

Abstract
Ligands that activate the serotonin 5-HT2C G protein-coupled receptor (GPCR) may be therapeutic for psychoses, addiction, and other neuropsychiatric disorders. Ligands that are antagonists at the closely related 5-HT2A GPCR also may treat neuropsychiatric disorders; in contrast, 5-HT2A activation may cause hallucinations. 5-HT2C-specific agonist drug design is challenging because 5-HT2 GPCRs share 80% transmembrane (TM) homology, same second messenger signaling, and no crystal structures are reported. To help delineate molecular determinants underlying differential binding and activation of 5-HT2 GPCRs, 5-HT2A, and 5-HT2C homology models were built from the β2-adrenergic GPCR crystal structure and equilibrated in a lipid phosphatidyl choline bilayer performing molecular dynamics simulations. Ligand docking studies at the 5-HT2 receptor models were conducted with the (2R, 4S)- and (2S, 4R)-enantiomers of the novel 5-HT2C agonist/5-HT2A/2B antagonist trans-4-phenyl-N,N-dimethyl-2-aminotetralin (PAT) and its 4′-chlorophenyl congners. Results indicate PAT–5-HT2 molecular interactions especially in TM domain V are important for the (2R, 4S) enantiomer, whereas, TM domain VI and VII interactions are more important for the (2S, 4R) enantiomer.
Keywords: serotonin 5-HT2A, 5-HT2C, GPCR, ligand–receptor interactions, homology modelling, docking, molecular dynamics, phosphatidyl choline bilayer, drug design
Introduction
The neurotransmitter serotonin (5-hydroxytryptamine, 5-HT) mediates some of its diverse physiological and psychological effects by activation of the 5-HT2 family of G protein-coupled receptors (GPCRs) that consists of the 5-HT2A, 5-HT2B, and 5-HT2C subtypes. Drugs that activate 5-HT2C receptors are in development for psychoses, addiction, and other neuropsychiatric disorders, as well as, obesity.[1,2] Meanwhile, most currently used antipsychotic drugs are antagonists at the 5-HT2A receptor.[2] In contrast, activation of 5-HT2A receptors is associated with hallucinogenic effects.[3] Moreover, activation of 5-HT2B receptors causes cardiopulmonary toxicity.[4] Thus, there is essentially no clinical tolerance for activation of 5-HT2A and 5-HT2B receptors. Development of 5-HT2C-specific agonist drugs, however, is challenging because 5-HT2 receptors share about 80% transmembrane (TM) sequence identity[5] and same second messenger signaling. Moreover, there are no three-dimensional (3D) crystal structures for any of the 5-HT2 GPCs.
G-protein coupled receptors have a similar 3D structure consisting of a bundle of seven TM alpha helices, connected by alternating intracellular and extracellular loops, with the N-terminus in the extracellular domain and C-terminus in the intracellular domain. About 900 GPCRs are known, however, crystal structures are known only for the following: bovine rhodopsin (bRho),[6–10] opsin,[11,12] human A2A adenosine receptor,[13] turkey β1 adrenoceptor,[14] human β2 adrenoceptor (β2AR) in an inactive state,[15–17] β2AR in a nanobody-stabilized active-state,[18] β2AR in complex with an irreversible agonist,[19,20] human dopamine D3 receptor in complex with a agonist,[21] and human H1 receptor in a complex with an antagonist at 3.1 Å (PDB code 3RZE).[22] The recent X-ray structure of an antagonist bound to the A2A receptor confirms ligands bind deep in the orthosteric site binding pocket.[23]
In the absence of crystal structures for most GPCRs, mutagenesis studies are used to characterize ligand–receptor molecular interactions and to validate GPCR receptor models and ligand docking studies. Thus, it is known that ligand binding at serotonin 5-HT2 and other monoaminergic GPCRs requires ionic interaction between a ligand positively charged amine moiety and the carboxylate of the fully conserved aspartate residue D3.32.[24–28] For example, we recently reported that mutation of D3.32 to alanine (D3.32A) in the 5-HT2C GPCR abolishes detectable binding of the radioligand [3H]-mesulergine.[29] This and other experimental results helped to validate ligand docking and molecular dynamics (MDs) results using a 5-HT2C receptor model built by homology to the human β2AR.[29,30]
In this work, we sought to compare overall receptor structures and ligand binding sites that could underlie ligand differential binding and activation of 5-HT2 GPCRs for drug development purposes. Accordingly, we built a 5-HT2A receptor homology model based on the structure the human β2AR (PDB code 2Rh1), using methods analogous to those used to obtain a 5-HT2C homology model.[30] Docking studies were carried on the model receptors for the (2R, 4S)- and (2S, 4R)-enantiomers of the novel 5-HT2C agonist/5-HT2A/2B antagonist trans-4-phenyl-N,N-dimethyl-2-aminotetralin (PAT)[24] and its 4′-chlorophenyl congners (4′-Cl-PAT). PAT–5-HT2 molecular interactions that lead to receptor subtype differential binding and activation are discussed relative to drug design.
