

Abstract
The Wiskott-Aldrich syndrome protein is an essential cytoskeleton regulator found in cells of the hematopoietic lineage and controls the motility of leukocytes. The impact of WAS gene deficiency on the mobilization of hematopoietic progenitor/stem cells in circulation has remained unexplored but information would be pertinent in the context of autologous gene therapy of Wiskott-Aldrich syndrome. The response to granulocyte-colony stimulating factor mobilization was investigated in a murine WAS knock-out model of the disease, by measuring hematologic parameters, circulation and engraftment of hematopoietic progenitor/stem cells. In the steady-state, adult WAS knock-out mice have B-cell lymphopenia, marked neutrophilia, increased counts of circulating hematopoietic progenitor cells and splenomegaly, presumably caused by the retention of hematopoietic progenitor cells due to high levels of splenic CXCL12. In spite of these anomalies, the administration of granulocyte-colony-stimulating factor mobilizes progenitor/stem cells in WAS knock-out mice to the same level and with the same kinetics as in wild-type control mice. Mobilized peripheral blood cells from WAS knock-out mice can be transduced and are able to engraft into lethally-irradiated hosts reconstituting multiple lineages of cells and providing more effective radio-protection than mobilized cells from wild-type control mice. Surprisingly, the homing and the peripheral blood recovery of B lymphocytes was influenced by the background of the host. Thus, in the absence of Wiskott-Aldrich syndrome protein, effective mobilization is achieved but partial correction may occur as a result of an abnormal hematopoietic environment.
Introduction
Wiskott-Aldrich syndrome (WAS) is a rare X-linked hereditary immunodeficiency disorder characterized by micro-thrombocytopenia, recurrent infections and eczema. It is associated with high incidences of auto-immunity and lymphoid malignancies, especially B-cell lymphomas. The disease is caused by a lack of WAS protein (WASp) which is a regulator of actin cytoskeleton reorganization expressed exclusively in cells of the hematopoietic lineage.1,2
Hematopoietic stem cell transplantation is the curative treatment for WAS when donors are available.3 Several groups, including ours, are also currently testing gene therapy as an alternative treatment of WAS for patients who do not have HLA-compatible donors.4 Gene therapy is based on the infusion of gene-modified autologous hematopoietic stem cells (HSC) using retroviral or lentiviral vectors.5–7 Current gene therapy protocols rely on the use of either bone marrow cells or mobilized peripheral blood cells to obtain CD34+ HSC that will be used for ex vivo gene transfer. Several procedures can be used to mobilize peripheral CD34+ HSC. Recombinant human granulocyte-colony-stimulating factor (G-CSF) analog (filgrastim or lenograstim) and more recently pegylated filgrastim (pegfilgrastim) have been utilized in autologous blood stem cell mobilization either alone or combined with chemotherapy.8
There are several mechanisms involved in hematopoietic stem cell mobilization9–12 and numerous studies support a major role of the CXCR4/CXCL12 axis in hematopoietic cell retention, trafficking and mobilization. G-CSF potently inhibits osteoblast activity in the bone marrow, reducing CXCL12 expression13 thereby decreasing retention of cells in the bone marrow and permitting their egress into the periphery. Novel strategies for mobilization are based directly on the use of a CXCR4 inhibitor (AMD3100/Plerixafor) which reversibly disrupts the interaction with CXCL12.14
WASp is an important regulator of chemokine responses in lymphocytes in particular through a chemokine-induced inside-out signaling pathway involving cdc42/WASp activation.15–18 WASp deficiency causes a marked defect in localization and trafficking of phagocytes and lymphocytes, including B cells, affecting their function.18,19 WASp-deficient murine B cells seem to express normal levels of CXCR4, CXCR5 and CCR7 but migrate poorly to CXCL12, CXCL13 and CCL19.20 The chemokine-induced migration defects and function of WASp-deficient murine B cells can be restored by WAS gene transfer.21,22 In the absence of WASp, hematopoietic bone marrow cells also respond poorly to CXCL12-induced chemotaxis and cdc42 activation and this hampers the migration of early HSC and progenitor cells from the liver to bone marrow during development.23 Thus, by affecting chemokine responses, WASp-deficiency may interfere with HSC mobilization. While WASp deficiency does not lead to hematopoietic defects resembling those observed in defective CXCL12/CXCR4 signaling,24 it is currently not known whether WASp-deficient HSC mobilize differently from normal cells. Two WAS patients were recently treated by gene therapy with autologous peripheral blood cells mobilized by G-CSF, showing engraftment and multi-lineage gene correction.25 While these findings demonstrate that WASp is not critically required for CD34+ cell mobilization, the process has not been characterized in detail. This prompted us to study, in mice, the response of WASp-deficient HSC to G-CSF by measuring mobilization and engraftment of the mobilized peripheral HSC in a lethally-irradiated host after gene transfer.
Design and Methods
Animals
All mice used for the bone marrow transplants were housed at the Genethon Animal Facility, Evry, France. 129Sv-WAS knock-out (WKO) mice, described elsewhere,26 were initially kindly provided to us by Dr. Snapper (Massachusetts General Hospital, Boston, MA, USA) and then bred at Genethon under specific pathogen-free conditions. Control wild-type (WT) 129 Sv/Ev mice were purchased from Iffa Credo (L’arbresle, France) and quarantined for at least 7 days prior to use.
