

Abstract
Children with high-risk acute lymphoblastic leukemia in first complete remission can benefit from allogeneic hematopoietic stem cell transplantation. We analyzed the outcome of 211 children with high-risk acute lymphoblastic leukemia in first complete remission who were given an allogeneic transplant between 1990 and 2008; the outcome of patients who, despite having an indication for transplantation and a suitable donor, did not receive the allograft for different reasons in the same time period was not analyzed. Sixty-nine patients (33%) were transplanted between 1990 and 1999, 58 (27%) between 2000 and 2005, and 84 (40%) between 2005 and 2008. A matched family donor was employed in 138 patients (65%) and an unrelated donor in 73 (35%). The 10-year probabilities of overall and disease-free survival were 63.4% and 61%, respectively. The 10-year cumulative incidences of transplantation-related mortality and relapse were 15% and 24%, respectively. After 1999, no differences in either disease-free survival or transplant-related mortality were observed in patients transplanted from unrelated or matched family donors. In multivariate analysis, grade IV acute graft-versus-host disease was an independent factor associated with worse disease-free survival. By contrast, grade I acute graft-versus-host disease and age at diagnosis between 1 and 9 years were favorable prognostic variables. Our study, not intended to evaluate whether transplantation is superior to chemotherapy for children with acute lymphoblastic leukemia in first complete remission and high-risk features, shows that the allograft cured more than 60% of these patients; in the most recent period, the outcome of recipients of grafts from matched family and unrelated donors was comparable.
Introduction
Nowadays, around 80–85% of children with acute lymphoblastic leukemia (ALL) can be definitively cured through the use of risk-oriented chemotherapy protocols.1–4 However, there are still subsets of children with ALL in whom the probability of event-free survival with chemotherapy remains unsatisfactory, because of the high chance of disease recurrence.
Over the last years, much research on childhood ALL has focused on the validation of biological and clinical prognostic variables able to identify patients at high risk of relapse. These characteristics predicting disease recurrence have varied over time. Currently, some biological risk factors conveying a dismal prognosis in childhood ALL can be identified already at diagnosis. They include early thymocyte precursor phenotype, cytogenetic anomalies such as t(9;22), t(4;11), intrachromosomal amplification of chromosome 21 and molecular lesions recently identified as predicting biological resistance to conventional treatment (e.g. Ikaros mutations or JAK2 mutations).5–8 Other prognostic factors, such as lack of achievement of complete remission at the end of induction therapy, reflect sensitivity of leukemia cells to treatment.9 Additionally, in the last years, response to induction treatment, measured by evaluation of minimal residual disease, was shown to have an even stronger predictive value on the risk of leukemia recurrence.10,12
Over the past decades, differences in definition of high-risk criteria and in frontline chemotherapy protocols among the childhood ALL cooperative groups have produced varying results, with reported cure rates ranging from 30% to 60%.1–4,12–15
The dynamic evolution of the results obtained with chemotherapy protocols has led to relevant modifications of the eligibility criteria for allogeneic hematopoietic stem cell transplantation (HSCT), so that, for instance, patients with B-cell precursor ALL and more than 100×109/L white blood cells at diagnosis not responding to the steroid pre-phase should not be offered transplantation.
Several retrospective studies suggested that allogeneic HSCT from an HLA-identical sibling improves the prognosis of high-risk ALL patients in first complete remission compared with further intensified chemotherapy protocols.16–20 In 1995, a prospective, cooperative randomized study was carried out through the collaboration between the International Berlin-Frankfurt-Münster (I-BFM) Study Group and the Pediatric Working Party of the European Blood and Marrow Transplantation (EBMT) Group with the aim of comparing the results of allogeneic HSCT from a compatible related donor with those of children with high-risk ALL in first complete remission diagnosed between 1995 and 2000 and treated with chemotherapy alone.21 In this study, 357 children were enrolled, 280 of whom received chemotherapy while the other 77 were given chemotherapy followed by related-donor HSCT, on the basis of genetic chance (i.e., availability of an HLA-identical donor). The 5-year disease-free survival was 40.6% in children allocated to chemotherapy and 56.7% in those given HSCT (P=0.02). Balduzzi et al. concluded that children with high-risk ALL in first complete remission benefit more from related-donor HSCT than from chemotherapy alone. The gap between the two strategies increases as the risk profile of the patient worsens. Indeed, children who were eligible for the study because of induction failure benefited more than others from related-donor availability.21
Based on the same cohort of patients they had previously analyzed, Balduzzi et al. recently discussed the impact of the time elapsed in first complete remission on prognosis, as well as the potential influence of waiting time to transplantation. The relative advantage of HSCT from compatible related donors in high-risk childhood ALL was found whatever the time elapsed from first complete remission to HSCT.22
The issue of whether patients with high-risk ALL in first complete remission should undergo HSCT from donors other than an HLA-identical sibling remains controversial. The outcome of unrelated donor HSCT has improved greatly in recent years, mostly because of high-resolution HLA-typing and improved prevention/treatment of graft-versus-host disease (GvHD).23,24
In order to further investigate the impact of allogeneic HSCT in high-risk ALL patients, we carried out a retrospective, multicenter study to analyze the outcome of 211 consecutive ALL pediatric patients who underwent HSCT from a related or unrelated donor for ALL in first complete remission and whose data were reported to the Italian Association of Pediatric Hematology and Oncology (AIEOP)-HSCT Registry between 1990 and 2008.
Design and Methods
Between November 1990 and June 2008, 211 patients aged over 12 months, with high-risk ALL in first complete remission treated with a frontline AIEOP protocol, underwent allogeneic HSCT in 19 Italian pediatric transplant centers.25,26 Among these 211 patients, 69 (33%) were given the allograft between 1990 and 1999, 58 (27%) between 2000 and 2005, and 84 (40%) between 2005 and 2008.