Methods
Model building
A homology model of the human serotonin 5-HT2A receptor was built based on the crystal structure of the β2AR/T4-lysozyme chimera (Protein Data bank entry 2RH1),[17] in similar method used to obtain the 5-HT2C homology model.[30] In brief, the 5-HT2A native sequence was aligned to the β2AR sequence using ClustalW multiple sequence alignment.[31,32] The inverse agonist, carazolol present in the β2AR crystal structure was removed, as well as T4, cholesterol and other molecules present in the β2AR/T4-lysozyme chimera. Point mutations were performed as needed and the gaps were analyzed, followed by the appropriate sequence additions and deletions to match the 5-HT2A receptor aminoacid sequence. The TM domains were built using the Biopolymer module of Sybyl-X 1.3.[33] The crude model of the unbound receptor was minimized using the Powell method implemented in Sybyl with Tripos force field [34] and AMBER charges.[35] The resulting model was equilibrated in a 1-palmitoyl-2-oleyl-sn-glycero phosphatidyl choline (POPC) bilayer.[36] The system was relaxed using the Tripos force field to a gradient 0.05 Kcal/Å mol, prior to MDs simulation in the POPC membrane. MD simulation conditions were time run 5 μs, time step 1 fs, with snapshots collected every 5 fs. Other parameters were the NVT (constant number of particles N, Volume V, and Temperature T) canonical ensemble, 300 K temperature, Boltzmann initial velocities, and nonbonded cutoff set at 8 Å. Constraints for alpha carbons in the TM domains were employed. Subsequently, the constraints were removed for a 1000 ps MD simulation run. The final unbound 5-HT2A homology model was obtained from the median structure after clustering analysis of the frames from the last 10 ps. of the MDs simulation, and optimized using the Tripos force field to a convergence of 0.05 Kcal/Å mol.
Ligands and docking
Synthesis, absolute configuration (based on X-ray crystal structure), and pharmacology of (2R, 4S)- and (2S, 4R)-PAT and 4′-Cl-PAT (Fig. 1) are reported elsewhere.[37,38] The ligand structures were built as monocations (protonated amines) using HyperChem 8.0 [39] and structures were optimized using PM3 model Hamiltonian to a gradient of 0.01 Kcal/Å mol.
Figure 1.
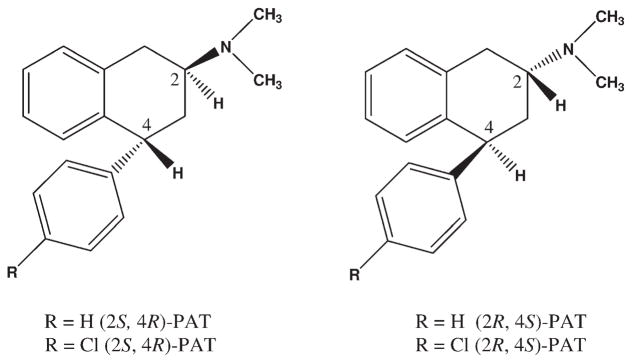
PATs used in 5-HT2A and 5-HT2C docking studies.
Ligands were pre-positioned in the binding pocket by performing rigid docking with the PatchDock server.[40] The low-energy-high-score solutions were analyzed to select the initial configuration, ensuring the essential interaction between the carboxylate oxygen of receptor residue D3.32 and the ligand protonated amine moiety.[20,22] The initial ligand-receptor complex configuration was used for flexible ligand docking with Flexidock in Sybyl-x 1.2.[33] Flexidock uses an algorithm to probe the conformational space defining possible interactions between the ligand and its putative binding site. The binding site was defined by assigning residue D3.32 as a definitive binding site interaction point, and including residues within a 7 Å radius. Structure preparation was performed prior to docking studies assigning AMBER[35] charges for the protein and Gesteiger– Marsili [41] charges for the ligand. Rotatable bonds in the ligand and the side chains of residues defining the receptor putative active site were screened for optimal positioning of the ligand and side chains in the conformational space; remaining residues were frozen during docking. Default FlexiDock parameters were set at 80,000-generation. The best docking solution, according to the highest FlexiDock score, was minimized using the Tripos force field to a gradient 0.05 Kcal/Å mol, prior to MDs simulation. The selected high-score pose of the docked ligand was subjected to a MD simulation run for 500 ps, with other parameters the same as above, to allow adjustment of the positions of side-chains and helices. The final structure of the ligand docked into the receptor was obtained from the average of last 10 ps of the dynamics simulation.
Results and Discussion
Homology modeling and 5-HT2A, 5-HT2C receptor comparison
The first step in the homology model building involves the sequence of the WT 5-HT2A receptor was aligned to the sequence of the template structure β2A receptor using ClustalW. Sequence alignment showing the TM domains is shown in Table 1, the 5-HT2C receptor sequence is also shown in the alignment. Conserved residues are indicated in bold, and reference residues are labeled according to the standard Ballesteros nomenclature for GPCRs.[42]
Table 1.
Alignment of 5-HT2C 5-HT2C and β2AR GPCRs sequences using ClustalW.

|
Conserved residues are indicated in bold. Reference residues are labeled according to Ballesteros nomenclature [42].