Mobilization protocol
Recombinant human G-CSF, (Neupogen, 30 MU or 0.3 mg/mL from Amgen, Thousand Oaks, CA, USA) was diluted in phosphate-buffered saline and administered by daily subcutaneous injection at a dose of 5 μg per mouse for 4 days (n=5 mice). Mice were analyzed 2 or 3 hours after each dose or the final G-CSF dose. Mobilization was measured in peripheral blood and by spleen histology as described in the Online Supplementary Design and Methods.
Transplantation
To obtain mobilized peripheral blood cells for transplantation, a total of 30 WT or 56 WKO male mice received G-CSF subcutaneously for 4 days. Two hours after the last injection, peripheral blood (500 μL per mouse) was collected from the retro-orbital sinus under sterile conditions into citrated (3.8%) tubes, then treated with Ammonium-Chloride-Potassium lysis buffer and washed to obtain the mobilized cells. To obtain bone marrow cells for transplantation, cells were collected by flushing the contents of femora and tibiae. Both mobilized peripheral blood cells and bone marrow cells were cultured for 1 day before transplantation to allow for overnight transduction of the cells as described in the Online Supplementary Design and Methods. For transplantation, between 1 to 2×106 mobilized peripheral blood cells or bone marrow cells were suspended in 200 μL of X-VIVO 20 and injected into the retro-orbital sinus of previously irradiated WT or WKO female recipients. Whole body irradiation was performed 2 to 3 hours before transplantation in ventilated Plexiglas containers using an X-ray source at a dose rate of 0.84 Gy/min for a total dose of 9.5 Gy (Faxitron CP160, Edimex, France). A fraction of the cells was not injected in the mice and expanded in culture for 7 days in the presence of cytokines or plated in methyl-cellulose to test transduction and hematopoietic activity.
Evaluation of transduction and engraftment in treated mice
The efficiency of transduction of the mobilized peripheral blood cells was evaluated by measuring vector copy number using quantitative real-time polymerase chain reaction (qPCR) and male donor cell engraftment into female recipient mice was evaluated using qPCR of a male Y sequence calibrated on a series of genomic DNA samples prepared from varying proportions of male/female blood cells, as previously described.22,27,28 Engraftment was evaluated in peripheral blood.
In vivo homing assay
The previously described assay23 is detailed in the Online Supplementary Design and Methods section.
Results
Increase of WKO progenitor cells in spleen and peripheral blood
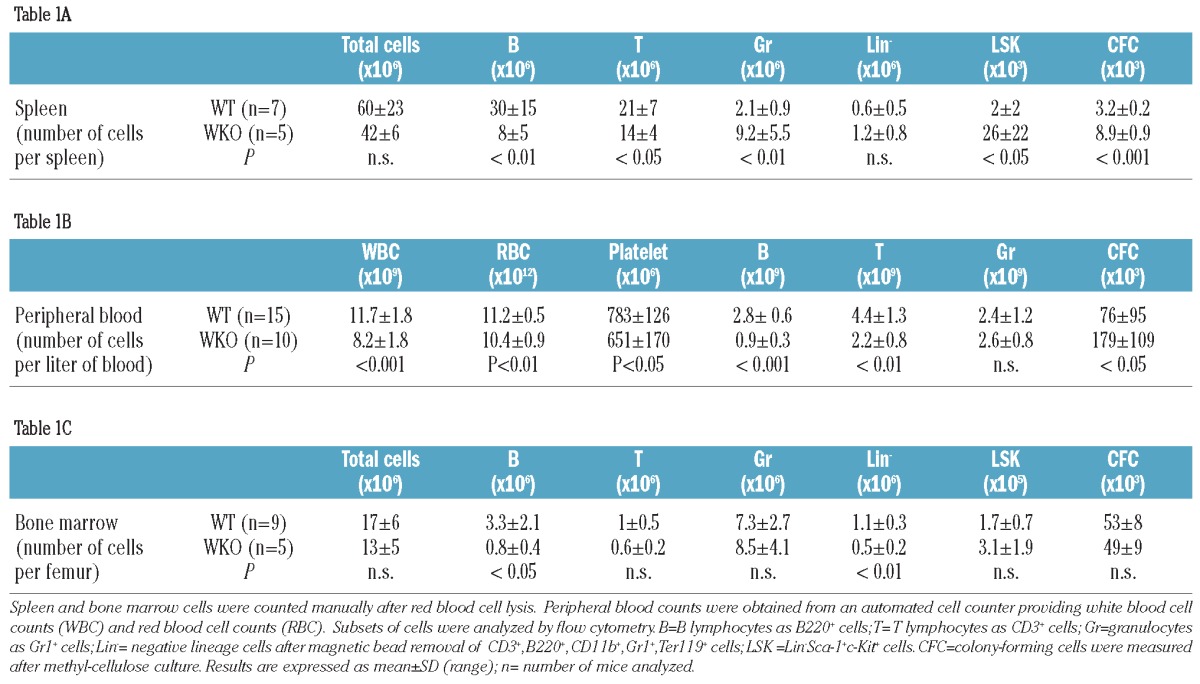
In steady-state specific pathogen-free conditions, the 129Sv WKO mice exhibit splenomegaly (Online Supplementary Figure S1A) with the average weight of their spleens being 82.6±19.7 mg compared to the 63.1±16.5 mg for the spleens of WT mice (P<0.05). The spleens of WT mice (Online Supplementary Figure S1B) have a normal parenchymal architecture, composed of white nodules embedded in the red pulp consisting of elongated splenic cords located between sinusoids. Splenomegaly in WKO mice is associated with smaller lymphoid nodules and increased areas of red pulp with an expansion of erythroid cells in the perifollicular zone and consequently a significant decrease of the white/red pulp area ratio as determined by histology (Online Supplementary Figure S1C: white pulp = 41±12% in WT versus 24±12% in WKO spleen; P<0.05). The reduction in splenic leukocytes was confirmed by flow cytometry analysis which revealed markedly reduced numbers of B and T lymphocytes in WKO mice (Table 1A, P<0.05). This contrasts with an increased number of granulocytes (WKO=9.2±5.5×106 cells/spleen versus WT= 2.1±0.9×106 cells/spleen, P<0.01) and of hematopoietic progenitor cells in the spleen as measured by the number of Lin− Sca-1+ c-kit+ (LSK) cells (WKO=26±22×103 LSK/spleen versus WT= 2±2×103 LSK/spleen, P<0.05) and by the number of colony-forming cells (CFC) (WKO = 8.9±0.9×103 CFC/spleen versus WT= 3.2±0.2×103 CFC/spleen; P<0.001, Table 1A). In the circulation, there is a moderate leukopenia (Table 1B) with a moderate