

Summary
Systemic chronic immune activation is considered today as the driving force of CD4+ T-cell depletion and acquired immunodeficiency syndrome (AIDS). A residual chronic immune activation persists even in HIV-infected patients in which viral replication is successfully inhibited by antiretroviral therapy, with the extent of this residual immune activation being associated with CD4+ T-cell loss. Unfortunately, the causal link between chronic immune activation and CD4+ T-cell loss has not been formally established. This article provides first a brief historical overview on how the perception of the causative role of immune activation has changed over the years and lists the different kinds of immune activation that have been observed to be characteristic for human immunodeficiency virus (HIV) infection. The mechanisms proposed to explain the chronic immune activation are multiple and are enumerated here, as well as the mechanisms proposed on how chronic immune activation could lead to AIDS. In addition, we summarize the lessons learned from natural hosts that know how to ‘show AIDS the door’, and discuss how these studies informed the design of novel immune modulatory interventions that are currently being tested. Finally, we review the current approaches aimed at targeting chronic immune activation and evoke future perspectives.
Keywords: HIV, SIV, pathogenesis, immune activation, inflammation, natural hosts
Introduction
Systemic chronic immune activation is considered today as the driving force of CD4+ T-cell depletion and acquired immunodeficiency syndrome (AIDS). However, the causal link between these two phenomena has not been formally established. In this article, we discuss how the perception of the causative role of immune activation has changed over the years, list the different kinds of immune activation described during human immunodeficiency virus (HIV) infection, enumerate the mechanisms proposed to explain the chronic immune activation, and propose how they could lead to acquired immunodeficiency syndrome (AIDS). In addition, we summarize the knowledge learned by studying natural simian immunodeficiency virus (SIV) hosts that learned how to ‘show AIDS the door’ and discuss how these studies informed the design of novel immune modulatory interventions that are currently being tested. Finally, we review the current approaches aimed at targeting chronic immune activation and evoke future perspectives. Chronic immune activation persists even in HIV-infected patients in which viral replication is successfully inhibited by antiretroviral therapy. However, since the pathogenic role of this residual immune activation is discussed in another review of this special issue on HIV Immunology, our manuscript mainly focuses on the natural history of HIV infection.
Historical review on immune activation in HIV
HIV was first isolated 30 years ago from a lymph node biopsy taken from a patient with generalized hyperplasic lymphadenopathy (1). The isolation of the virus was attempted on cells from lymph node, based on the presumption that the amount of virus would be higher in this organ. Lymphadenopathy was a recognized symptom of patients with AIDS, and it was already acknowledged by clinicians as a sign of immune activation in AIDS patients. Since it had been shown that HIV is cytopathic in vitro, uses CD4 as the major receptor and predominantly replicates in CD4+ T lymphocytes, it was obvious to speculate that HIV causes the progressive depletion of CD4+ T cells by direct cytopathic effects. Yet even early on, pathogenic models that hypothesized indirect mechanisms and immune system activation as important contributors to progressive immunodeficiency were proposed (2). For example, the levels of inflammatory markers in the plasma during the asymptomatic phase, such as of neopterin, were demonstrated to be predictive for the development of AIDS (3, 4). Moreover, high levels of activated CD8+ T cells were shown to predictive of shorter survival (5).
A few years later, however, support emerged for direct virus involvement in CD4+ T-cell loss. In particular, the analysis of viral dynamics under HAART demonstrated that the asymptomatic phase of HIV infection is not silent but rather characterized by a massive and continuous viral replication, with as many as 50 billion viruses produced every day in the body of an infected individual (6, 7). This view was further supported by reports highlighting the gastrointestinal tract as a major site of viral replication in HIV and SIV infections and the associated massive and rapid CD4+ T-cell depletion in this tissue (8, 9).