The TM alignment shows the very close similarity between the 5-HT2A and -HT2C GPCRs.
The β2AR has been used to build homology models of several related GPCRs including the 5-HT2C receptor; the latter homology model was validated by site-directed mutagenesis and ligand binding studies.[29,30] Despite the low overall sequence identity between the β2A and both 5-HT2A and 5-HT2C receptors (~30%), the majority of highly conserved residues among these GPCRs are in the TM helices, thus allowing for accurate alignment.
Conserved residue sequences among 5-HT2 receptors and the β2AR receptor in the TM domains were used to verify the alignment, comprising: GNXLVI motif in TM I, including the reference residue N1.50, TNYF, SLAXAD motifs in TM II, including the reference residue D2.50, DVL motif in TM III, including the essential residue D3.32, TASI, and DRY motifs, also in TM III, KA motif and reference residue W4.50 in TM IV, FXXPLXIM motif in TM V, including P5.50, WXPFFIXNI motif in TM VI, including residues W6.48, and reference residue P6.50, and WIGY and NPLXY motifs in TM VII, including reference residue P7.50.
The alignment in Table 1 was used to generate the 3D model of the WT 5-HT2A, as described above, suing the method used to build the 5-HT2C homology models based on β2AR crystal structure 2Rh1. The resulting TM bundle structure of the WT 5-HT2A receptor is shown in Figure 2, generated with PyMOL 1.3.[43] TMH are spectrum-color-coded, from blue for TM I, to red for TM VII. In Figure 2, panel A shows the TM bundle oriented with the extracellular domain on top and the intracellular domain at the bottom. Panel B is the view from the extracellular domain down into the receptor cavity.
Figure 2.
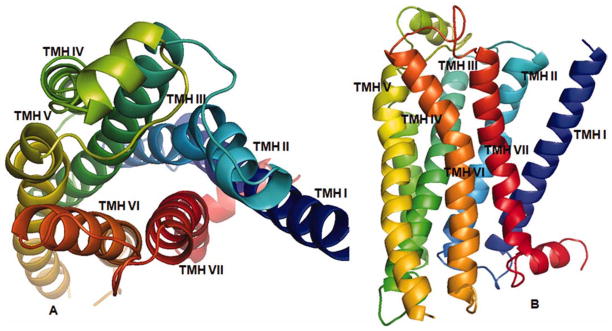
3D 5-HT2A model structure: Panel A: TM domains with the extracellular domain on top, and the intracellular domain at the bottom. Panel B: View from the extracellular domain showing the binding site cavity. [Color figure can be viewed in the online issue, which is available at wileyonlinelibrary.com.]
The resulting 5-HT2A model was analyzed using PDBsum in PROCHECK 3.6.2.[44,45] Figure 3 shows the Ramachandran plot for the 5-HT2A model; PROCHECK statistics are reported in Table 2. Analysis of the Ramachandran plot results show 97.3% of residues are in the allowed regions, 84.1% are in the most favored regions (Psi angle values vary −180 to 0°; Phi angle values vary 0 to 180°; bottom-left quadrant, Fig. 3), and 12.4% are in additional allowed regions; supplementary 0.8% are in generously allowed regions, confirming the quality of the model.
Figure 3.

Ramachandran Plot of the β2AD-based 5-HT2A homology model generated with PROCHECK. [Color figure can be viewed in the online issue, which is available at wileyonlinelibrary.com.]
Table 2.
Recheck statistics of TM residues in the 5-HT2A homology model.
| Number of residues | % | |
|---|---|---|
| Most favored regions | 217 | 84.1 |
| Additional allowed regions | 32 | 12.4 |
| Generously allowed regions | 2 | 0.8 |
5-HT2A–5-HT2C receptor structure analysis
Superposition of the 5-HT2A and -5HT2C GPCR structures was performed by selecting all alpha carbons as superposition criteria, in Sybyl-x 1.2.[33] The root-mean-square deviation (RMSD) 1.879 Å overall suggests close similarity between these structures (Fig. 4, Panel A).
Figure 4.
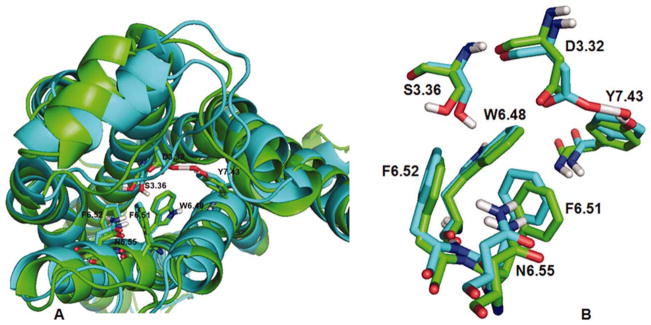
Comparison of 5-HT2A (green ribbons) and 5-HT2C (blue ribbons) GPCR models. Panel A: GPCRs are oriented with extracellular domains at top and intracellular domains at bottom, overall RMSD = 1.88 Å. Panel B: Superimposition conserved residues in the binding pocket is shown, RMSD = 0.49 Å. [Color figure can be viewed in the online issue, which is available at wileyonlinelibrary.com.]