lymphopenia caused by a decrease of both B and T lymphocyte counts. The percentage of WKO granulocytes was markedly higher than that of WT granulocytes (WKO=41±8% versus WT= 19±9%) (Online Supplementary Table S2) but due to the leukopenia, the total granulocyte count in the blood of WKO mice was comparable to that in WT mice (Table 1B). In the steady-state, there was also a significant increase in circulating CFC numbers in WKO mice (WKO = 179±109×103 CFC/L of blood versus WT= 76±95×103/L of blood, P<0.05, Table 1B). As already documented, hematopoiesis is functional at the central level in WKO mice. Bone marrow analysis showed a decrease in B lymphocyte counts as previously described28 (WKO=0.8±0.4×106 cells/femur versus WT=3.3±2.1×106 cells/femur, P<0.05) reflected also by a decrease in numbers of lineage-negative (Lin−) cells (Table 1C), but granulocyte and progenitor cell counts were comparable to those in WT mice. Altogether, the comparative analysis of WKO and WT mice revealed that WASp deficiency causes perturbations in the peripheral blood compartment, in particular a notable lymphopenia and increased circulating levels of hematopoietic progenitor cells.
Table 1.
Phenotype comparison between WT and WKO mice.
Mobilization of WKO progenitors
To characterize the effects of human G-CSF on the mobilization of hematopoietic cells in WKO mice, we measured blood levels of Gr1+ granulocytes, c-Kit+ hematopoietic progenitor cells29,30 and progenitor cell counts following daily administration of the G-CSF for 4 days. This protocol induced rapid and significant increases in the percentages and absolute numbers of granulocytes in circulation in both WKO and WT mice (Figure 1A,B; steady-state versus G-CSF-treated P<0.001). The rate of induction of granulocytes was similar in WT and WKO mice. Treatment with human G-CSF also increased circulating CD45+ c-Kit+ cells to comparable levels in WKO and WT mice (Figure 1C). The presence of circulating hematopoietic progenitor cells was confirmed by functional tests. Increased numbers of blood CFC were measured starting on day 3 of human G-CSF administration (Figure 1D). As shown in Table 1B, WKO mice had significantly higher levels of circulating CFC in the steady-state and these levels were increased by three consecutive daily administrations of G-CSF (Figure 1Dd), but did not differ significantly from those in WT mice on day 3 and 4 of G-CSF administration implying that only constitutive hematopoietic progenitor mobilization is greater.
Figure 1.

Kinetics of granulocyte and hematopoietic progenitor cell mobilization in peripheral blood after G-CSF administration. G-CSF was administered to WT or WKO mice at a dose of 5 μg/mouse every day, for 4 days, subcutaneously. Graphs represent peripheral blood granulocytes measured as percentage (A) or absolute count (B), c-Kit+ cells (C) or CFC (D) hematopoietic progenitor cells. Data from three mice by group, *P value < 0.05, **P value < 0.01, ***P value < 0.001
The weight and morphology of spleens were monitored after mobilization because WKO mice have splenomegaly and it has been documented, in both mice and humans, that human G-CSF administration may potentially cause splenic rupture.30–33 As previously described after human G-CSF administration30,33 the weight (Online Supplementary Figure S1A, P<0.01) and size (data not shown) of the spleens increased with hypertrophy of the red pulp (Online Supplementary Figure S1B, P<0.01). This resulted in luminal dilatation of trabecular vessels filled with large numbers of erythrocytes. In WKO mice, we also observed an increase in the weight of the spleen following human G-CSF administration (Online Supplementary Figure S1A) but the distribution between white and red pulp remained similar to that in non-treated WKO animals (Online Supplementary Figure S1B, n.s.). No splenic rupture was detected in any of the WT or WKO mice that received G-CSF in this study (n=86 mice).
In summary, hG-CSF administration over 4 days is able to mobilize WKO progenitor cells, increasing CFC levels 7-fold compared to the steady state, without adverse events such as splenic rupture.