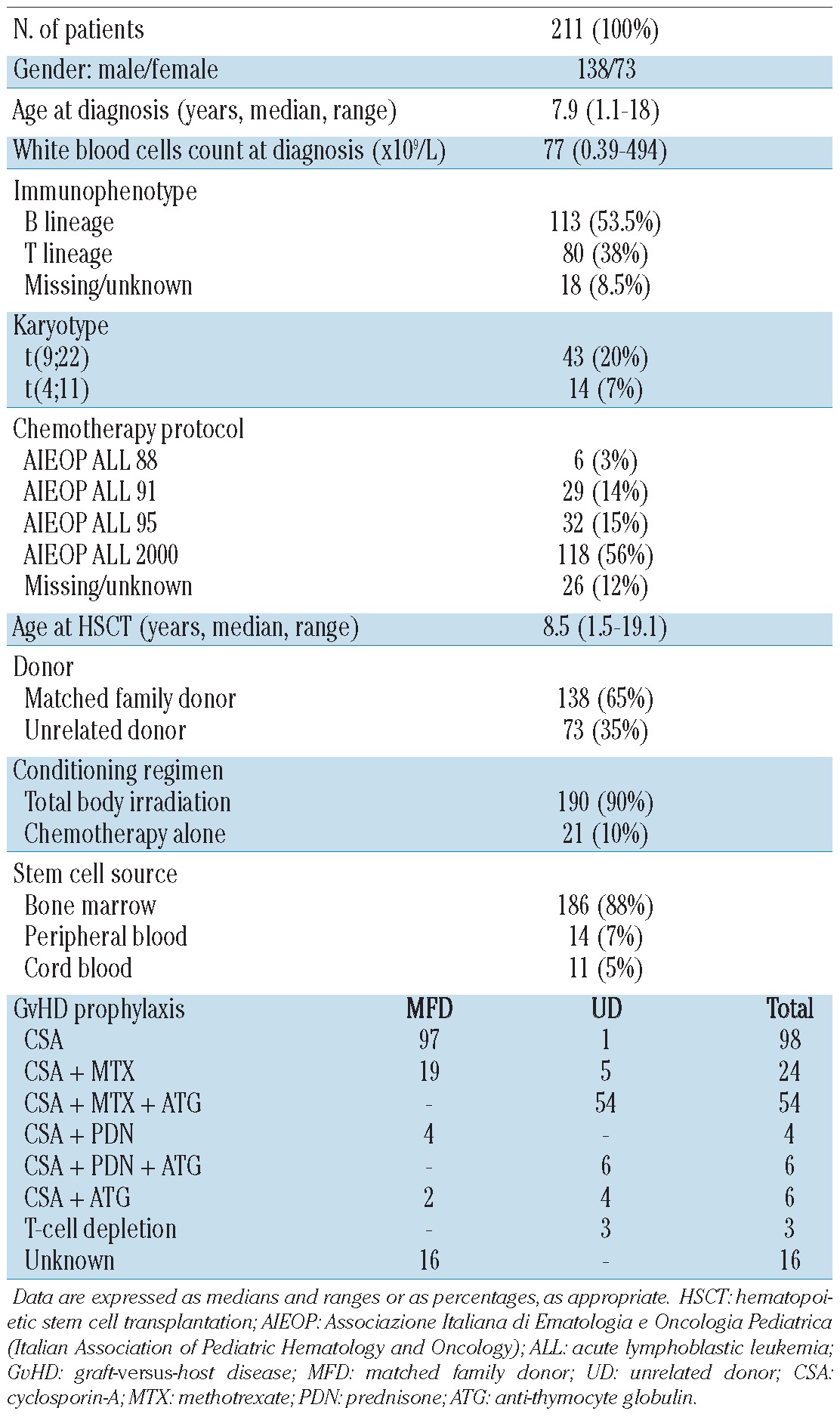
The clinical characteristics of the patients are reported in Table 1. In all donor-recipient pairs, histocompatibility was determined by serology for HLA-A and HLA-B antigens and by DNA typing for the HLA-DRB1 locus. In all patients transplanted with a graft from an unrelated donor, HLA-DRB1 typing was performed by a high-resolution allelic technique. After 1998, all class I and class II HLA alleles (i.e. HLA-A, HLA-B, HLA-C, DRB1, and DQβ1) were typed by a high-resolution DNA technique.
Table 1.
Characteristics of the 211 patients enrolled in the study and their transplants.
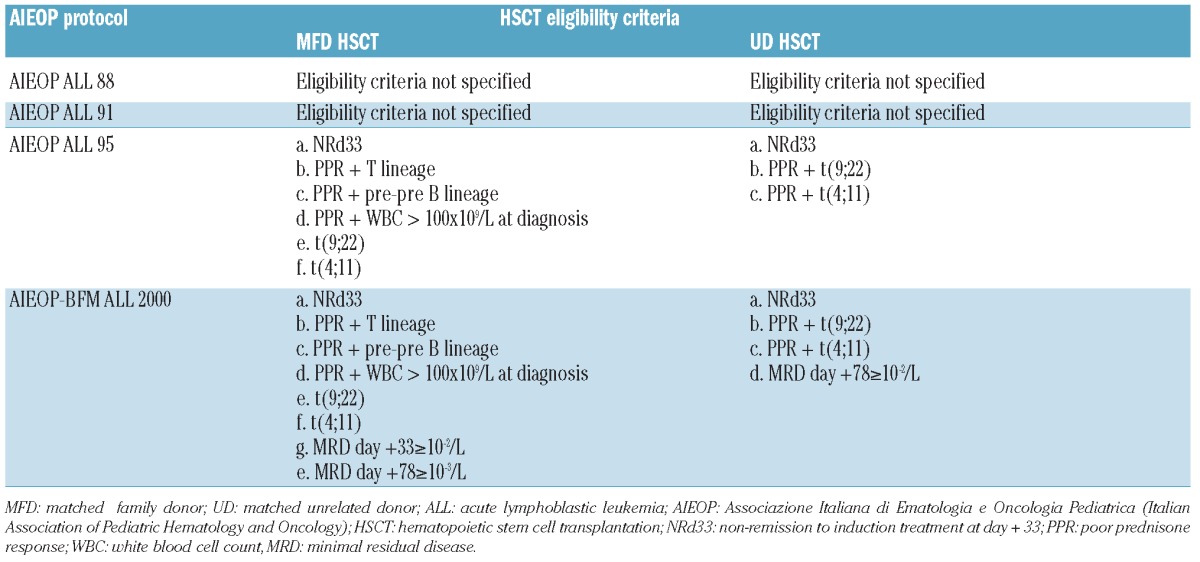
Eligibility criteria for HSCT are detailed in Table 2. In general, patients with an estimated probability of event-free survival below 40% in the various time periods of the study were considered to be eligible to receive HSCT. The 35 patients enrolled in protocols AIEOP-ALL88 and AIEOP-ALL91 who received HSCT had been considered eligible for allografting mainly because of a poor prednisone response and either hyperleukocytosis or a T-cell phenotype.
Table 2.
Eligibility criteria for transplantation with a graft from a matched family donor or unrelated donor in high-risk ALL.
All parents or guardians signed the appropriate informed consent, approved by the local ethics committee or Institutional Review Board.
Minimal residual disease analysis and risk group stratification
Minimal residual disease was investigated in patients enrolled in the AIEOP/BFM ALL-2000 protocol at two consecutive follow-up time-points, namely on day 33 (TP1) and day 78 (TP2) after the beginning of induction therapy, as described elsewhere.27–34 Technical details are reported in the Online Supplementary Material. Patients enrolled in the AIEOP-BFM 2000 protocol were stratified into different risk groups considering also the results of minimal residual disease evaluation.26,35 For reliable standard risk ratification, minimal residual disease negativity at TP1 and TP2 was required. Patients with high amounts of residual disease (≥10−3) at TP2 were stratified in the minimal residual disease-high-risk group. Minimal residual disease-intermediate risk stratification was feasible in the case that minimal residual disease was detected at either one or both time-points, but at a level <10−3 at TP2.
Statistical analysis
The criteria used for defining complete remission, neutrophil and platelet engraftment, acute and chronic GvHD, transplant-related mortality, relapse, overall survival, and disease-free survival are reported in the Online Supplementary Material.36,37
Overall and disease-free survival were calculated according to the Kaplan-Meier method. Acute and chronic GvHD occurrence, transplant-related mortality and relapse incidence were expressed as cumulative incidence curves, in order to adjust the analysis for competing risks. The significance of differences between the disease-free survival curves was estimated by the log–rank test (Mantel–Cox), while, in univariate analyses, Gray’s test was used to assess differences between relapse incidence and transplant-related mortality.38 A multivariate analysis on disease-free survival was performed using the Cox proportional regression model,39 while the proportional sub-distribution hazard regression model was used to perform multivariate analyses of relapse incidence and transplant-related mortality. P values <0.05 were considered to be statistically significant. Further details on statistical analyses are presented in the Online Supplementary Material.
Results
The median follow-up was 6.8 years (range, 1.6–18.9) for surviving patients, and 0.9 years for patients who died (range, 0.2–4.3).