Additional comparison of the structures was performed by aligning the alpha carbons of binding pocket residues conserved in each of the structures, that is, D3.32, S3.36, W6.48, F6.51, F6.52, N6.55, and Y7.43. The RMSD result was 0.4932 Å, indicating the binding pocket of these structures are very analogous (Fig. 4, Panel B).
Ligand affinity and docking at 5-HT2A AND 5-HT2C receptors
Affinity of (2S, 4R) and (2R, 4S)-PAT and 4′-Cl-PAT at 5-HT2A and 5-HT2C GPCRs
The experimentally determined affinity values (Ki) for (2R, 4S)- and (2S, 4R)-PAT and 4′-Cl-PAT at human WT 5-HT2A and 5-HT2C receptors[24,38] are summarized in Table 3.
Table 3.
Affinity of PAT and 4′-Cl-PAT at 5-HT2A and 5-HT2C GPCRs.
| Ligand |
Ki ± SEM (nM)
|
|
|---|---|---|
| 5-HT2A | 5-HT2C | |
| (2R, 4S)-PAT | 460 ± 47 | 500 ± 70 |
| (2S, 4R)-PAT | 95 ± 7.2 | 27 ± 2.6 |
| (2R, 4S)-4′-Cl-PAT | 42 ± 6.0 | 45 ± 7.0 |
| (2S, 4R)-4′-Cl-PAT | 240 ± 32 | 130 ± 16 |
Stereochemistry apparently plays an important role in binding of PAT and 4′-Cl-PAT at 5-HT2A and 5-HT2C receptors. Moreover, electronic as well as steric parameters of the 4-phenyl-2-aminotetralin scaffold impact stereoselective 5-HT2 binding and function. For example, at both 5-HT2A and 5-HT2C receptors, higher affinity is associated with the (2S, 4R)-configuration of PAT whereas it is the (2R, 4S)-enantiomer of 4′-Cl-PAT that has higher affinity (Table 3). Regarding function, agonist activity at 5-HT2C receptors is observed for both PAT and 4′-Cl-PAT, regardless of stereochemistry. At 5-HT2A receptors, however, function appears to be sensitive to stereochemistry and perhaps other electronic and steric parameters associated with the 4-phenyl-2-aminotetralin scaffold; thus, agonist activity is observed only for (2R, 4S)-PAT (Table 3).
Docking of (2S, 4R-) and (2R, 4S)-PAT at 5-HT2A and 5-HT2C GPCRs
To help interpret the experimental data in Table 3, ligand docking studies were undertaken to delineate ligand– receptor molecular interactions for (2S, 4R)- and (2R, 4S)-PAT at the 5-HT2A and 5-HT2C receptor models. Figure 5 shows the more active (2S, 4R)-enantiomer of PAT docked at each model.
Figure 5.
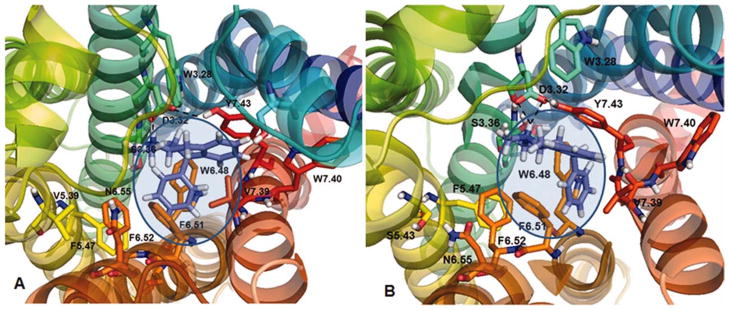
(2S, 4R)-PAT docked at 5-HT2A (panel A) and 5-HT2C (panel B) receptor models. The binding pocked is showed as shaded area. Dashed lines indicate ionic and HB interactions. [Color figure can be viewed in the online issue, which is available at wileyonlinelibrary.com.]
At the 5-HT2A model (Fig. 5A), the protonated amine group of (2S, 4R)-PAT could form a bifurcated hydrogen bond (HB) with the carboxylate oxygen atoms of 5-HT2A residue D3.32 (bonding distance 2.02–2.30 Å), which also could form an HB with the para-hydroxy group of Y7.43 (bonding distance 1.40 Å). The C(4) phenyl group of (2S, 4R)-PAT docked close to the F6.52 phenyl sidechain (bonding distance 4.2 Å), however, the orientation does not favor π-π interactions.
At the 5-HT2C model (Fig. 5B), the protonated amine group of (2S, 4R)-PAT could form a bifurcated HB with the carboxylate of D3.32 (bonding distance 1.77–2.22 Å), over a shorter distance than was observed at the 5-HT2A model. The 5-HT2C D3.32 moiety also could form an HB with the para-hydroxy group of Y7.43 (bonding distance 1.56 Å), as is the case for 5-HT2A, and, also could form an HB with the hydroxy group of S3.36 (bonding distance 1.58 Å). Moreover, at the 5-HT2C receptor model (in contrast to the 5-HT2A model), the aromatic part of the PAT tetrahydronaphthalene system docked close and parallel to the aromatic ring of Y7.32 (bonding distance 3.7 Å; Fig. 5B), which could facilitate π–π interactions. Meanwhile, the (2S, 4R)-PAT C(4) phenyl moiety docked in a hydrophobic pocket formed by 5-HT2C residues W6.48, F6.51, and F6.52 (Fig. 5B), as observed in previously studies.[29,30] Thus, in comparison to the 5-HT2A receptor, there are additional aromatic and hydrophobic interactions possible between (2S, 4R)-PAT and the 5-HT2C receptor that likely account for its three-times higher affinity at 5-HT2C compared to 5-HT2A receptors (Table 3).