Restoration of lethally irradiated WT and WKO mice transplanted with mobilized WKO cells
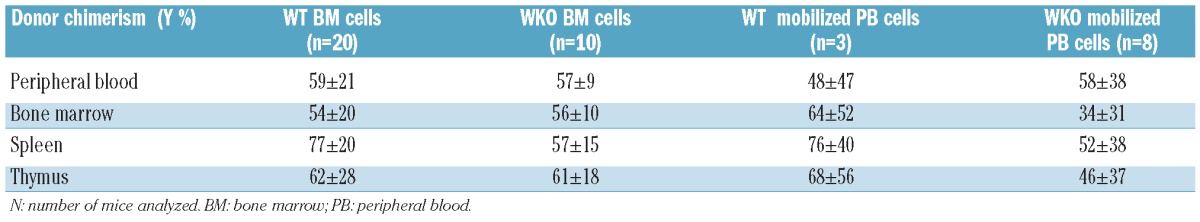
The hematopoietic reconstitution potential of mobilized peripheral blood cells was demonstrated in transplantation experiments using lethally-irradiated (9.5 Gy) recipient mice. Peripheral blood cells collected after 4 days of mobilization were compared against steady-state bone marrow in different backgrounds. Between 1 and 2×106 cells from mobilized peripheral blood cells or bone marrow cells were injected into the mice. Progenitor cell content in the transplanted bone marrow cells or mobilized peripheral blood cells was evaluated (Online Supplementary Table S3) and it was found that for 1×106 bone marrow cells or mobilized peripheral blood cells, a higher level of hematopoietic progenitor cells (CFC, LSK and c-Kit+) was obtained in WKO mice than in the WT mice. As previously described,27 untransplanted lethally-irradiated mice died within 2 weeks. In contrast, 75–100% of mice transplanted with bone marrow cells from WKO or WT mice were still alive after 4 months (Figure 2). Bone marrow cells provided significantly higher survival (P<0.05) than mobilized peripheral blood cells. The survival data are coherent with the number of progenitor cells determined in the different groups of engrafted mice and, interestingly, WKO mobilized peripheral blood cells provided better survival than WT mobilized peripheral blood cells (P<0.05) which is consistent with higher levels of circulating hematopoietic progenitor cells in mobilized WKO mice than in WT mice. After 2 to 4 months, donor chimerism was measured in different compartments using a calibrated qPCR, showing equivalent engraftment with bone marrow and mobilized peripheral blood cells obtained from either WT or WKO mice (Table 2). Collectively, these data confirm the presence of functional hematopoietic stem cells in mobilized peripheral blood cells obtained from WKO mice.
Figure 2.
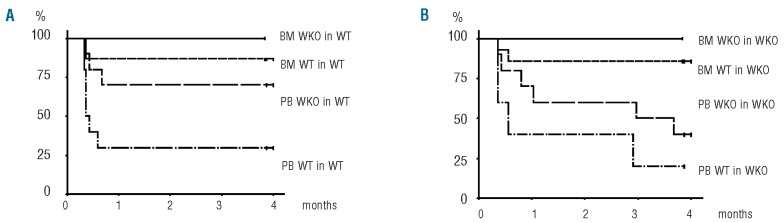
Effects of WASp and origin of cells on the survival of lethally-irradiated mice following transplantation. WT or WKO mice were lethally irradiated and transplanted with bone marrow (BM) or mobilized peripheral blood cells (PB) from either WT or WKO mice. Survival is represented by Kaplan-Meier curves in lethally irradiated WT (A) or WKO (B) hosts. The following number of mice were included in the analysis: BM WT in WT (n=15); BM WKO in WT (n=5); PB WT in WT (n=10); PB WKO in WT (n=10); BM WT in WKO (n=14); BM WKO in WKO (n=5); PB WT in WKO (n=5); PB WKO in WKO (n=10). Differences between curves were analyzed by the log-rank test.
Table 2.
Engraftment of hematopoietic progenitor/stem cells from various sources in lethally irradiated WT or WKO mice. Quantification of male cells by qPCR amplification of Y chromosome sequences was normalized to mouse TTN gene and calibrated on known mixtures of male and female cells. Results are expressed as percentage mean±SD (range).
Lentiviral vector-mediated gene transfer in WKO mobilized peripheral blood cells
We confirmed that mobilized peripheral blood cells from WKO mice could be efficiently gene-modified with a lentiviral vector coding for GFP and engrafted. In vitro, the transduction of mobilized peripheral blood cells from WKO mice was as effective as that of bone marrow cells as shown in cells expanded for 7 days (Online Supplementary Table S4) and in the CFC population (data not shown). In vivo, GFP-transduced mobilized peripheral blood cells from WKO mice engrafted successfully and generated sustained levels of GFP-expressing cells in various hematopoietic tissues (Online Supplementary Figure S2B), as did transduced bone marrow cells (data not shown). Lymphoid and myeloid reconstitution was observed in several tissues with transduced mobilized peripheral blood cells from WKO mice (Online Supplementary Figure S2C), as did mobilized peripheral blood cells from WT mice (data not shown).
