, Christensen EI. CUBN dysfunction causes abnormal metabolism of the steroid hormone 25(OH) vitamin D3. Proc. Natl. Acad. Sci. USA. 2001;98:13895–13900. doi: 10.1073/pnas.241516998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Kozyraki R, Fyfe J, Verroust PJ, Jacobsen C, Dautry-Varsat A, Gburek J, Willnow TE, Christensen EI, Moestrup SK. Megalin-dependent CUBN-mediated endocytosis is a major pathway for uptake of transferrin in polarized epithelia. Proc. Natl. Acad. Sci. USA. 2001;98:12491–12496. doi: 10.1073/pnas.211291398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Birn H, Fyfe J, Jacobsen C, Mounier F, Verroust PJ, Orskov H, Willnow TE, Moestrup SK, Christensen EI. CUBN is an albumin binding protein important for renal albumin reabsorption. J. Clin. Invest. 2000;105:1353–1361. doi: 10.1172/JCI8862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Kozyraki R, Fyfe J, Kristiansen M, Gerdes C, Jacobsen C, Cui S, Christensen EI, Aminoff M, de la Chapelle A, Krahe R, Verroust PJ, Moestrup SK. The intrinsic factor vitamin B12 receptor, CUBN, is a novel high affinity apolipoprotein A-1 receptor facilitating endocytosis of high-density lipoprotein. Nat. Med. 1999;5:656–661. doi: 10.1038/9504. [DOI] [PubMed] [Google Scholar]
- [27].Kumar P, Henikoff S, Ng PC. Predicting the effects of coding non-synonymous variants on protein function using the SIFT algorithm. Nat. Protoc. 2009;4:1073–1081. doi: 10.1038/nprot.2009.86. [DOI] [PubMed] [Google Scholar]
- [28].Xu D, Fyfe JC. Cubilin expression and posttranslational modification in the canine gastrointestinal tract. Am. J. Physiol. Gastrointest. Liver Physiol. 2000;279:G748–G756. doi: 10.1152/ajpgi.2000.279.4.G748. [DOI] [PubMed] [Google Scholar]
- [29].Hooper CW, Whipple GH. Blood regeneration after simple anemia: I. Curve of regeneration influenced by dietary factors. Am. J. Physiol. 1918;45:573–575. [Google Scholar]
- [30].Kristiansen M, Aminoff M, Jacobsen C, de La Chapelle A, Krahe R, Verroust PJ, Moestrup SK. CUBN P1297L mutation associated with hereditary megaloblastic anemia 1 causes impaired recognition of intrinsic factor-vitamin B12 by CUBN. Blood. 2000;96:405–409. [PubMed] [Google Scholar]
- [31].Wahlstedt-Fröberg V, Pettersson T, Aminoff M, Dugué B, Gräsbeck R. Proteinuria in CUBN-deficient patients with selective vitamin B12 malabsorption. Pediatr. Nephrol. 2003;18:417–421. doi: 10.1007/s00467-003-1128-y. [DOI] [PubMed] [Google Scholar]
- [32].Tanner SM, Li Z, Bisson R, Acar C, Oner C, Oner R, Cetin M, Abdelaal MA, Ismail EA, Lissens W, Krahe R, Broch H, Gräsbeck R, de la Chapelle A. Genetically heterogeneous selective intestinal malabsorption of vitamin B12: Founder effects, consanguinity, and high clinical awareness explain aggregations in Scandinavia and the Middle East. Hum. Mutat. 2004;23:327–333. doi: 10.1002/humu.20014. [DOI] [PubMed] [Google Scholar]
- [33].Carmel R. The disappearance of cobalamin absorption testing: a critical diagnostic loss. J. Nutr. 2007;137:2481–2484. doi: 10.1093/jn/137.11.2481. [DOI] [PubMed] [Google Scholar]