In comparison to (2S, 4R)-PAT, the (2R, 4S)-PAT enantiomer binds with lower affinity at both 5-HT2A and 5-HT2C receptors (Table 3) and docking results are helpful to explain the observed experimental data. For example, when (2R, 4S)-PAT was docked at the 5-HT2A receptor model (not shown), its protonated amine moiety could form an HB with D3.32 at a slightly longer distance (1.80–3.37 Å) than was observed for (2S, 4R)-PAT, however, the (2R, 4S)-PAT C(4) phenyl ring and tetrahydronaphthyl system docked relatively far (7.0 Å) from 5-HT2A binding pocket aromatic residues W6.48, F6.51, and F6.52, presumably, precluding significant binding interactions with these residues. When (2R, 4S)-PAT was docked at the 5HT2C model (not shown), its protonated amine moiety was relatively far (2.45 Å) from the receptor D3.32 residue, and its C(4) phenyl moiety was far (6.6–7.0 Å) from the 5-HT2C binding pocket aromatic residues W6.48, F6.51, and F6.52, precluding hydrophobic or π–π interactions. Overall, in comparison to the (2S, 4R)-PAT enantiomer, docking results reveal a significant lack of potential binding interactions between the (2R, 4S)-PAT enantiomer and the 5-HT2A and 5-HT2C receptor models, that likely explains the lower affinity of this enantiomer at both receptors (Table 3).
Docking of (2S, 4R) and (2R, 4S)-4′-Cl-PAT at 5-HT2A and 5-HT2C GPCRs
For comparison to the PAT binding and docking results reported above, (2S, 4R)-Cl-PAT (Fig. 6) and (2R, 4S)-4′-Cl-PAT (Fig. 7) were each docked at the 5-HT2A and 5-HT2C receptor models; for clarity, only the binding pocket amino acid residues of the receptor are shown. When (2S, 4R)-4′-Cl-PAT was docked at the 5-HT2A model (Fig. 6A), it was apparent that a bifurcated HB could form with the D3.32 carboxylate (bonding distance 1.83–1.96 Å), which, could also form an HB with the indol sidechain of W3.28 (bonding distance 1.78 Å), but, not with Y7.38. When (2S, 4R)-4′-Cl-PAT was docked at the 5-HT2C model (Fig. 6B), a single HB could form with D3.32 (bonding distance 1.77 Å), which, also could form an HB with the indol side-chain of W3.28 (bonding distance 1.78 Å) and with the hydroxy sidechain of Y7.43 (bonding distance 1.36 Å). There were no apparent interactions observed between the 4′-Cl-substituent of (2S, 4R)-4′-Cl-PAT and the 5-HT2A or 5-HT2C receptor models.
Figure 6.
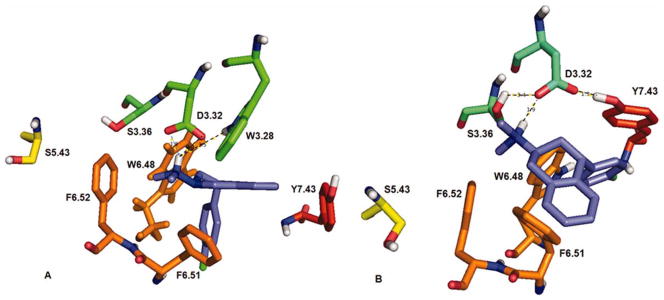
(2S, 4R)-4′-Cl-PAT docked at 5-HT2A (panel A) and 5-HT2C (panel B) receptor models. Only the binding pocket amino acid residues of the receptor are shown for clarity. [Color figure can be viewed in the online issue, which is available at wileyonlinelibrary.com.]
Figure 7.

(2R, 4S)-4′-Cl-PAT docked at 5-HT2A (panel A) and 5-HT2C (panel B) receptor models. Only the binding pocket amino acid residues of the receptor are shown for clarity. [Color figure can be viewed in the online issue, which is available at wileyonlinelibrary.com.]
When (2R, 4S)-4′-Cl-PAT was docked at the 5-HT2A model (Fig. 7A), it was apparent that the protonated amine could form an HB with the carboxylate sidechain of D3.32 (bonding distance 1.75 Å) over a shorter distance than was found for the (2S, 4R)-4′-Cl-PAT enantiomer. Also, D3.32 could form an HB with the hydroxy group of Y7.43 (bonding distance 1.49 Å). The 4′-Cl-substituent of (2R, 4S)-4′-Cl-PAT could interact with the hydroxy sidechain of S5.43 (bonding distance 2.50 Å).