Hematologic restoration is dependent of the background of the recipient mice
The reconstitution capacity of mobilized peripheral blood cells versus bone marrow was further examined 2 to 4 months after transplantation, by comparing multiple lineages of cells in various tissues and analyzing the impact of WASp deficiency on the graft or on the recipient properties (Figure 3 and Online Supplementary Table S2). Overall, mobilized peripheral blood cells and bone marrow cells behaved similarly to each other to reconstitute B and T lymphoid cells when tested in the same context. The lack of WASp had little impact on T lymphocyte recovery which was comparable whether or not WT or WKO grafts were transplanted into WT or WKO recipients (Online Supplementary Table S2). Levels of mature CD3+ cells as well as CD4+CD8+ double positive immature thymocytes were relatively comparable in all cases (Online Supplementary Table S2). This is coherent with the fact that in the steady-state, adult WT and WKO mice have comparable T-cell counts. On the other hand, the lack of WASp in donor cells was detrimental for B-lymphocyte reconstitution as seen particularly clearly in the case of bone marrow transplants (Figure 3A,B,C and Online Supplementary Table S2). B-cell levels were particularly low when WKO bone marrow or mobilized peripheral blood cells were transplanted into WKO mice. B-cell recovery was also impaired in WT mice recipients, the defect being particularly visible in the blood compartment following transplantation of WKO bone marrow cells (Figure 3A). Thus, WASp deficiency causes an intrinsic defect in B lymphoid recovery confirming, in part, our own prior results.28 In contrast to the lymphoid compartment, we observed a clear impact of mobilization on granulocyte recovery after transplantation. The transplantation of mobilized peripheral blood cells from WKO mice provided higher counts of granulocytes in the blood and spleen compared to transplantation of bone marrow, although this was not seen in the bone marrow compartment (Figure 3D,E,F). The lack of WASp in the transplanted cells also increased the percentages of granulocytes in blood and spleen (Online Supplementary Table S2). All parameters considered, the highest levels of granulocytes in the spleen were obtained with WKO mobilized peripheral blood cells transplanted into WKO mice (Figure 3F).
Figure 3.

B lymphocyte and granulocyte reconstitution following transplantation. In the same experimental set-up as in Figure 2, B lymphocyte and granulocyte counts were measured in peripheral blood (PB) (A, D), bone marrow (BM) (B, E), and spleen (C, F) of mice between 2 to 3 months (A, D) and 4 months (B, C, E, F) after transplantation. *P value < 0.05, **P value < 0.01, ***P value < 0.001.
Unexpectedly, the perturbations in B lymphocyte and granulocyte recovery were partly determined extrinsically by the background of recipient mice. Indeed, the transplantation of WT bone marrow cells generated significantly lower B lymphocyte counts in the blood of WKO recipient mice than in the blood of WT recipient mice (Figure 3A). The transplantation of mobilized peripheral blood cells from WKO mice generated significantly higher levels of granulocytes in WKO mice than in WT recipient mice (Figure 3F). These data suggest that WASp plays an important role in determining the properties of the hematopoietic micro-environment.
Mechanisms involve homing and chemokine levels
To explore why the recipient background may cause differences in B-cell and granulocyte recovery particularly in the periphery, we examined the homing of cells and the levels of the stromal-derived CXCL12 chemokine in the hematopoietic environment. The homing capacity of mature bone marrow cells, including B cells and granulocytes, to the spleen was measured in irradiated mice using a competitive homing assay. As shown in Figure 4A, total bone marrow cells, B cells and granulocytes homed less effectively in the spleen of WKO recipients (n=5) than in the spleen of WT mice (n=8). The B-cell homing defect was particularly visible with WKO cells in WKO recipients. We speculate that this reduced migration of mature B lymphocytes to the spleen could partially explain the B lymphopenia observed in transplanted as well as in steady-state WKO animals. In contrast, it is not possible to explain the augmentation of granulocyte levels following transplantation of WKO mobilized peripheral blood cells (Figure 3F) by increased homing of WKO granulocytes to the spleen, as the opposite is observed (Figure 4A). Because spleens of WKO mice have elevated levels of hematopoietic progenitor/stem cells (LSK and CFC) (Table 1A), it is tempting to speculate that spleens of WKO mice may either produce or retain granulocytes. We found enhanced expression of CXCL12 mRNA in the spleens of WKO mice compared to the expression in WT mice in the steady-state (Figure 4B, P<0.01) while CXCL12 mRNA expression in WKO bone marrow was slightly reduced compared to that in WT mice (Figure 4C; P=0.07). Such dysregulated levels of CXCL12 indicate an abnormal hematopoietic microenvironment in WKO mice, thereby providing a potential molecular mechanism for hematopoietic anomalies observed in the steady-state as well as following transplantation in these mice.
Figure 4.

Involvement of cellular homing and chemokine levels. The homing capacity of bone marrow (BM) cells, including B lymphocyte and granulocyte subsets, to the spleen following BM transplantation was measured (A). Lethally irradiated WT (n=8) or WKO (n=5) mice were transplanted with a mixture of whole BM cells consisting of an equal number of WT cells stained with CFSE and of WKO cells stained with PKH26. After 24 h, spleens were harvested from the indicated recipient mice (bottom line) and the percentages of total cells, B220 lymphocytes and Gr1+ granulocytes were determined and normalized to that of WT cells in WT mice. (B) Quantification of CXCL12 mRNA in spleen cells (B) and in BM (C) in the steady-state (n=5). *P value < 0.05, **P value < 0.01, ***P value < 0.001.
Discussion
In this study we show for the first time in mice that the lack of the cytoskeleton regulator WASp does not affect the G-CFS-induced mobilization of hematopoietic progenitor/stem cells and enables the collection of a substantial hematopoietic graft capable of engrafting in lethally-irradiated hosts, providing radio-protection and restoring myeloid and lymphoid lineages in various tissues for several months. WKO mice had abnormal hematologic parameters in the steady-state but mobilized with the same kinetics and as effectively as WT mice. These findings may have practical implications for the treatment of patients with WAS. Gene correction of autologous cells is currently being tested as an alternative to allogeneic bone marrow transplantation in WAS patients.4,6,7,25 Our results show that WASp-deficient cells can be mobilized with G-CSF and transduced effectively without preventing their engraftment capacity. WAS patients may develop splenomegaly, as was observed in WKO mice. In mice we have not observed splenic rupture following G-CSF administration suggesting that this risk may not be particularly increased in WAS. Collectively, these results support the use of G-CSF-mobilized leukapheresed CD34+ cells to serve as a source of autologous gene-corrected cells in gene therapy trials of WAS.