Engraftment and graft-versus-host disease
Neutrophil and platelet engraftment occurred at a median of 16 (range, 8–44) and 24 (range, 11–97) days after HSCT, respectively. Neutrophil engraftment was obtained after 14.5 (range, 8–38) days for recipients of grafts from matched family donors and 19 (range, 11–44) days for recipients of unrelated donor grafts (P<0.0001). Platelet engraftment for matched family donor and unrelated donor transplant recipients was reached after a median time of 25.5 (range 11–81) and 25 (range, 12–97) days, respectively (P=NS). Engraftment of children given cord blood cells was delayed (data not shown).
Fifty-two patients did not develop acute GvHD (25%), 67 patients had grade I GvHD (32%), 62 patients grade II (29%), 19 patients grade III (9%) and 11 patients developed grade IV acute GvHD (5%).
The cumulative incidence of grade III–IV acute GvHD was 12% [95% confidence intervals (95% CI), 8–17]. It was 10% (95% CI, 6–17) and 15% (95% CI, 9–26) for matched family donor and unrelated donor transplant recipients, respectively (P=NS) and 12% (95% CI, 8–18) and 27% (95% CI, 10–72) for bone marrow and peripheral blood HSCT recipients, respectively (P=NS). No case of grade III–IV acute GvHD was recorded in patients receiving cord blood cells.
Considering recipient age, a higher incidence of grade III–IV acute GvHD was observed in patients older than 15 years (35%; 95% CI, 19–67) than in patients aged between 1–9 years (10%; 95% CI, 6–17) or 10–14 years (9%; 95% CI, 4–19) (P=0.0084).
One hundred forty-three patients did not develop chronic GvHD (68%), 25 patients developed limited chronic GvHD (12%), and 27 patients had extensive chronic GvHD (13%). No data were available for the remaining 16 patients (7%). The overall cumulative incidence of chronic GvHD was 27% (95% CI, 21–34).
Overall and disease-free survival
The 10-year overall survival probability was 63.4% (95% CI, 57–70) (Figure 1). The 10-year probability of disease-free survival for all patients analyzed in the study was 61% (95% CI, 54–68) (Figure 1). In univariate analyses, factors influencing disease-free survival probability were the patient’s age at diagnosis, and acute GvHD severity (Online Supplementary Table S1, Figure 2A).
Figure 1.
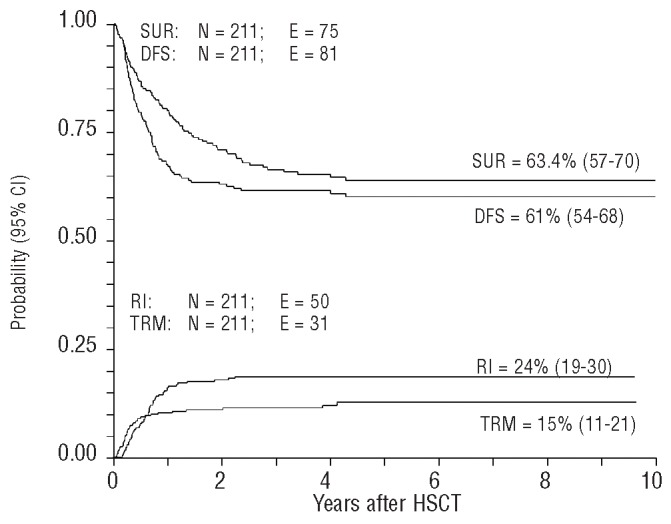
Overall survival (SUR), disease-free survival (DFS), transplant-related mortality (TRM) and relapse incidence (RI) of the whole population analyzed. TRM and RI are expressed as cumulative incidence curves, in order to adjust the analysis for competing risks.
Figure 2.
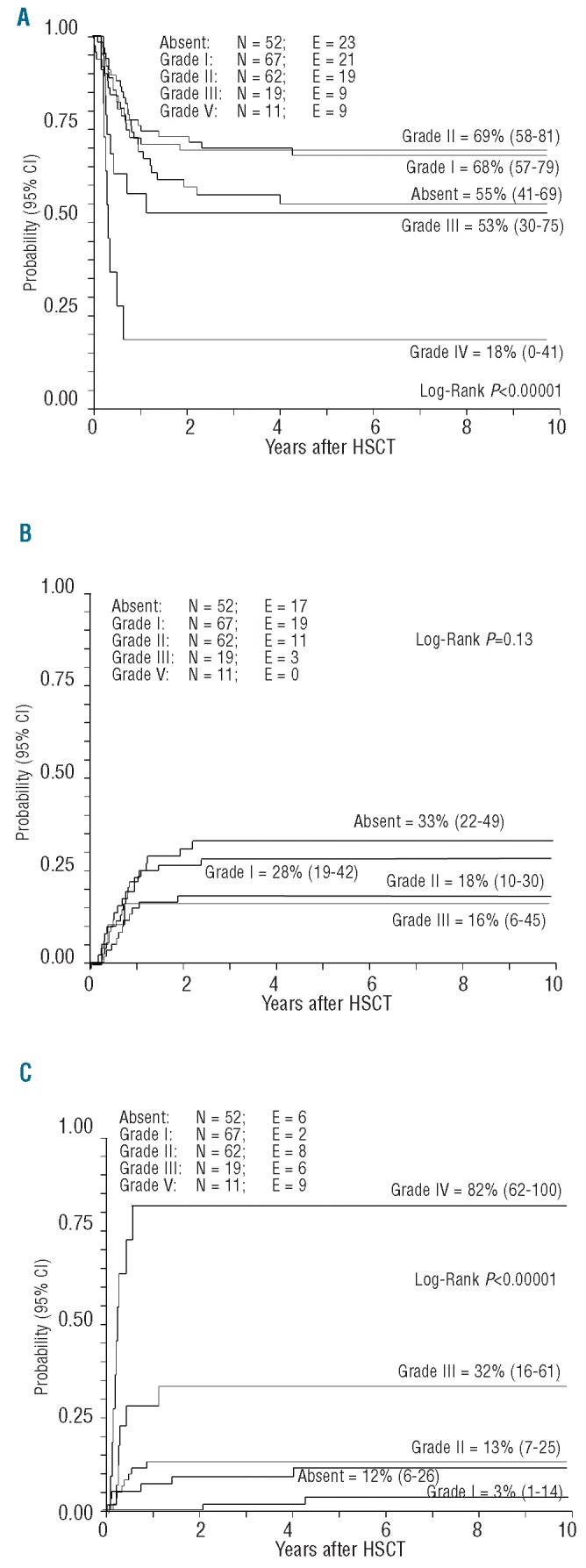
(A) Disease-free survival, (B) relapse incidence and (C) transplantation-related mortality according to severity of acute GvHD.
Children aged between 1 and 9 years at diagnosis had a better disease-free survival than those aged between 10–14 years, or older than 15 years: 68% (95% CI, 60–76), 55% (95% CI, 43–67), and 34% (95% CI, 11–57), respectively, P=0.026). The type of donor had an impact on disease-free survival only among patients transplanted between 1990 and 1999: matched family donor, 65% (95% CI, 53–77); unrelated donor, 33% (95% CI, 3–64), P=0.06). There were no differences between recipients of matched family donor or unrelated donor grafts for patients who underwent HSCT after 2000 (Online Supplementary Table S1 and Figure 3A-C).
Figure 3.