Meanwhile, tools were developed to quantify viral load in the natural hosts of SIV, African nonhuman primates (NHPs) that do not progress to AIDS, i.e. sooty mangabeys (SMs), African green monkeys (AGMs), and mandrills (see ‘Learn from the experts’ section below). Natural hosts showed sustained high levels of plasma viremia, high viral load in gut and an early, massive intestinal CD4+ T-cell depletion (10, 11). While viral replication pattern and cytopathicity were similar in nonpathogenic and pathogenic infections, natural hosts did not show high chronic immune activation (10, 11). These observations have been central to refocus attention on the role of immune activation in HIV infection. Indeed, more recent studies are in full support of immune activation being the major driving force of CD4+ T-cell loss and AIDS: (i) the levels of activated CD8+ T cells were shown to be more closely associated with a shorter survival than viral load or CD4+ T cells (12); (ii) comparative studies on HIV-2-infected individuals are consistent with a direct causal relationship between immune activation and CD4+ T-cell depletion but show only an indirect relationship of these parameters with the virus replication rate (13); (iii) a few HIV-1-infected individuals do not progress to AIDS despite displaying high viremia (14, 15); (iv) successfully HAART-treated patients with undetectable viremia still show higher levels of T-cell activation than healthy controls, with the extent of this residual immune activation being associated with increased morbidity and mortality (16); and (v) elite controllers of HIV (HIC), i.e. individuals who spontaneously control viral replication to below detection levels, show levels of T-cell activation higher than healthy donors and even higher than HAART-suppressed individuals (17). Of note, those HIC with higher T-cell activation display slow but progressive CD4+ T-cell loss and can develop AIDS. The fact that antiviral drugs do not target immune activation and that a residual immune activation persists despite successfully suppressed viremia highlights the importance of identifying, and then targeting, the mechanisms that cause immune activation.
Immune activation in distinct cells and tissues
Markers of abnormal immune activation during HIV infection
The first descriptions of HIV-associated immune activation were based on clinical observations (lymphadenopathy) and analyses of blood samples collected from chronically HIV-infected individuals. These analyses revealed increased levels of markers of activation (HLA-DR, CD38) and proliferation (Ki-67) on both CD4+ and CD8+ T cells (Table 1). Increased numbers of HLA-DR+, CD38+ CD8+ T lymphocytes were associated with a fall in CD4+ T-cell counts and the development of AIDS (18). Later on, even in HIC, higher CD4+ and CD8+ T-cell activation was associated with lower CD4+ T-cell counts (17). For each individual, the level of T-cell activation stabilizes for a period of time after the acute infection. This time point of stabilization is established early in infection (generally 6 months post-acute infection) and called ‘set-point’. Interestingly, the T-cell activation at the set-point, and even at earlier time points, is predictive of the rate at which CD4+ T cells are lost over time (5, 19, 20).
Table 1. Biomarkers predicting progression towards AIDS independently of CD4+ T cells and viral load.
The table lists the parameters of T cell activation, inflammation and coagulation quantified in the blood that have been shown to predict the levels of disease progression markers at set-point (CD4 counts, viremia, T cell activation) and/or disease progression independently of CD4+ T cells and viral load. For parameters predictive of disease progression, it is not further specified whether they refer to CD4+ T cell counts or AIDS-related events. The parameters that are solely increased in progressors or are simply associated with viremia and/or CD4+ T cell levels but were not identified as predictors are not listed here.
| STAGE OF INFECTION |
MARKER | PREDICTION |
|---|---|---|
| acute | IP-10 | CD4+ T cell levels (set-point) Rapid disease progression |
| acute | IL-7, IL-12p40, IL-1α, eotaxin, GM-CSF | CD4+ T cell loss (< 350) |
| acute | IL-7, IL-12p40, IL-12p70, IFN-γ, IL-15 | Viremia (set-point) |
| acute | TGF-β1, IL-18 | T cell activation (setpoint) |
|
| ||
| post-acute | CD8+ T cell activationa | disease progression |
|
| ||
| chronic | T cell activation and proliferationb | disease progression |
| chronic | neopterin | disease progression |
| chronic | beta2-microglobulin | disease progression |
| chronic | D-dimer | disease progression |
| chronic | CRPc | disease progression |
| chronic | TNF-α | disease progression |
| chronic | TNF-RIId | disease progression |
| chronic | IL-6 | disease progression |
| chronic | LPS | disease progression |
| chronic | sCD14e | disease progression |
| chronic | IL-10 | disease progression |
| chronic | TGF-β1 | disease progression |
CD8+ T cells expressing CD38 and HLA-DR.