The study of hematopoietic mobilization in WKO mice provided some new insights into the role of WASp in hematologic homeostasis. First, we observed a spontaneous distribution of hematopoietic progenitor/stem cells in the periphery of WKO mice, as if mice were spontaneously mobilized. Normally, the levels of peripheral CFC in the circulation are very low, but WKO mice spontaneously exhibit high levels of circulating blood CFC and very high counts of LSK hematopoietic progenitor/stem cells in the spleen. Such high numbers of hematopoietic progenitor cells in the spleen of WKO mice may lead to increased granulocytes and erythroid cells in this organ, reducing mononuclear cell counts and inverting the ratio of white and red pulp, as observed. In spite of basal differences, WT and WKO mice both reacted to G-CSF administration by increasing the numbers of CFC and c-kit+ cells in the blood, apparently to the same levels. However, mobilized peripheral blood cells from WKO mice seemed to provide better radioprotection than mobilized peripheral blood cells from WT mice suggesting that there were perhaps qualitative differences between these grafts. Possibly, G-CSF administration to WKO mice caused the effective mobilization of an already existing pool of LSK hematopoietic progenitor/stem cells from the spleen which, combined with the pre-existing circulating cells, could have provided an additional benefit in the context of transplantation.
The chemokine CXCL12 plays a central role in HSC trafficking and the mechanism of mobilization by G-CSF involves a reduction in CXCL12 expression in the osteoclastic bone marrow stem cell niche.13 Inversely, and as described for the maintenance of HSC in the bone marrow stromal cell niche,34 the high level of hematopoietic progenitor/stem cells in the spleen of WKO mice is probably caused by retention linked to high levels of local CXCL12 mRNA in this organ. Perhaps the retention of progenitor cells in the spleen of WKO mice is compounded by the lack of WASp impairing progenitor/HSC motility and egress, as described for WASp-deficient B cells and dendritic cells.18,35 Indeed, we documented reduced homing of bone marrow cells to the spleen, further emphasizing that cells lacking WASp are less motile. Altogether, these data suggest that disrupting CXCR4/SDF1a interactions with AMD3100/plerixafor may be useful in the case of WASp deficiency to mobilize not only HSC in bone marrow but also additional hematopoietic progenitor cells in the periphery.
Surprisingly, our results reveal that the lack of WASp modified the characteristics of the stem cell niche or of the stromal environment. Indeed, the background of the mice determined the efficiency of hematologic recovery and this was particularly visible with B-cell recovery following transplantations. As previously described,28 the lack of WASp caused an intrinsic defect in B-cell recovery which can be restored upon transplantation of WT progenitor/stem cells but not to the full extent in WKO recipient mice. Intrinsic B-cell defects in WASp deficiency have only been recently fully appreciated19 and include defective B-cell homing to the spleen36 as we report here. Such defective B-cell homing may partly explain the marked B lymphopenia that is observed in the steady-state in adult WKO mice. More surprisingly, we observed B-cell extrinsic defects as WT B lymphocytes also failed to home properly to the spleen of WKO mice. To our knowledge this constitutes the first evidence that the hematopoietic microenvironment is perturbed in WKO mice. Perhaps these observations reflect multiple consequences of perturbations in CXCL12. This chemokine regulates the migration of various types of cells, including progenitor cells and B cells, but also regulates hematopoiesis, including B-cell lymphopoiesis.24,37 The cause of dysregulated expression of CXCL12 in WKO mice is not known and deserves further investigation, but is beyond the scope of this paper. It is also likely that additional mechanisms perturb the homeostasis of B lymphocytes and stem/progenitor cells in the absence of WASp. Because WASp is expressed only in the hematopoietic lineage,38 this suggests that the effects of the lack of WASp on the microenvironment are probably indirect. A likely component of this microenviromental defect could be macrophages/osteoclasts because these cells play an essential role in the remodeling of the hematopoietic niche but are also known to be severely affected by the lack of WASp.39 One possible consequence for WAS gene therapy is that the engraftment of gene-corrected WASp-deficient cells could be suboptimal if such defects in the microenvironment were also found in man.
Acknowledgments
The authors would like to thank Khalil Seye, Maxime Ferrand, Severine Charles for technical help, Bernard Gjata and Isabelle Adamski for spleen histology, Daniel Stockholm for spleen quantification, and the Imaging-Cytometry facility.
Footnotes
The online version of this article has a Supplementary Appendix.
Funding
The work was supported in part by funds from the CONSERT European project (LSHB-CT-2004-005242), a PhysioWAS grant to AG (ANR-07-MRAR-022-02), the French Muscular Dystrophy Association (AFM) and also by equipment funds from ‘Region Ile-de-France’, ‘Conseil General de l’Essonne’, ‘Genopole Recherche’ of Evry, and the University of ‘Evry Val d’Essonne’.
Authorship and Disclosures
Information on authorship, contributions, and financial & other disclosures was provided by the authors and is available with the online version of this article at www.haematologica.org.