When (2R, 4S)-4′-Cl-PAT was docked to the 5-HT2C model (Fig. 7B), its protonated amine moiety could form an HB to D3.32 (bonding distance 1.60 Å), which could also form an HB with Y7.43 (bonding distance 1.48 Å). As observed for the 5-HT2A receptor, the 4′-Cl-substituent of (2R, 4S)-4′-Cl-PAT also oriented close to the hydroxyl moiety of 5-HT2C residue S5.43 (bonding distance 2.50 Å). Thus, at both 5-HT2A and 5-HT2C receptors, the interaction of the 4′-Cl-substituent of (2R, 4S)-4′-Cl-PAT with the S5.43 residue, together, with the potential for a close and presumably tight HB with D3.32, likely explains the higher affinity of this enantiomer over the (2S, 4R)-4′-Cl-PAT enantiomer at 5-HT2A and 5-HT2C receptors (Table 3).
Conclusions
Computational studies were performed to build βAR-based homology models of the 5-HT2A and 5-HT2C GPCRs for ligand docking studies, to characterize 3D (stereochemical) molecular determinants for binding of drug candidates. Ligand–receptor interactions in the binding pocket were analyzed and compared to the experimentally determined affinity values (Ki) at both 5-HT2A and 5-HT2C GPCRs. The higher affinity of (2S, 4R)-PAT at 5-HT2C versus 5-HT2A receptors was rationalized in light of modeling results indicating the ligand could form significant π–π interactions with the 5-HT2C Y7.43 residue, but, not with the corresponding 5-HT2A Y7.43 residue. The observed reversed stereoselective affinity of PAT and 4′-Cl-PAT at 5-HT2A and 5-HT2C receptors (Table 3) was rationalized in view of docking results indicating that (2R, 4S)-4′-Cl-PAT could form a close and presumably tight HB with the D3.32 residue of both receptors. Meanwhile, the corresponding (2R, 4S)-PAT enantiomer docked relatively far away from the 5-HT2 residue D3.32 compared to the other ligands, and, the same is true for its C(4) phenyl ring and tetrahydronaphthyl system regarding interactions with 5-HT2 binding pocket aromatic residues W6.48, F6.51, and F6.52.
It has been suggested that 5-HT2 residue Y7.43 plays a role in stabilizing the negative charge of the D3.32 carboxylate, and, this interaction is more or less, depending on the ligand structure and conformation/activation state of the receptor.[29,30] In this work, we found that binding of PAT analogs to the 5-HT2A or 5-HT2C receptor decreased the distance between the receptor Y7.43 hydroxy moiety and D3.32 carboxylate moiety; the exception was (2S, 4R)-4′-Cl-PAT, where no closer interaction between these residues was observed upon binding of the ligand. It is not yet clear how D3.32–Y7.43 interaction upon ligand binding may impact ligand affinity or resulting functional activity.
Homology modeling incorporating MD simulations leads to improved results when all-atom MD simulations and relatively long simulation times with sufficiently accurate force fields are used.[46] Thus, in the present work we used all-atom MD simulations of the receptor embedded in a pre-equilibrated POPC system to emulate the membrane, with simulation times of 5 μs. We have obtained improved accuracy regarding the predictions of such models, for example, the β2-AR based-homology model of the human histamine H1 receptor, built using a protocol analogous to the one resented in this work, was compared with the human H1 crystal structure, with resulting RMSD 2.91 Å.[47] This result suggests the present methodology produces reliably predictable homology models, useful for the study of drug–receptor interaction for drug design purposes.
Acknowledgments
Contract grant sponsor: National Institutes of Health; Contract/grant numbers: RO1 DA023928, DA030989, and MH081193.