Proliferation has in most cases been defined by the proportion of cells expressing Ki-67.
CRP : high sensitivity C-reactive protein.
TNF-RII := tumor necrosis factor-a receptor II p75.
sCD14 : soluble CD14, a marker of monocyte activation and indirect marker of microbial translocation.
The expressions of the activation markers CD38 and HLA-DR are also increased on HIV-specific CD8+ T cells and, although to a lesser extent, on CD8+ T cells specific for other viruses (21). CD38 expression on HIV-specific as well as EBV-, CMV-, and FLU-specific CD8+ T cells correlates with HIV viral load (21).
This persistent pathological level of T-cell activation is associated with inflammation, microbial translocation (MT) and increased levels of coagulation markers (Table 1). The first plasma proteins documented to be increased in chronic HIV infection were neopterin, β2-microglobulin (β2M), tumor necrosis factor-α (TNF-α), soluble TNF receptor II (TNFRII), soluble interleukin-2R and interferon-γ, with β2M generally being the most stable marker (3, 22-24). Many other proteins, including anti-inflammatory cytokines IL-10 and TGF-β1, have been shown to also be increased in the plasma of chronically HIV-1-infected individuals and to predict the rate of progression towards AIDS (25). More and more data point towards an association between monocyte/macrophage activation and disease progression. In viremic, untreated patients, the numbers of pro-inflammatory CD14+CD16+ monocytes are increased (26, 27). In SIV-infected MAC, massive turnover of peripheral monocytes predicted progression towards AIDS better than viral load (28). Less is known on HIV-2, but sCD14, a marker of monocyte activation and an indirect marker of microbial translocation, is also negatively correlated with CD4+ T-cell counts in HIV-2 infected individuals (29).
In acute infection, a massive increase of cytokines, called cytokine storm, is characteristic of HIV-1 infection (30). The levels of some of these are predictive of the rate of CD4+ T-cell loss and of T-cell activation levels, and plasma levels of the IFN-inducible protein-10 (IP-10) during acute infection are predictive of rapid disease progression (31).
Cellular sources of factors associated with chronic immune activation
Most of the cytokines involved in chronic immune activation and inflammation during HIV-1 and SIVmac infections can be produced by multiple cell types. Not all of them have been defined. It has been reported that (i) T cells are the major sources of IP-10 (32); (ii) TGF-β is mostly produced by T-regulatory (Treg) cells (33); and (iii) plasmacytoid dendritic cells (pDCs) are the predominant IFN-I secreting cells, although other cells might serve as another important source in later stages of infection (34, 35). Blood mononuclear cells from viremic patients produce more TNF-α in response to lipopolysaccharide (LPS) than those from virologically suppressed patients, with M-DC8+ monocytes being predominantly responsible for this overproduction (27). Excessive production of TNF-α could act at mucosal sites to compromise the integrity of the epithelial barrier (see ‘Proposed mechanisms inducing chronic immune activation’ section below).
Other cells of the immune system, such as B cells, neutrophils, or natural killer T (NKT) cells, contribute to the inflammatory or immunosuppressive milieu, as well as secreting chemokines able to modify cellular distributions. Finally, some non-immune cells, such as cervical epithelial cells, are capable of secreting chemokines after exposure to HIV-1 (36, 37).
Immune activation in tissues
The large majority of studies on immune activation were performed in blood, as this compartment is the most accessible. Studies that could assess immune activation in tissues, particularly at earlier phases of infection, are rare (38). They consistently showed that HIV-1 and SIVmac infections are characterized by marked hyperplasia in lymph nodes with significant expansion of plasma B cells (39). Moreover, all stages of infection are characterized by infiltrations of CD8+ T cells into germinal centers (40). The T-follicular helper (Tfh) subset is also expanded according to several recent studies (see ‘Proposed mechanisms inducing chronic immune activation’ section below). The T zone of lymph nodes is marked by infiltrations of CD163+ macrophages, polymorphonuclear leukocytes, as well as TGF-β and IL-10-producing Treg cells (33, 41).