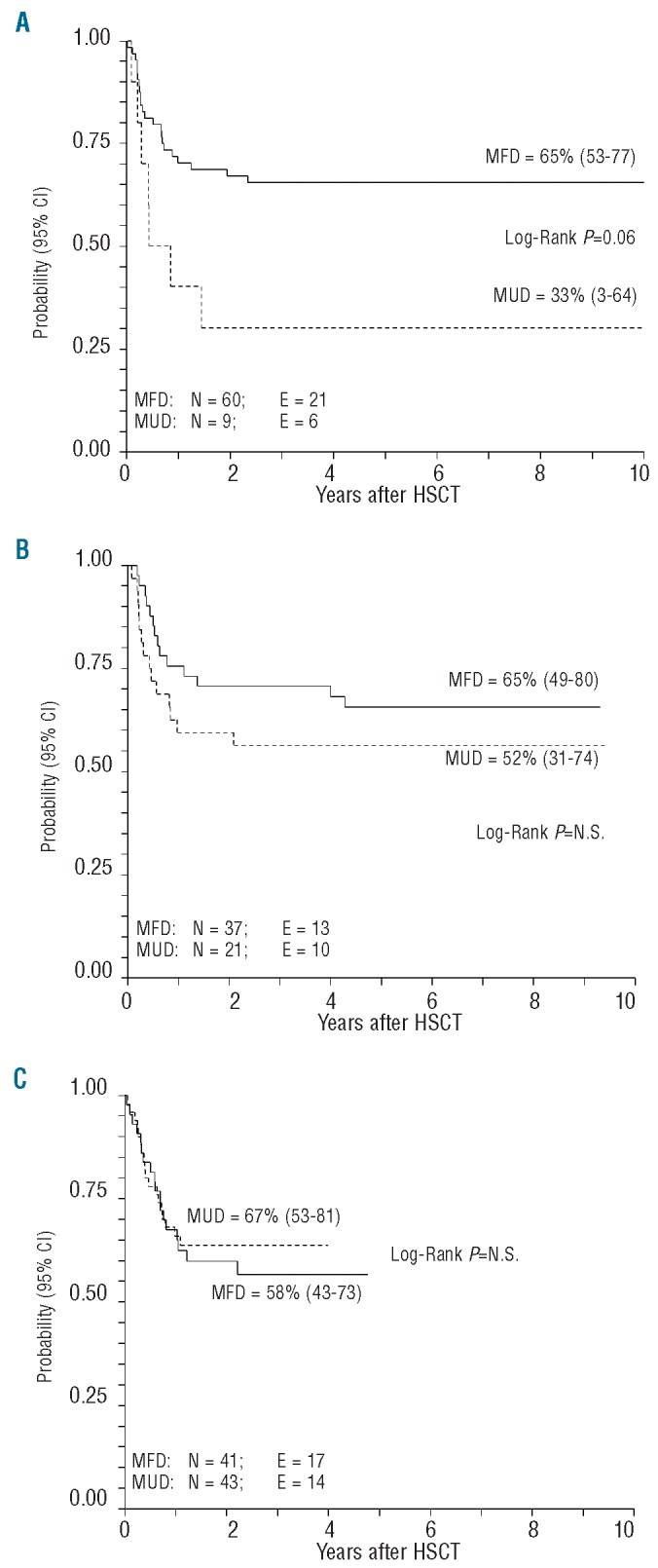
Disease-free survival according to year of transplantation and donor type: matched unrelated donor (MUD) or matched family donor (MFD). (A) 1990–1999. (B) 2000–2004. (C) 2005–2008.
In univariate analyses, the disease-free survival rate was higher in patients with grade 0-II acute GvHD than in those with grade III–IV acute GvHD: 65% (95% CI, 58–62) versus 40% (95% CI, 22–58), respectively; (P=0.002). Other variables had no impact on disease-free survival (see Online Supplementary Table S1 for details).
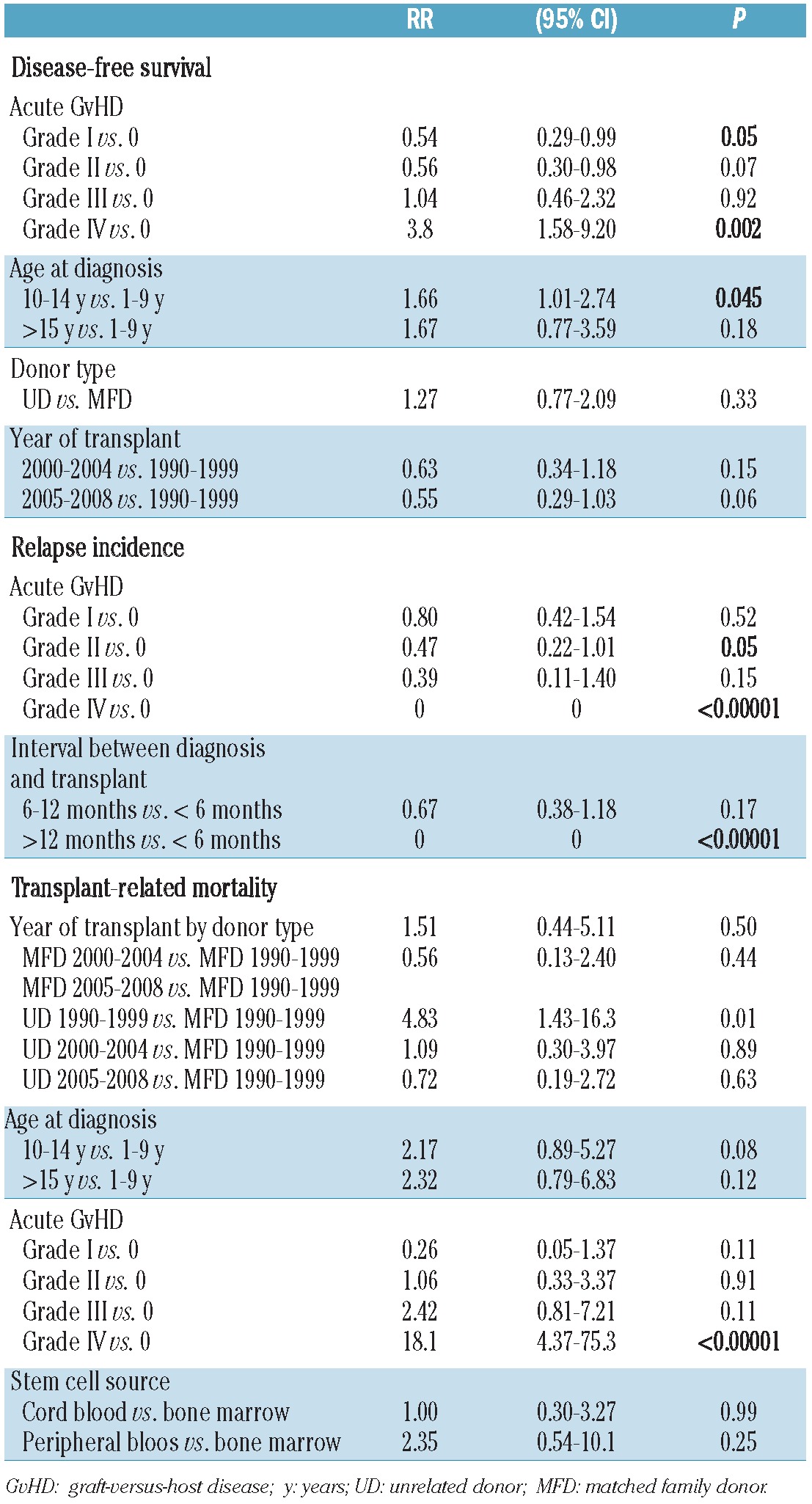
In multivariate analyses, the occurrence of grade IV acute GvHD [RR=3.8 (95% CI, 1.58–9.20), P=0.002] was an independent factor associated with a worse disease-free survival rate. In contrast, the occurrence of grade I acute GvHD was an independent favorable prognostic variable for disease-free survival [RR=0.54 (95% CI, 0.29–0.99), P=0.05) as was age at diagnosis between 1 and 9 years (P=0.045) (Table 3).