References
- 1.Thrasher AJ, Burns SO. WASP: a key immunological multitasker. Nat Rev Immunol. 2010;10(3):182–92 [DOI] [PubMed] [Google Scholar]
- 2.Bosticardo M, Marangoni F, Aiuti A, Villa A, Grazia Roncarolo M. Recent advances in understanding the pathophysiology of Wiskott-Aldrich syndrome. Blood. 2009;113 (25):6288–95 [DOI] [PubMed] [Google Scholar]
- 3.Pai SY, Notarangelo LD. Hematopoietic cell transplantation for Wiskott-Aldrich syndrome: advances in biology and future directions for treatment. Immunol Allergy Clin North Am. 2010;30(2):179–94 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Galy A, Thrasher AJ. Gene therapy for the Wiskott-Aldrich syndrome. Curr Opin Allergy Clin Immunol. 2011;11(6):545–50 [DOI] [PubMed] [Google Scholar]
- 5.Boztug K, Dewey RA, Klein C. Development of hematopoietic stem cell gene therapy for Wiskott-Aldrich syndrome. Curr Opin Mol Ther. 2006;8(5):390–5 [PubMed] [Google Scholar]
- 6.Merten OW, Charrier S, Laroudie N, Fauchille S, Dugue C, Jenny C, et al. Large scale manufacture and characterisation of a lentiviral vector produced for clinical ex vivo gene therapy application. Hum Gene Ther. 2011;18(5):479–87 [DOI] [PubMed] [Google Scholar]
- 7.Scaramuzza S, Biasco L, Ripamonti A, Castiello MC, Loperfido M, Draghici E, et al. Preclinical safety and efficacy of human CD34(+) cells transduced with lentiviral vector for the treatment of Wiskott-Aldrich syndrome. Mol Ther. 2013;21(1):175–84 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kobbe G, Bruns I, Fenk R, Czibere A, Haas R. Pegfilgrastim for PBSC mobilization and autologous haematopoietic SCT. Bone Marrow Transplant. 2009;43(9):669–77 [DOI] [PubMed] [Google Scholar]
- 9.Lapid K, Vagima Y, Kollet O, Lapidot T. Egress and mobilization of hematopoietic stem and progenitor cells. StemBook [Internet]. Cambridge (MA): Harvard Stem Cell Institute; 2008–2009. March 15 [PubMed] [Google Scholar]
- 10.Katayama Y, Battista M, Kao WM, Hidalgo A, Peired AJ, Thomas SA, et al. Signals from the sympathetic nervous system regulate hematopoietic stem cell egress from bone marrow. Cell. 2006;124(2):407–21 [DOI] [PubMed] [Google Scholar]
- 11.Kollet O, Dar A, Shivtiel S, Kalinkovich A, Lapid K, Sztainberg Y, et al. Osteoclasts degrade endosteal components and promote mobilization of hematopoietic progenitor cells. Nat Med. 2006;12(6):657–64 [DOI] [PubMed] [Google Scholar]
- 12.Hoggatt J, Pelus LM. Many mechanisms mediating mobilization: an alliterative review. Curr Opin Hematol. 2011;18(4):231–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Semerad CL, Christopher MJ, Liu F, Short B, Simmons PJ, Winkler I, et al. G-CSF potently inhibits osteoblast activity and CXCL12 mRNA expression in the bone marrow. Blood. 2005;106(9):3020–7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dar A, Schajnovitz A, Lapid K, Kalinkovich A, Itkin T, Ludin A, et al. Rapid mobilization of hematopoietic progenitors by AMD3100 and catecholamines is mediated by CXCR4-dependent SDF-1 release from bone marrow stromal cells. Leukemia. 2011;25(8):1286–96 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Haddad E, Zugaza JL, Louache F, Debili N, Crouin C, Schwarz K, et al. The interaction between Cdc42 and WASP is required for SDF-1-induced T-lymphocyte chemotaxis. Blood. 2001;97(1):33–8 [DOI] [PubMed] [Google Scholar]
- 16.Westerberg L, Greicius G, Snapper SB, Aspenstrom P, Severinson E. Cdc42, Rac1, and the Wiskott-Aldrich syndrome protein are involved in the cytoskeletal regulation of B lymphocytes. Blood. 2001;15;98(4):1086–94 [DOI] [PubMed] [Google Scholar]
- 17.Stabile H, Carlino C, Mazza C, Giliani S, Morrone S, Notarangelo LD, et al. Impaired NK-cell migration in WAS/XLT patients: role of Cdc42/WASp pathway in the control of chemokine-induced beta2 integrin high-affinity state. Blood. 2010;115(14): 2818–26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Burns S, Cory GO, Vainchenker W, Thrasher AJ. Mechanisms of WASp-mediated hematologic and immunologic disease. Blood. 2004;104(12):3454–62 [DOI] [PubMed] [Google Scholar]
- 19.Recher M, Burns SO, de la Fuente MA, Volpi S, Dahlberg C, Walter JE, et al. B cell-intrinsic deficiency of the Wiskott-Aldrich syndrome protein (WASp) causes severe abnormalities of the peripheral B-cell compartment in mice. Blood. 2012;119(12):2819–28 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Westerberg L, Larsson M, Hardy SJ, Fernandez C, Thrasher AJ, Severinson E. Wiskott-Aldrich syndrome protein deficiency leads to reduced B-cell adhesion, migration, and homing, and a delayed humoral immune response. Blood. 2005;105(3):1144–52 [DOI] [PubMed] [Google Scholar]