Utilizing the longitudinal collection of mucosal tissues and the comparison between pathogenic and nonpathogenic SIV infections, studies on nonhuman primates showed that the level of Ki-67 expressing T cells was increased in both MAC and natural hosts during the acute phase of infection, but only remained sustained throughout the chronic phase in pathogenic SIVmac infection (42). In chronically HIV-infected patients with high proviral loads, pDCs accumulate in the spleen and traffic to mucosal sites (34, 43, 44). In the brain, patients with encephalitis can, for example, display intraparenchymal infiltrations by CD8+ lymphocytes (45). Thus, HIV infection is associated with a modification of the distribution of immune cells. This is probably due to increased and sustained expression of chemokines in the respective tissues where the cells accumulate, but also to the modification of the expression of homing receptors. Indeed, in SIVmac infection, pDCs capable of producing pro-inflammatory cytokines significantly upregulated the gut-homing marker integrin α4β7 (34, 43, 44), and in vivo blockade of α4β7 dampened pDC recruitment to the colorectum and resulted in reduced immune activation. Remarkably, upregulation of β7-integrin expression on circulating pDCs was observed in HIV-infected humans but not in chronically SIV-infected SMs that show low levels of immune activation.
Collectively, these findings clearly illustrate that HIV infection is characterized by an immune activation status that encompasses many cells and tissues, with T-cell- and monocyte/macrophage-associated markers as well as inflammatory soluble plasma molecules being predictive of disease progression. Although the general consensus is for a link between inflammation and T-cell activation, the exact mechanisms binding these two phenomena still need to be clearly defined.
Proposed mechanisms inducing chronic immune activation
In the previous section, we discussed how extended and generalized chronic immune activation is in the setting of HIV infection. That being established, the next burning question is what mechanisms contribute to chronic immune activation during HIV infection.
Unfortunately, and despite intense research efforts, there is no clear response to this question. Given the complexity of the interaction between HIV and the host immune system, there are multiple molecular and cellular mechanisms by which HIV infection, at least in theory, can induce immune activation. To make things even more complicated, it is possible that several of the proposed mechanisms synergistically contribute to cause aberrant chronic immune activation. Moreover, it is conceivable, and in our opinion very likely, that the relative contribution of the different mechanisms changes significantly in different subsets of HIV-infected individuals, in different phases of HIV-infection (early vs. chronic vs. late), and in naive versus HAART-treated patients. In this section, we discuss the mechanisms that are considered key players in chronic immune activation in the literature (Fig. 1). For each of these mechanisms, we summarize the available experimental data supporting or questioning their contribution.
Fig. 1. Proposed contributors to HIV-associated chronic immune activation.

There are multiple molecular and cellular mechanisms by which HIV infection could induce generalized immune activation. Among these, as summarized in this cartoon, HIV replication; immunomodulatory functions of viral proteins and immunes response to the virus; immune responses to reactivated infections; loss of mucosal integrity with consequent microbial translocation; altered balance of critical CD4+ T-cell subsets; increased homeostatic proliferation in response to CD4+ T-cell depletion; increased production of pro-inflammatory molecules. Importantly, each mechanism may feed to the others, thus creating an uncontrolled positive feedback. Furthermore, it is likely that the relative contribution of each varies among HIV-infected individuals or in distinct stages of HIV-infection, as well as in naïve versus HAART-treated patients. Adapted from Steven Deeks, XIX International AIDS Conference, 2012.
HIV replication and immune response to the virus
The most obvious cause of immune activation in the context of HIV infection is the direct innate and adaptive immune responses against the virus and its antigens. Not only are HIV antigens recognized by, and thus activate, T cells expressing virus-specific T-cell receptors and B cells bearing virus-specific surface immunoglobulins, but HIV components also bind to pattern recognition receptors, such as the Toll-like receptors 7 and 9 (46-49). In addition, during its process of entry and fusion, HIV might activate target cells by signaling through CD4 and its coreceptors, such as CCR5.