Table 3.
Multivariate analyses of variables influencing the probability of disease-free survival, and the cumulative incidence of transplant-related mortality, and relapse.
Relapse incidence
The median time from HSCT to relapse was 8 months (range, 2–28). The overall relapse incidence was 24% (95% CI, 19–30) (Figure 1). In univariate analyses, patients who underwent HSCT between 6 and 12 months or within 6 months after diagnosis had a higher risk of relapse than those transplanted more than 12 months after diagnosis: 32% (95% CI, 23–46), 22% (95% CI, 16–31), and 0%, respectively; (P=0.02).
Moreover, patients with grade 0-I acute GvHD [30% (95% CI, 23–40)] had a higher relapse incidence than patients who developed grade II–IV acute GvHD [15% (95% CI, 9–25); P=0.013].
None of the other variables analyzed was associated with an increased incidence of relapse (Online Supplementary Table S2, Figure 2B). In multivariate analysis, both the time-interval between diagnosis and HSCT and the occurrence of grade II–IV acute GvHD remained independent prognostic variables for relapse incidence (Table 3).
Transplant-related mortality
The overall cumulative incidence of transplant-related mortality was 15% (95% CI, 11–21) (Figure 1). In univariate analyses, factors significantly associated with an increased risk of transplant-related mortality were age at diagnosis and transplantation, year of transplantation by donor type, stem cell source and occurrence of acute or chronic GvHD (Online Supplementary Table S3).
In detail, transplant-related mortality was lower for children aged 1–9 years at diagnosis as compared to those aged 10–14 years or older than 15 years [8% (95% CI, 4–15), 18% (95% CI, 11–31), and 54% (95% CI, 34–84), respectively, P<0.00001]. Among the patients older than 15 years, we observed a marked reduction of transplant-related mortality on the basis of the period of transplant (1990–1999: 100%, 2000–2004: 65%, and 2005–2008: 16%, P=0.03) (Online Supplementary Table S3).
It is noteworthy that the impact of the type of donor on transplant-related mortality was only observed for patients transplanted between 1990 and 1999 [unrelated donor: 44% (95% CI, 21–92), matched family donor: 8% (95% CI, 4–19), P=0.0043)]. There were no differences in transplant-related mortality between patients transplanted with grafts from matched family donors or unrelated donors after 2000. A higher transplant-related mortality was observed in patients receiving peripheral blood stem cells than in patients transplanted with either cord blood or bone marrow stem cells [45% (95% CI, 24–87), 21% (95% CI, 8–58) and 13% (95% CI, 9–19), P=0.0062].
The transplant-related mortality rates of patients with grade III and IV acute GvHD were 32% (95% CI, 16–61) and 82% (95% CI, 62–100), respectively, versus 13% (95% CI, 7–25), 3% (95% CI, 1–14) and 12% (95% CI, 6–26) for patients with grade II, I and 0 acute GvHD, respectively (P<0.00001) (Figure 2C).
None of the other variables analyzed was associated with an increased incidence of transplant-related mortality (Online Supplementary Table S3). When the Cox proportional hazard regression model was applied, the strongest predictors of transplant-related mortality were grade IV acute GvHD [RR 18.1 (95% CI, 4.37–75.3), P<0.00001] and unrelated donor for HSCT performed between 1990 and 1999 [RR 4.83 (95% CI, 1.43–16.3), P=0.01] (Table 3).
The causes of transplant-related mortality according to year of transplant and donor type are reported in Online Supplementary Table S4.
Minimal residual disease
Overall, among the patients who had been enrolled in the AIEOP-BFM ALL-2000 protocol, 100 had minimal residual disease results available at TP1 and TP2. On the basis of these results, 27 out of these 100 patients were classified as being at intermediate risk and 73 at high-risk.
The influence of minimal residual disease on disease-free survival, transplant-related mortality and relapse incidence was then analyzed. The disease-free survival was 74% (95% CI, 58–91) for intermediate-risk patients and 60% (95% CI, 48–72) for high-risk children (P=0.369) (Online Supplementary Table S1). Likewise, minimal residual disease had no apparent effect on relapse incidence [intermediate-risk: 19% (95% CI, 9–42), high-risk: 23% (95% CI, 15–35), P=0.77] or transplant-related mortality [intermediate risk: 8% (95% CI, 2–29), high-risk: 17% (95% CI 10–30), P=0.53] (Online Supplementary Tables S2 and S3).
Discussion
The prognosis of patients with childhood ALL has improved dramatically over the past quarter of a century. However, despite advances in the diagnosis and treatment of childhood ALL, there are still subgroups of patients who, after obtaining a first complete remission, have a high risk of failing to benefit from current multiagent chemotherapy regimens and, thus, require alternative treatment strategies, such as allogeneic HSCT, to prevent disease relapse.40,41
We analyzed a large series of pediatric and adolescent patients with ALL, whose disease was defined as high-risk according to the criteria of the different AIEOP protocols running at the time of the allograft, and who underwent either related or unrelated HSCT while in first complete remission, from 1990 to 2008. This is a retrospective, multicenter study analyzing the outcome of 211 consecutive ALL pediatric patients who received the allograft, while we did not address the issue of patients who, despite having an indication for HSCT and a suitable donor, did not receive the allograft because of an event occurring before the procedure or precluding its performance. Furthermore, it should be emphasized that this study was not intended to compare the results of HSCT with those obtained with conventional chemotherapy.
We found a 10-year disease-free survival rate of 61% in the whole cohort of 211 patients. This result compares favorably with those in other published studies on children with high-risk ALL in first complete remission treated with allogeneic HSCT. Balduzzi et al. performed an international prospective study showing that high-risk ALL children in first complete remission who underwent matched family donor HSCT had a 16% benefit in 5-year disease-free survival compared with those who received chemotherapy alone (56.7% versus 40.6%, P=0.02). Similarly, the Children’s Oncology Group reported a 5-year disease-free survival of 58.6% in a small cohort of high-risk ALL patients in first complete remission who received HSCT from a family member.41 It is noteworthy that the probability of disease-free survival of our children with t(9;22) approached 60%, a figure comparable to that of Philadelphia chromosome-negative ALL patients and should be considered remarkable having been mainly obtained in patients treated in an era before the introduction of tyrosine kinase inhibitors.
Our results indicate that after 1999 transplant outcomes are similar in recipients of grafts from unrelated donor or matched family donors. Specifically, there were no obvious differences in disease-free survival, and most importantly, in the risk of transplant-related mortality. These results are in agreement with previously published findings by the Italian pediatric group in children or adolescents with ALL in second complete remssion24,42 and suggest that indications for HSCT in first complete remission should be similar for patients with an HLA-identical sibling or a matched unrelated donor.
We observed that the group of patients 10–14 years old had a worse 10-year disease-free survival rate compared to those 1–9 years old [RR 1.66 (95% CI 1.01–2.74), P=0.045]. The lower disease-free survival rate in the 10–14-year olds was mainly due to a slightly higher transplant-related mortality [RR 2.17 (95% CI 0.89–5.27), P=0.08], while the relapse incidence was comparable. The transplant-related mortality of adolescents (i.e. patients above 15 years of age) was high, although a marked improvement was observed over time. Thus, HSCT in ALL in first remission in patients over 15 years old must be carefully weighed, considering the pros and cons of alternative treatments.