- 21.Bosticardo M, Draghici E, Schena F, Sauer AV, Fontana E, Castiello MC, et al. Lentiviral-mediated gene therapy leads to improvement of B-cell functionality in a murine model of Wiskott-Aldrich syndrome. J Allergy Clin Immunol. 2011; 127:(6)1376–84.e5 [DOI] [PubMed] [Google Scholar]
- 22.Marangoni F, Bosticardo M, Charrier S, Draghici E, Locci M, Scaramuzza S, et al. Evidence for long-term efficacy and safety of gene therapy for Wiskott-Aldrich syndrome in preclinical models. Mol Ther. 2009;17(6):1073–82 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lacout C, Haddad E, Sabri S, Svinarchouk F, Garcon L, Capron C, et al. A defect in hematopoietic stem cell migration explains the nonrandom X-chromosome inactivation in carriers of Wiskott-Aldrich syndrome. Blood. 2003;102(4):1282–9 [DOI] [PubMed] [Google Scholar]
- 24.Nagasawa T, Hirota S, Tachibana K, Takakura N, Nishikawa S, Kitamura Y, et al. Defects of B-cell lymphopoiesis and bone-marrow myelopoiesis in mice lacking the CXC chemokine PBSF/SDF-1. Nature. 1996; 382(6592):635–8 [DOI] [PubMed] [Google Scholar]
- 25.Boztug K, Schmidt M, Schwarzer A, Banerjee PP, Diez IA, Dewey RA, et al. Stem-cell gene therapy for the Wiskott-Aldrich syndrome. N Engl J Med. 2010;363 (20):1918–27 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Snapper SB, Rosen FS, Mizoguchi E, Cohen P, Khan W, Liu CH, et al. Wiskott-Aldrich syndrome protein-deficient mice reveal a role for WASP in T but not B cell activation. Immunity. 1998;9(1):81–91 [DOI] [PubMed] [Google Scholar]
- 27.Charrier S, Stockholm D, Seye K, Opolon P, Taveau M, Gross DA, et al. A lentiviral vector encoding the human Wiskott-Aldrich syndrome protein corrects immune and cytoskeletal defects in WASP knockout mice. Gene Ther. 2005;12(7):597–606 [DOI] [PubMed] [Google Scholar]
- 28.Zanta-Boussif MA, Charrier S, Brice-Ouzet A, Martin S, Opolon P, Thrasher AJ, et al. Validation of a mutated PRE sequence allowing high and sustained transgene expression while abrogating WHV-X protein synthesis: application to the gene therapy of WAS. Gene Ther. 2009;16(5):605–19 [DOI] [PubMed] [Google Scholar]
- 29.Ogawa M, Matsuzaki Y, Nishikawa S, Hayashi S, Kunisada T, Sudo T, et al. Expression and function of c-kit in hemopoietic progenitor cells. J Exp Med. 1991;174(1): 63–71 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Szumilas P, Barcew K, Baskiewicz-Masiuk M, Wiszniewska B, Ratajczak MZ, Machalinski B. Effect of stem cell mobilization with cyclophosphamide plus granulocyte colony-stimulating factor on morphology of haematopoietic organs in mice. Cell Prolif. 2005;38(1):47–61 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Falzetti F, Aversa F, Minelli O, Tabilio A. Spontaneous rupture of spleen during peripheral blood stem-cell mobilisation in a healthy donor. Lancet. 1999;353(9152):555. [DOI] [PubMed] [Google Scholar]
- 32.Nakayama T, Kudo H, Suzuki S, Sassa S, Mano Y, Sakamoto S. Splenomegaly induced by recombinant human granulocyte-colony stimulating factor in rats. Life Sci. 2001;69(13):1521–9 [DOI] [PubMed] [Google Scholar]
- 33.Yannaki E, Psatha N, Athanasiou E, Karponi G, Constantinou V, Papadopoulou A, et al. Mobilization of hematopoietic stem cells in a thalassemic mouse model: implications for human gene therapy of thalassemia. Hum Gene Ther. 2010;21(3):299–310 [DOI] [PubMed] [Google Scholar]
- 34.Sugiyama T, Kohara H, Noda M, Nagasawa T. Maintenance of the hematopoietic stem cell pool by CXCL12-CXCR4 chemokine signaling in bone marrow stromal cell niches. Immunity. 2006;25(6):977–88 [DOI] [PubMed] [Google Scholar]
- 35.de Noronha S, Hardy S, Sinclair J, Blundell MP, Strid J, Schulz O, et al. Impaired dendritic cell homing in vivo in the absence of Wiskott-Aldrich syndrome protein. Blood. 2004;105(4):1590–7 [DOI] [PubMed] [Google Scholar]
- 36.Westerberg LS, Dahlberg C, Baptista M, Moran CJ, Detre C, Keszei M, et al. Wiskott-Aldrich syndrome protein (WASP) and N-WASP are critical for peripheral B-cell development and function. Blood. 2012;119(17): 3966–74 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Nagasawa T. Microenvironmental niches in the bone marrow required for B-cell development. Nat Rev Immunol. 2006;6(2):107–16 [DOI] [PubMed] [Google Scholar]
- 38.Munoz P, Toscano MG, Real PJ, Benabdellah K, Cobo M, Bueno C, et al. Specific marking of hESCs-derived hematopoietic lineage by WAS-promoter driven lentiviral vectors. PLoS One. 2012;7(6):e39091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Monypenny J, Chou HC, Banon-Rodriguez I, Thrasher AJ, Anton IM, Jones GE, et al. Role of WASP in cell polarity and podosome dynamics of myeloid cells. Eur J Cell Biol. 2011;90(2–3):198–204 [DOI] [PMC free article] [PubMed] [Google Scholar]