Fully supporting the important contribution of HIV replication, immune activation and inflammation correlate with the level of viremia and are significantly lower in HIV-infected patients who control viral replication spontaneously (HIC) or by HAART. Although the direct contribution of HIV replication to chronic immune activation is well recognized, several lines of evidence indicate that high levels of HIV replication are neither sufficient nor necessary to induce pathological levels of immune activation. First, the frequency of activated T cells largely exceeds the frequency of HIV-infected CD4+ T cells and HIV-specific CD4+ and CD8+ T cells (21, 50, 51). Furthermore, other cells types, including B cells, NK cells, pDCs, and monocytes, show increased levels of activation, turnover, and/or cell death (28, 52, 53). Second, levels of immune activation predict CD4+ T-cell decline better than, and independently of, viral load (12, 19). Third, in ART-treated HIV-infected patients as well as in HIC, the main markers of immune activation and inflammation are still significantly higher than in HIV-uninfected individuals despite undetectable viral loads (16, 54). Thus, inhibition of viral replication is still associated with non-physiologic levels of immune activation. Fourth, high viral loads are not sufficient to induce pathogenic levels of immune activation, as indicated in studies of natural hosts for SIV, such as SMs and AGMs. Indeed, SIV infection in natural hosts is non pathogenic and does not result in chronic immune activation despite viral loads similar to those present in chronically HIV-infected individuals (10, 11) (see ‘Proposed mechanisms for the pathogenic role of HIVassociated chronic immune activation’ section below). Further evidence comes from studies of immune activation in a rare subset of HIV-infected individuals named virologic non progressors (VNPs). VNPs maintain a high and remarkably stable CD4+ T-cell count for many years despite levels of viral replication similar to those found in non-controllers. Despite high viral loads, VNPs show a profile of non-activation, remarkably similar to those found in SIV-infected natural hosts (15, Klatt N, personal communication). Consistent with all these findings, it has been confirmed that viral load is only an indirect contributor to the rate of progression to AIDS, that immune activation predicts changes in CD4+ T cells stronger and independent of viral load, and that the effect of anti-retroviral therapy in increasing CD4+ T-cell counts better correlates with the decrease in immune activation than the suppression of viral load (17, 55-57). Collectively, the body of available data clearly supports a direct role of HIV replication in inducing chronic immune activation but also indicates that high levels of HIV replication are neither sufficient nor necessary to induce pathological levels of immune activation.
Viral proteins and immune activation
HIV gene products, such as Env, Tat, and Nef, have been proposed to be involved in HIV-induced immune activation. The Nef protein of HIV-1 has lost the ability to downmodulate the CD3-TCR complex from the surface of infected T cells (58). As a consequence, HIV-1 Nef may directly contribute to immune activation by rendering infected CD4+ T cells highly sensitive to re-stimulation through the T-cell receptor. The potential contribution of this mechanism is supported by the finding that the Nef proteins of SIV-infected SMs and AGMs can downmodulate the CD3-TCR complex from the cell surface, thus making infected CD4+ T cells more refractory to further antigenic stimulation (59). However, Nef from HIV-2 and SIVmac can also downmodulate CD3-TCR from the cell surface. To better define the role of this viral feature on immune activation in vivo, further studies in which AGMs will be infected with SIVagm containing an ‘HIV-1-like’ Nef are under investigation (F. Kirchhoff, personal communication).