References
- 1.Jensen NH, Cremers TI, Sotty F. Sci World J. 2010;10:1870. doi: 10.1100/tsw.2010.180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bubar MJ, Cunningham KA. Prog Brain Res. 2008;172:319. doi: 10.1016/S0079-6123(08)00916-3. [DOI] [PubMed] [Google Scholar]
- 3.Nichols DE. Pharmacol Ther. 2004;101:131. doi: 10.1016/j.pharmthera.2003.11.002. [DOI] [PubMed] [Google Scholar]
- 4.(a) Fitzgerald LW, Burn TC, Brown BS, Patterson JP, Corjay MH, Valentine PA, Sun JH, Link JR, Abbaszade I, Hollis JM, Largent BL, Hartig PR, Hollis GF, Meunier PC, Robichaud AJ, Robertson DW. Mol Pharmacol. 2000;57:75. [PubMed] [Google Scholar]; (b) Launay JM, Herve P, Peoc’h K, Tournois C, Callebert J, Nebigil CG, Etienne N, Drouet L, Humbert M, Simonneau G, Maroteaux L. Nat Med. 2000;8:1129. doi: 10.1038/nm764. [DOI] [PubMed] [Google Scholar]; (c) Rothman RB, Baumann MH, Savage JE, Rauser L, McBride A, Hufeisen SJ, Roth BL. Circulation. 2000;102:2836. doi: 10.1161/01.cir.102.23.2836. [DOI] [PubMed] [Google Scholar]
- 5.Kroeze WK, Kristiansen K, Roth BL. Curr Top Med Chem. 2002;2:507. doi: 10.2174/1568026023393796. [DOI] [PubMed] [Google Scholar]
- 6.Palczewski K, Kumasaka T, Hori T, Behnke CA, Motoshima H, Fox BA, Trong IL, Teller DC, Okada T, Stenkamp RE, Yamamoto M, Miyano M. Science. 2000;289:793. doi: 10.1126/science.289.5480.739. [DOI] [PubMed] [Google Scholar]
- 7.Li J, Edwards PC, Burghammer M, Villa C. J Mol Biol. 2004;343:1409. doi: 10.1016/j.jmb.2004.08.090. [DOI] [PubMed] [Google Scholar]
- 8.Okada T, Sugihara M, Bondar AN, Elstner M, Entel P, Buss V. J Mol Biol. 2004;342:571. doi: 10.1016/j.jmb.2004.07.044. [DOI] [PubMed] [Google Scholar]
- 9.Okada T, Fujiyoshi Y, Silow M, Navarro J, Landau EM, Shichida Y. Proc Natl Acad Sci USA. 2002;99:5982. doi: 10.1073/pnas.082666399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Teller DC, Okada T, Behnke CA, Palczewski K, Stenkamp RE. Biochemistry. 2001;40:7761. doi: 10.1021/bi0155091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Park JH, Scheerer P, Hofmann KP, Choe HW, Ernst OP. Nature. 2008;454:183. doi: 10.1038/nature07063. [DOI] [PubMed] [Google Scholar]
- 12.Scheerer P, Park JH, Hildebrand PW, Kim YJ, Krauss N, Choe HW, Hofmann KP, Ernst OP. Nature. 2008;455:497. doi: 10.1038/nature07330. [DOI] [PubMed] [Google Scholar]
- 13.Jaakola VP, Griffith MT, Hanson MA, Cherezov V, Chien EYT, Lane JR, Ijzerman AP, Stevens RC. Science. 2008;322:1211. doi: 10.1126/science.1164772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Warne T, Serrano-Vega MJ, Baker JG, Moukhametzianov R, Edwards PC, Henderson R, Leslie AGW, Tate CG, Schertler GFX. Nature. 2008;454:486. doi: 10.1038/nature07101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.(a) Cherezov V, Rosenbaum DM, Hanson MA, Rasmussen SGF, Thian FS, Kobilka TS, Choi HJ, Kuhn P, Weis WI, Kobilka BK, Stevens RC. Science. 2007;318:1258. doi: 10.1126/science.1150577. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Rosenbaum DM, Cherezov V, Hanson MA, Rasmussen SGF, Thian FS, Kobilka TS, Choi HJ, Yao XJ, Weis WI, Stevens RC, Kobilka BK. Science. 2007;318:1266. doi: 10.1126/science.1150609. [DOI] [PubMed] [Google Scholar]
- 16.Hanson MA, Cherezov V, Griffith MT, Roth CB, Jaakola VP, Chien EY, Velasquez J, Kuhn P, Stevens RC. Structure. 2008;16:897. doi: 10.1016/j.str.2008.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Rasmussen SG, Choi HJ, Rosenbaum DM, Kobilka TS, Thian FS, Edwards PC, Burghammer M, Ratnala VR, Sanishvili R, Fischetti RF, Schertler GF, Weis WI, Kobilka BK. Nature. 2007;450:383. doi: 10.1038/nature06325. [DOI] [PubMed] [Google Scholar]
- 18.Rassmussen SG, Choi HJ, Fung JJ, Pardon E, Casarosa P, Chae PS, DeVree BT, Rosembaum DM, Thian FS, Kobilka TS, Schnapp A, Konetzki I, Sunahara RK, Gellman SH, Pautsch A, Steyaert J, Weis WI, Kobilka BK. Nature. 2011;469:175. doi: 10.1038/nature09648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Rosembaum DM, Zhang C, Lyons JA, Holl R, Aragao D, Arlow DH, Rasmussen SG, Choi HJ, DeVree BT, Sunahara RK, Chae PS, Gellman SH, Dror RO, Shaw DE, Weis WI, Caffrey M, Gmeiner P, Kobilka BK. Nature. 2011;469:236. doi: 10.1038/nature09665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Congreve M, Langmea CJ, Mason JS, Marshall FH. J Med Chem. 2011;54:4283. doi: 10.1021/jm200371q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chien EYT, Liu W, Zhao Q, Katritch V, Han GW, hanson MA, Shi L, Newman AH, Javitch JA, Cherezov V, Stevens RC. Science. 