The effect of the conditioning regimen on various transplantation outcomes in children and adults with ALL was recently analyzed by the International Bone Marrow Transplant Registry.43,44 Among the patients with high-risk ALL in first complete remission, a comparison of various conditioning regimens, all including total body irradiation combined with different cytotoxic drugs, did not show any significant differences in transplant-related mortality, relapse incidence and disease-free survival. In our cohort of patients, we did not observe any differences in disease-free survival, relapse incidence or early transplant-related mortality between patients given either total body irradiation or chemotherapy-based regimens (90% versus 10% of patients), although the latter group included a limited number of children. This observation needs to be confirmed by larger prospective randomized studies.
We also analyzed the impact of risk stratification based on minimal residual disease at day +33 and +78 after diagnosis on HSCT outcome. Minimal residual disease has already proven to be the most sensitive method for evaluating treatment response and, thus, an important prognostic factor for a better stratification of patients in different risk classes in front-line treatment protocols.45,46 Recently, the AIEOP-BFM ALL 2000 multicenter trial demonstrated the predictive role of minimal residual disease on relapse occurrence, the 5-year disease-free survival being 92.3%, 77.6%, and 50.1% for B-cell precursor ALL and 91.1%, 80.6%, and 49.8% for T-cell precursor ALL in patients defined as being at standard, intermediate or high risk according to the minimal residual disease findings.26,35 On these bases, the determination of minimal residual disease also allows a better selection of those patients who need the immunological effect of an allograft to be cured. In accordance with the AIEOP-BFM criteria, after 2000, the presence of minimal residual disease ≥10−2 at TP1 or ≥10−3 at TP2, as well as minimal residual disease ≥10−2 at TP2, were considered independent indications for matched family donor or unrelated donor HSCT, respectively. In this study, 27 and 73 out of the 100 patients investigated had minimal residual disease-intermediate or high risk features, respectively. The 27 with intermediate risk on the basis of minimal residual disease were given the allograft because of the presence of other characteristics rendering them classifiable as high-risk. The probability of disease-free survival and relapse incidence of patients at intermediate or high risk on the bases of minimal residual disease were comparable. Thus, although the number of patients investigated was limited, our results seem to suggest that HSCT could reduce the effects of minimal residual disease, evaluated during the first 11 weeks of treatment, on patients’ outcome. Unfortunately, we do not have data on pre-transplant minimal residual disease, a variable shown to influence patients’ outcome profoundly.10,47,48 It is reasonable to hypothesize that, nowadays, pre-transplant minimal residual disease is one of the most important prognostic factors and that it is more important than intermediate risk and high-risk minimal residual disease determinations performed several months prior to HSCT.
Low-intensity GvHD prophylaxis has been reported to facilitate better immune control of leukemia cells, this resulting in a decreased risk of relapse.49 The benefit offered by the occurrence of GvHD in terms of reduction of disease recurrence can, however, be offset by a higher incidence of transplant-related mortality. In our cohort, considering patients who did not have acute GvHD as the reference group, a lower relapse incidence was observed in patients who developed grade II or grade IV acute GvHD, although the latter had a transplant-related mortality of 82%. In multivariate analysis, a significantly better probability of disease-free survival was found only in patients who developed grade I acute GvHD, while grade II acute GvHD was associated with a trend towards better disease-free survival. Collectively, these data suggest that only mild GvHD can have a favorable impact on the final outcome.
We found an inverse correlation between the time elapsed from diagnosis to HSCT and the risk of relapse. Specifically, no relapses were reported in patients who underwent HSCT more than 12 months after diagnosis, while the relapse incidence was higher in those transplanted within 6 or 12 months from diagnosis. The most plausible explanation for this finding might be that patients who received HSCT more than 12 months after diagnosis could have already been cured by chemotherapy. However, we cannot rule out that longer chemotherapy consolidation might have had a positive impact on post-HSCT relapse risk.
The higher transplant-related mortality observed in our patients given peripheral blood progenitors confirms previously published data from Eapen et al.50 Thus, bone marrow must be preferentially chosen over peripheral blood as the source of stem cells for children with either malignant or non-malignant disorders, although, for the unrelated donor, the ultimate choice of the stem cell source pertains to the donor. In the case the unrelated donor chooses to donate peripheral blood stem cells, the risk of performing transplantation must be evaluated carefully.
In this study we demonstrated that remarkable progress in terms of reduction of transplant-related mortality has been achieved over the years, especially for transplants performed after 2000. Specifically, we observed no deaths from bleeding complications in patients transplanted after 2000, which is reasonably attributable to the improvement of supportive care (Online Supplementary Table S4).
In conclusion, our data indicate that allogeneic HSCT is a suitable option for a subgroup of children with ALL in first complete remission with well-defined high-risk features. Since the outcome of recipients of grafts from matched family donors or unrelated donors was comparable in the most recent period, the indications for transplantation from either an HLA-identical sibling or an unrelated volunteer should now be considered identical. Overall, these indications changed significantly over time and are now more restricted than those adopted in past years.
Footnotes
The online version of this article has a Supplementary Appendix.
Funding
This work was partly supported by a special grant (5 per mille) from AIRC (Associazione Italiana Ricerca sul Cancro) to FL and by the Oncology Network of Piedmont and Valle d’Aosta and Compagnia di San Paolo Foundation (Turin) to FF.
Authorship and Disclosures
Information on authorship, contributions, and financial & other disclosures was provided by the authors and is available with the online version of this article at www.haematologica.org.