Immune response to reactivated infections
Since HIV infection induces immunodeficiency, a second obvious mechanism contributing to chronic immune activation is represented by opportunistic infections and environmental pathogens (Fig. 1). This mechanism may be of particular importance in the late phase of HIV-infection but may also occur during acute infection. The vast majority of HIV-infected individuals contain other chronic viral infections, and highly prevalent latent viruses, such as cytomegalovirus (CMV) and Epstein-Barr virus (EBV), reactivate more frequently during HIV infection due to the depletion of CD4+ T cells. Through stimulation of a large numbers of cells, opportunistic pathogens may play a central role in sustaining chronic activation of the immune system in HIV-infection (21). Consistently, chronic CMV infection or CMV reactivation have been found to promote immune activation, inflammation, the release of angiogenic growth factors, and hypercoagulability; in addition, a role for CMV-specific CD4+ T cells in inducing HIV-associated atherosclerosis has been recently described (60). Moreover, it has been shown that in HIV-infected women, CMV antibody titers are increased and associated with subclinical cardiovascular disease (61). The contribution of CMV to immune activation and inflammation is also evident in HAART-treated patients with sustained virological suppression, in which CMV-induced immune response associates with chronic immune activation and CMV viremia predicts increased mortality (62). As perhaps the strongest piece of evidence for CMV (and/or other herpesvirus) replication being an important mechanism of immune activation in HIV-infected individuals, a recent clinical study showed that Valganciclovir significantly reduced the levels of activated (CD38+HLA-DR+) CD8+ T cells in HAART-treated HIV-infected individuals with incomplete CD4+ T-cell reconstitution (63). Thus, an intervention able to suppress a hypothesized cause of immune activation did indeed decrease chronic immune activation in ART-treated HIV-infected individuals.
Loss of mucosal integrity of the gastrointestinal tract and microbial translocation Another potential key contributor to HIV-associated chronic immune activation is the loss of immune function in the gastrointestinal (GI) tract. Many studies have shown that in HIV-infected humans and SIV-infected MACs, CD4+ T cells, particularly those expressing CCR5, are severely depleted from GI tract in the first weeks of infection (9, 64, 65). Due to the large surface area of the GI tract, this massive depletion of CD4+ T cells reflects loss of most of the CD4+ T cells within the body and associates with loss of intestinal epithelial cells, disruption of tight junctions and compromised integrity of the mucosal intestinal barrier.
It is commonly accepted that loss of mucosal integrity favors translocation of bacterial and fungal products – including peptidoglycan, lipoteichoic acid, LPS, flagellin, ribosomal DNA and unmethylated CpG-containing DNA from the intestinal lumen to the systemic circulation (66). Through the stimulation of several TLRs, these microbial products may activate and induce production of pro-inflammatory cytokines, such as TNF-α, IL-6, IL-1β, and type I interferons, in various immune cell types (23, 24, 38). While essential to the host in the setting of acute infection, these responses may significantly contribute to aberrant immune activation in chronic HIV infection. Overproduction of TNF, for example, can be particularly deleterious for the integrity of the epithelial barrier of the GI tract by inducing enterocyte apoptosis (67), and therapeutic interventions that reduced TNF levels in vivo resulted in improved integrity of the structural barrier of the GI tract in individuals with Crohn’s disease (68).
An association between HIV infection and high LPS levels has been described in the vast majority of studies in chronic HIV infection (69). Although it is widely accepted that during HIV infection the numerous insults to the mucosal immune system result in microbial translocation, the relative contribution of microbial translocation to immune activation is not completely understood and still a matter of intense discussion. In our opinion, data generated in studies of natural hosts for SIV are among the strongest evidence supporting the role of microbial translocation in maintaining immune activation. Indeed, SIV-infected SMs and AGMs, that avoid chronic immune activation, do not show increased level of LPS in plasma or in the colon (42, 70, 71). Thus, microbial translocation is specific for progressive HIV/SIV infection. In addition, systemic injections of LPS increased immune activation in SIV-infected AGMs, thus supporting the conclusion that maintenance of mucosal integrity and lack of microbial translocation is an important mechanism for the absence of chronic immune activation in natural hosts for SIV (72). In a recent study aimed at reducing microbial translocation, MAC were treated with rifaximine (a luminal antibiotic) and Sulfasalazine (an anti-inflammatory drug used in patients with inflammatory bowel disease). This treatment was effective in reducing plasma levels of LPS, and treated SIV-infected macaques showed significantly lower levels of immune activation and cardiovascular complications compared to control animals (73). Finally, in a recent study exogenous administration of Interleukin (IL)-21 significantly reduced plasma level of LPS and sCD14 in SIV-infected MAC. Although there were no differences in the activation or proliferation levels of circulating T cells, reduced LPS levels correlate with lower levels of several markers of systemic activation, including sTNFR II, Neopterine, D-Dimer (Paiardini M, unpublished data). However, it should be noted that not all currently generated evidence supports the working model in which microbial translocation is a key cause of chronic immune activation. For example, some studies in chronic HIV infection described immune activation in the absence of increased level of LPS (69). Despite these few contrasting findings, we believe that the bulk of evidence is consistent with the working model in which loss of CD4+ T cells, compromised mucosal integrity and microbial translocation are critical contributors to chronic immune activation in HIV infection, both in the natural history of infections as well as in the context of ART-treatment (as discussed in a different article in this issue of Immunological Reviews).