2010;330:1091. doi: 10.1126/science.1197410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Schimamura T, Shiroishi M, Weyand S, Tsujimoto H, Winter G, Katritch V, Abagyan R, Cherezov V, Liu W, Won Han G, Kobayashi T, Stevens RC, Iwata S. Nature. 2011;475:65. doi: 10.1038/nature10236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Congreve M, Andrews SP, Dore AS, Hollenstein K, Hurrell E, Langmead CJ, Mason JS, Ng IW, Tehan B, Zhukov A, Weir M, Marshall FH. J Med Chem. 2012;55:1898. doi: 10.1021/jm201376w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Booth RG, Fang L, Huang Y, Wilczynski A, Sivendran S. Eur J Pharmacol. 2009;615:1. doi: 10.1016/j.ejphar.2009.04.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bucholtz EC, Brown RL, Tropsha A, Booth RG, Wyrick SD. J Med Chem. 1999;42:3041. doi: 10.1021/jm980428x. [DOI] [PubMed] [Google Scholar]
- 26.Kristiansen K, Dahl SG. Eur J Pharmacol. 1996;306:195. doi: 10.1016/0014-2999(96)00180-x. [DOI] [PubMed] [Google Scholar]
- 27.Kristiansen K, Kroeze WK, Willins DL, Gelber EI, Savage JE, Glennon RA, Roth BL. J Pharmacol Exp Ther. 2000;293:735. [PubMed] [Google Scholar]
- 28.Booth RG, Fang L, Wilczynski A, Sivendren S, Sun Z, Travers S, Bruysters M, Sansuk K, Leurs R. Inflam Res. 2008;57(suppl 1):S43. doi: 10.1007/s00011-007-0621-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Canal C, Cordova-Sintjago TC, Villa NY, Fang LJ, Booth RG. Eur J Pharmacol. 2011;673:1. doi: 10.1016/j.ejphar.2011.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Cordova-Sintjago T, Villa N, Canal C, Booth R. Int J Quantum Chem. 2011;112:140. doi: 10.1002/qua.23231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Thompson JD, Higgins DG, Gibson TJ. Nucleic Acids Res. 1994;22:4673. doi: 10.1093/nar/22.22.4673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Larkin MA, Blackshields G, Brown NP, Chenna R, McGettigan PA, McWilliam H, Valentin F, Wallace IM, Wilm A, Lopez R, Thompson JD, Gibson TJ, Higgins DG. Bioinformatics. 2007;23:2947. doi: 10.1093/bioinformatics/btm404. [DOI] [PubMed] [Google Scholar]
- 33.SYBYL-X 1.2. Tripos International; 1699 South Hanley Rd., St. Louis, Missouri, 63144, USA: [Google Scholar]
- 34.Clark M, Cramer RD, III, van Opdenhosch N. J Comput Chem. 1989;10:982. [Google Scholar]
- 35.Cornell WD, Cieplak P, Bayly CI, Gould IR, Merz KM, Jr, Ferguson DM, Spellmeyer DC, Fox T, Caldwell JW, Kollman PA. J Am Chem Soc. 1995;117:5179. [Google Scholar]
- 36.Heller H, Schaefer M, Schulten K. J Phys Chem. 1993;97:8343. [Google Scholar]
- 37.Wyrick SD, Booth RG, Myers AM, Owens CE, Kula NS, Baldessarini RJ, McPhail AT, Mailman RB. J Med Chem. 1993;36:2542. doi: 10.1021/jm00069a013. [DOI] [PubMed] [Google Scholar]
- 38.Sakhuja R, Kondobolu K, Canal CE, Cordova-Sintjago T, Kim MS, Sun Z, Vincek AS, Travers S, Abboud KA, Booth RG. J Med Chem. (submitted for publication) [Google Scholar]
- 39.HyperChem (TM) Professional 8.0. Hypercube, Inc; 1115 NW 4th Street, Gainesville, Florida 32601, USA: [Google Scholar]
- 40.Schneidman-Duhovny D, Inbar Y, Nussinov R, Wolfson HJ. Nucleic Acids Res. 2006;33:W363. doi: 10.1093/nar/gki481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Gasteiger J, Marsili M. Tetrahedron. 1980;36:3219. [Google Scholar]
- 42.(a) Ballesteros JA, Jensen AD, Liapakis G, Rasmussen SG, Shi L, Gether U, Javitch JA. J Biol Chem. 2001;276:29171. doi: 10.1074/jbc.M103747200. [DOI] [PubMed] [Google Scholar]; (b) Ballesteros JA, Shi L, Javitch JA. Mol Pharmacol. 2001;60:1. [PubMed] [Google Scholar]
- 43.The PyMOL Molecular Graphics System, Version 1.3. Schrödinger, LLC; [Google Scholar]
- 44.Laskowski RA, MacArthur MW, Moss DS, Thornton JM. J Appl Crystallogr. 1993;26:283. [Google Scholar]
- 45.Morris AL, MacArthur MW, Hutchinson EG, Thornton JM. Proteins. 1992;12:345. doi: 10.1002/prot.340120407. [DOI] [PubMed] [Google Scholar]
- 46.Raval A, Piana S, Eastwood MP, Dror RO, Shaw DE. Proteins Struct Funct Bioinform. 2012 doi: 10.1002/prot.24098. [DOI] [PubMed] [Google Scholar]
- 47.Cordova-Sintjago T, Fang L, Bruysters M, Leurs R, Booth R. 243rd ACS national meeting; San Diego, California. 2012. MEDI 214. [Google Scholar]