References
- 1.Silverman LB, Gelber RD, Dalton VK, Asselin BL, Barr RD, Clavell LA, et al. Improved outcome for children with acute lymphoblastic leukemia: results of Dana-Farber Consortium Protocol 91-01. Blood. 2001;97(5):1211–8 [DOI] [PubMed] [Google Scholar]
- 2.Conter V, Arico M, Valsecchi MG, Rizzari C, Testi A, Miniero R, et al. Intensive BFM chemotherapy for childhood ALL: interim analysis of the AIEOP-ALL 91 study. Associazione Italiana Ematologia Oncologia Pediatrica. Haematologica. 1998;83(9):791–9 [PubMed] [Google Scholar]
- 3.Gaynon PS, Steinherz PG, Bleyer WA, Ablin AR, Albo VC, Finklestein JZ, et al. Improved therapy for children with acute lymphoblastic leukemia and unfavorable presenting features: a follow-up report of the Children’s Cancer Group Study CCG-106. J Clin Oncol. 1993;11(11):2234–42 [DOI] [PubMed] [Google Scholar]
- 4.Arico M, Valsecchi MG, Conter V, Rizzari C, Pession A, Messina C, et al. Improved outcome in high-risk childhood acute lymphoblastic leukemia defined by prednisone-poor response treated with double Berlin-Frankfurt-Muenster protocol II. Blood. 2002;100(2):420–6 [DOI] [PubMed] [Google Scholar]
- 5.Coustan-Smith E, Mullighan CG, Onciu M, Behm FG, Raimondi SC, Pei D, et al. Early T-cell precursor leukaemia: a subtype of very high-risk acute lymphoblastic leukaemia. Lancet Oncol. 2009;10(2):147–56 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Arico M, Schrappe M, Hunger SP, Carroll WL, Conter V, Galimberti S, et al. Clinical outcome of children with newly diagnosed Philadelphia chromosome-positive acute lymphoblastic leukemia treated between 1995 and 2005. J Clin Oncol. 2010;28(31): 4755–61 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chen IM, Harvey RC, Mullighan CG, Gastier-Foster J, Wharton W, Kang H, et al. Outcome modeling with CRLF2, IKZF1, JAK, and minimal residual disease in pediatric acute lymphoblastic leukemia: a Children’s Oncology Group study. Blood. 2012;119(15):3512–22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Moorman AV. The clinical relevance of chromosomal and genomic abnormalities in B-cell precursor acute lymphoblastic leukaemia. Blood Rev. 2012;26(3):123–35 [DOI] [PubMed] [Google Scholar]
- 9.Schrappe M, Hunger SP, Pui CH, Saha V, Gaynon PS, Baruchel A, et al. Outcomes after induction failure in childhood acute lymphoblastic leukemia. N Engl J Med. 2012;366(15):1371–81 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bader P, Kreyenberg H, Henze GH, Eckert C, Reising M, Willasch A, et al. Prognostic value of minimal residual disease quantification before allogeneic stem-cell transplantation in relapsed childhood acute lymphoblastic leukemia: the ALL-REZ BFM Study Group. J Clin Oncol. 2009;27(3):377–84 [DOI] [PubMed] [Google Scholar]
- 11.von Stackelberg A, Volzke E, Kuhl JS, Seeger K, Schrauder A, Escherich G, et al. Outcome of children and adolescents with relapsed acute lymphoblastic leukaemia and non-response to salvage protocol therapy: a retrospective analysis of the ALL-REZ BFM Study Group. Eur J Cancer. 2010;47(1):90–7 [DOI] [PubMed] [Google Scholar]
- 12.Schrappe M, Reiter A, Ludwig WD, Harbott J, Zimmermann M, Hiddemann W, et al. Improved outcome in childhood acute lymphoblastic leukemia despite reduced use of anthracyclines and cranial radiotherapy: results of trial ALL-BFM 90. German-Austrian-Swiss ALL-BFM Study Group. Blood. 2000;95(11):3310–22 [PubMed] [Google Scholar]
- 13.Schrappe M, Arico M, Harbott J, Biondi A, Zimmermann M, Conter V, et al. Philadelphia chromosome-positive (Ph+) childhood acute lymphoblastic leukemia: good initial steroid response allows early prediction of a favorable treatment outcome. Blood. 1998;92(8):2730–41 [PubMed] [Google Scholar]
- 14.Uckun FM, Nachman JB, Sather HN, Sensel MG, Kraft P, Steinherz PG, et al. Clinical significance of Philadelphia chromosome positive pediatric acute lymphoblastic leukemia in the context of contemporary intensive therapies: a report from the Children’s Cancer Group. Cancer. 1998;83(9):2030–9 [PubMed] [Google Scholar]
- 15.Pui CH, Gaynon PS, Boyett JM, Chessells JM, Baruchel A, Kamps W, et al. Outcome of treatment in childhood acute lymphoblastic leukaemia with rearrangements of the 11q23 chromosomal region. Lancet. 2002;359(9321):1909–15 [DOI] [PubMed] [Google Scholar]
- 16.Chessells JM, Bailey C, Wheeler K, Richards SM. Bone marrow transplantation for high-risk childhood lymphoblastic leukaemia in first remission: experience in MRC UKALL X. Lancet. 1992;340(8819):565–8 [DOI] [PubMed] [Google Scholar]
- 17.Saarinen UM, Mellander L, Nysom K, Ringden O, Schroeder H, Glomstein A, et al. Allogeneic bone marrow transplantation in first remission for children with very high-risk acute lymphoblastic leukemia: a retrospective case-control study in the Nordic countries. Nordic Society for Pediatric Hematology and Oncology (NOPHO). Bone Marrow Transplant. 1996;17(3):357–63 [PubMed] [Google Scholar]
- 18.Uderzo C, Valsecchi MG, Balduzzi A, Dini G, Miniero R, Locatelli F, et al. Allogeneic bone marrow transplantation versus chemotherapy in high-risk childhood acute lymphoblastic leukaemia in first remission. Associazione Italiana di Ematologia ed Oncologia Pediatrica (AIEOP) and the Gruppo Italiano Trapianto di Midollo Osseo (GITMO). Br J Haematol. 1997;96(2):387–94 [DOI] [PubMed] [Google Scholar]
- 19.Wheeler KA, Richards SM, Bailey CC, Gibson B, Hann IM, Hill FG, et al. Bone marrow transplantation versus chemotherapy in the treatment of very high-risk childhood acute lymphoblastic leukemia in first remission: results from Medical Research Council UKALL X and XI. Blood. 2000;96(7):2412–8 [PubMed] [Google Scholar]
- 20.Sharathkumar A, Saunders EF, Dror Y, Grant R, Greenberg M, Weitzman S, et al. Allogeneic bone marrow transplantation vs chemotherapy for children with Philadelphia chromosome-positive acute lymphoblastic leukemia. Bone Marrow Transplant. 2004;33(1):39–45 [DOI] [PubMed] [Google Scholar]
- 21.Balduzzi A, Valsecchi MG, Uderzo C, De Lorenzo P, Klingebiel T, Peters C, et al. Chemotherapy versus allogeneic transplantation for very-high-risk childhood acute lymphoblastic leukaemia in first complete remission: comparison by genetic randomisation in an international prospective study. Lancet. 2005;366(9486):635–42 [DOI] [PubMed] [Google Scholar]
- 22.Balduzzi A, De Lorenzo P, Schrauder A, Conter V, Uderzo C, Peters C, et al. Eligibility for allogeneic transplantation in very high risk childhood acute lymphoblastic leukemia: the impact of the waiting time. Haematologica. 2008;93(6):925–9 [DOI] [PubMed] [Google Scholar]
- 23.Woolfrey AE, Anasetti C, Storer B, Doney K, Milner LA, Sievers EL, et al. Factors associated with outcome after unrelated marrow transplantation for treatment of acute lymphoblastic leukemia in children. Blood. 2002;99(6):2002–8 [DOI] [PubMed] [Google Scholar]
- 24.Locatelli F, Zecca M, Messina C, Rondelli R, Lanino E, Sacchi N, et al. Improvement over time in outcome for children with acute lymphoblastic leukemia in second remission given hematopoietic stem cell transplantation from unrelated donors. Leukemia. 2002;16(11):2228–37 [DOI] [PubMed] [Google Scholar]