- [34].Nexø E, Hoffmann-Lücke E. Holotranscobalamin, a marker of vitamin B-12 status: analytical aspects and clinical utility. Am. J. Clin. Nutr. 2011;94:359S–365S. doi: 10.3945/ajcn.111.013458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Tanner SM, Zhongyuan L, Perko JD, Oner C, Cetin M, Altay C, Yurtsever Z, David KL, Faivre L, Ismail EA, Gräsbeck R, de la Chapelle A. Hereditary juvenile cobalamin deficiency caused by mutations in the intrinsic factor gene. Proc. Natl. Acad. Sci. USA. 2005;102:4130–4133. doi: 10.1073/pnas.0500517102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Coelho D, Kim JC, Miousse IR, Fung S, du Moulin M, Buers I, Suormala T, Burda P, Frapolli M, Stucki M, Nürnberg P, Thiele H, Robenek H, Höhne W, Longo N, Pasquali M, Mengel E, Watkins D, Shoubridge EA, Majewski J, Rosenblatt DS, Fowler B, Rutsch F, Baumgartner MR. Mutations in ABCD4 cause a new inborn error of vitamin B12 metabolism. Nat. Genet. 2012;44:1152–1155. doi: 10.1038/ng.2386. [DOI] [PubMed] [Google Scholar]
- [37].Kim JC, Lee N-C, Hwu PW-L, Chien YH, Fahiminiya S, Majewski J, Watkins D, Rosenblatt DS. Late onset of symptoms in an atypical patient with cblJ inborn error of vitamin B12 metabolism: Diagnosis and novel mutation revealed by exome sequencing. Mol. Genet. Metab. 2012;107:664–668. doi: 10.1016/j.ymgme.2012.10.005. [DOI] [PubMed] [Google Scholar]
- [38].Rutsch F, Gailus S, Miousse IR, Suormala T, Sagné C, Toliat MR, Nürnberg G, Wittkampf T, Buers I, Sharifi A, Stucki M, Becker C, Baumgartner M, Robenek H, Marquardt T, Höhne W, Gasnier B, Rosenblatt DS, Fowler B, Nürnberg P. Identification of a putative lysosomal cobalamin exporter altered in the cblF defect of vitamin B12 metabolism. Nat. Genet. 2009;41:234–239. doi: 10.1038/ng.294. [DOI] [PubMed] [Google Scholar]
- [39].Beedholm-Ebsen R, van de Wetering K, Hardlei T, Nexø E, Borst P, Moestrup SK. Identification of multidrug resistance protein 1 (MRP1/ABCC1) as a molecular gate for cellular export of cobalamin. Blood. 2010;115:1632–1639. doi: 10.1182/blood-2009-07-232587. [DOI] [PubMed] [Google Scholar]
- [40].Shah NP, Beech CM, Sturm AC, Tanner SM. Investigation of the ABC transporter MRP1 in selected patients with presumed defects in vitamin B12 absorption. Blood. 2011;117:4397–4398. doi: 10.1182/blood-2010-12-322750. [DOI] [PubMed] [Google Scholar]
- [41].Fenton WA, Gravel RA, Rosenblatt DS. Disorders of propionate and methylmalonate metabolism. In: Scriver CR, Beaudet AL, Sly WS, Valle D, editors. The Metabolic & Molecular Basis of Inherited Disease. 8th ed McGraw-Hill; New York: 2001. pp. 2165–2193. [Google Scholar]
- [42].Kervestin S, Jacobson A. NMD: a multifaceted response to premature translational termination. Nat. Rev. Mol. Cell. Biol. 2012;13:700–712. doi: 10.1038/nrm3454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Hu PY, Waheed A, Sly WS. Partial rescue of human carbonic anhydrase II frameshift mutation by ribosomal frameshift. Proc. Natl. Acad. Sci. USA. 1995;92:2136–2140. doi: 10.1073/pnas.92.6.2136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Ovunc B, Otto EA, Vega-Warner V, Saisawat P, Ashraf S, Ramaswami G, Fathy HM, Schoeb D, Chernin G, Lyons RH, Yilmaz E, Hildebrandt F. Exome sequencing reveals cubilin mutation as a single-gene cause of proteinuria. J. Am. Soc. Nephrol. 2011;22:1815–1820. doi: 10.1681/ASN.2011040337. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.