- 25.Conter V, Arico M, Basso G, Biondi A, Barisone E, Messina C, et al. Long-term results of the Italian Association of Pediatric Hematology and Oncology (AIEOP) Studies 82, 87, 88, 91 and 95 for childhood acute lymphoblastic leukemia. Leukemia. 2010;24 (2):255–64 [DOI] [PubMed] [Google Scholar]
- 26.Conter V, Bartram CR, Valsecchi MG, Schrauder A, Panzer-Grumayer R, Moricke A, et al. Molecular response to treatment redefines all prognostic factors in children and adolescents with B-cell precursor acute lymphoblastic leukemia: results in 3184 patients of the AIEOP-BFM ALL 2000 study. Blood. 2010;115(16):3206–14 [DOI] [PubMed] [Google Scholar]
- 27.Szczepanski T, Pongers-Willemse MJ, Langerak AW, Harts WA, Wijkhuijs AJ, van Wering ER, et al. Ig heavy chain gene rearrangements in T-cell acute lymphoblastic leukemia exhibit predominant DH6-19 and DH7-27 gene usage, can result in complete V-D-J rearrangements, and are rare in T-cell receptor alpha beta lineage. Blood. 1999; 93(12):4079–85 [PubMed] [Google Scholar]
- 28.van Dongen JJ, Macintyre EA, Gabert JA, Delabesse E, Rossi V, Saglio G, et al. Standardized RT-PCR analysis of fusion gene transcripts from chromosome aberrations in acute leukemia for detection of minimal residual disease. Report of the BIOMED-1 Concerted Action: investigation of minimal residual disease in acute leukemia. Leukemia. 1999;13(12):1901–28 [DOI] [PubMed] [Google Scholar]
- 29.van Dongen JJ, Langerak AW, Bruggemann M, Evans PA, Hummel M, Lavender FL, et al. Design and standardization of PCR primers and protocols for detection of clonal immunoglobulin and T-cell receptor gene recombinations in suspect lymphoprolifera-tions: report of the BIOMED-2 Concerted Action BMH4-CT98-3936. Leukemia. 2003; 17(12):2257–317 [DOI] [PubMed] [Google Scholar]
- 30.Germano G, Songia S, Biondi A, Basso G. Rapid detection of clonality in patients with acute lymphoblastic leukemia. Haematologica. 2001;86(4):382–5 [PubMed] [Google Scholar]
- 31.Verhagen OJ, Willemse MJ, Breunis WB, Wijkhuijs AJ, Jacobs DC, Joosten SA, et al. Application of germline IGH probes in real-time quantitative PCR for the detection of minimal residual disease in acute lymphoblastic leukemia. Leukemia. 2000;14(8):1426–35 [DOI] [PubMed] [Google Scholar]
- 32.van der Velden VH, Willemse MJ, van der Schoot CE, Hahlen K, van Wering ER, van Dongen JJ. Immunoglobulin kappa deleting element rearrangements in precursor-B acute lymphoblastic leukemia are stable targets for detection of minimal residual disease by real-time quantitative PCR. Leukemia. 2002;16 (5):928–36 [DOI] [PubMed] [Google Scholar]
- 33.van der Velden VH, Hochhaus A, Cazzaniga G, Szczepanski T, Gabert J, van Dongen JJ. Detection of minimal residual disease in hematologic malignancies by real-time quantitative PCR: principles, approaches, and laboratory aspects. Leukemia. 2003;17(6):1013–34 [DOI] [PubMed] [Google Scholar]
- 34.van Dongen JJ, Seriu T, Panzer-Grumayer ER, Biondi A, Pongers-Willemse MJ, Corral L, et al. Prognostic value of minimal residual disease in acute lymphoblastic leukaemia in childhood. Lancet. 1998;352(9142):1731–8 [DOI] [PubMed] [Google Scholar]
- 35.Schrappe M, Valsecchi MG, Bartram CR, Schrauder A, Panzer-Grumayer R, Moricke A, et al. Late MRD response determines relapse risk overall and in subsets of childhood T-cell ALL: results of the AIEOP-BFM-ALL 2000 study. Blood. 2011;118(8):2077–84 [DOI] [PubMed] [Google Scholar]
- 36.Przepiorka D, Weisdorf D, Martin P, Klingemann HG, Beatty P, Hows J, et al. 1994 Consensus Conference on Acute GVHD Grading. Bone Marrow Transplant. 1995;15 (6):825–8 [PubMed] [Google Scholar]
- 37.Shulman HM, Sullivan KM, Weiden PL, McDonald GB, Striker GE, Sale GE, et al. Chronic graft-versus-host syndrome in man. A long-term clinicopathologic study of 20 Seattle patients. Am J Med. 1980;69(2):204–17 [DOI] [PubMed] [Google Scholar]
- 38.Kaplan EL, Meier P. Non parametric estimation from incomplete observation. J Am Stat Assoc. 1958;53:457–81 [Google Scholar]
- 39.Cox DR. Regression models and life tables. J R Stat Soc. 1972;34:187–202 [Google Scholar]
- 40.Kersey JH. Fifty years of studies of the biology and therapy of childhood leukemia. Blood. 1997;90(11):4243–51 [PubMed] [Google Scholar]
- 41.Satwani P, Sather H, Ozkaynak F, Heerema NA, Schultz KR, Sanders J, et al. Allogeneic bone marrow transplantation in first remission for children with ultra-high-risk features of acute lymphoblastic leukemia: a Children’s Oncology Group study report. Biol Blood Marrow Transplant. 2007;13(2): 218–27 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Dini G, Zecca M, Balduzzi A, Messina C, Masetti R, Fagioli F, et al. No difference in outcome between children and adolescents transplanted for acute lymphoblastic leukemia in second remission. Blood. 2011;118(25):6683–90 [DOI] [PubMed] [Google Scholar]
- 43.Marks DI, Forman SJ, Blume KG, Perez WS, Weisdorf DJ, Keating A, et al. A comparison of cyclophosphamide and total body irradiation with etoposide and total body irradiation as conditioning regimens for patients undergoing sibling allografting for acute lymphoblastic leukemia in first or second complete remission. Biol Blood Marrow Transplant. 2006;12(4):438–53 [DOI] [PubMed] [Google Scholar]
- 44.Marks DI. Conditioning regimens for acute lymphoblastic leukaemia allografts. Bone Marrow Transplant. 2007;39(6):377. [DOI] [PubMed] [Google Scholar]
- 45.Jacquy C, Delepaut B, Van Daele S, Vaerman JL, Zenebergh A, Brichard B, et al. A prospective study of minimal residual disease in childhood B-lineage acute lymphoblastic leukaemia: MRD level at the end of induction is a strong predictive factor of relapse. Br J Haematol. 1997;98(1):140–6 [DOI] [PubMed] [Google Scholar]
- 46.Cazzaniga G, Biondi A. Molecular monitoring of childhood acute lymphoblastic leukemia using antigen receptor gene rearrangements and quantitative polymerase chain reaction technology. Haematologica. 2005;90(3):382–90 [PubMed] [Google Scholar]
- 47.Ruggeri A, Michel G, Dalle JH, Caniglia M, Locatelli F, Campos A, et al. Impact of pre-transplant minimal residual disease after cord blood transplantation for childhood acute lymphoblastic leukemia in remission: an Eurocord, PDWP-EBMT analysis. Leukemia. 2012;26(12):2455–61 [DOI] [PubMed] [Google Scholar]
- 48.Leung W, Pui CH, Coustan-Smith E, Yang J, Pei D, Gan K, et al. Detectable minimal residual disease before hematopoietic cell transplantation is prognostic but does not preclude cure for children with very-high-risk leukemia. Blood. 2012;120(2):468–72 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Locatelli F, Zecca M, Rondelli R, Bonetti F, Dini G, Prete A, et al. Graft versus host disease prophylaxis with low-dose cyclosporine-A reduces the risk of relapse in children with acute leukemia given HLA-identical sibling bone marrow transplantation: results of a randomized trial. Blood. 2000;95(5):1572–9 [PubMed] [Google Scholar]
- 50.Eapen M, Horowitz MM, Klein JP, Champlin RE, Loberiza FR, Ringden O, et al. Higher mortality after allogeneic peripheral-blood transplantation compared with bone marrow in children and adolescents: the Histocompatibility and Alternate Stem Cell Source Working Committee of the International Bone Marrow Transplant Registry. J Clin Oncol. 2004;22(24):4872–80 [DOI] [PubMed] [Google Scholar]