Loss of specific CD4+ T-cell subsets
CD4+ T cells, the main target of HIV and SIV, are a heterogeneous population of immune cells based on phenotype, function, and anatomic distribution. CD4+ T cells can be phenotypically classified in broad subsets of naïve, central memory, transitional memory and effector memory cells based on their differentiation status, and into Th1, Th2, Th17, Tfh, and regulatory T-cell subsets based on their function, cytokine, and transcriptional profile. This list is not exhaustive, with more subsets being better characterized (stem T cells, Th22, Th9) while we are writing. HIV/SIV infections are associated with major perturbations of the different CD4+ T-cell subsets. Interestingly, some CD4+ T-cell subsets are perturbed differently in natural and non-natural hosts for HIV/SIV, thus suggesting they may play an important role in the progressive or nonprogressive outcome of the infection, as well as in the establishment and/or maintenance of immune activation. A description of the main CD4+ T-cell subsets that are differentially regulated in natural and non-natural hosts is provided below.
Th17 cells
In both humans and MAC, the HIV/SIV-induced depletion of intestinal CD4+ T cells preferentially involves Th17 cells, a lineage of CD4+ T cells characterized by the production of IL-17 and IL-22 (66, 74-76). IL-17 and IL-22 function in vivo to promote recruitment of neutrophils to areas of bacterial infection, epithelial regeneration, production of tight junction proteins and antibacterial defensins. Therefore, Th17 cells are thought to be crucial for mucosal immunity, and their specific loss may render chronically HIV/SIV-infected subjects less capable of maintaining the physical and immunological integrity of the mucosal barrier. While the mechanisms underlying the preferential loss of Th17 cells remain unclear, the importance of this loss is highlighted by several pieces of evidence. First, Th17 cells are relatively preserved in the GI tract of chronically SIV-infected SMs and AGMs (66, 77), which, as noted above, avoid microbial translocation and chronic immune activation, as well as in HIC and long-term nonprogressors (78-80). Second, the severity of Th17 cell depletion correlates with microbial translocation, chronic immune activation, and disease progression in HIV/SIV-infected subjects (74, 76, 81, 82), as well as with effective CD4+ T-cell restoration in gut-associated lymphoid tissue of HIV-infected patients on HAART (83). Third, a recent study showed that the size of the Th17 cell compartment prior to infection limited viral replication in SIV-infected RMs (84). Finally, we recently found that in SIV-infected RMs, preservation of intestinal Th17 cells associates with reduced levels of intestinal T cell activation and plasma LPS (Paiardini, unpublished data), thus confirming directly in vivo a molecular link between loss of Th17 cells and microbial translocation in pathogenic SIV-infection. Although Th17 cells are the predominant source of IL-17 and IL-22, recent studies have demonstrated that these cytokines can be produced also by subsets of CD8+ T cells and innate lymphocytes (ILCs) (85). As described for Th17 cells, the frequencies of CD8+ T cells and ILCs that produce IL-17 and/or IL-22 are significantly lower within the GI tracts of chronically SIV-infected macaques (75, 86, 87). The fact that neither CD8+ T cells nor ILCs express CD4 on their surface suggests that the general loss of IL-17 and/or IL-22 producing cells within the GI tracts of SIV-infected macaques is likely due to complex mechanisms and not simply related to preferential infection by HIV/SIV.
Central memory CD4+ T cells
Central memory CD4+ T cells (TCM) express CD62L and CCR7, reside in the lymph nodes and other inductive lymphoid tissues, and show limited effector functions but strong proliferation in response to antigenic re-stimulation. CD4+ TCM are of particular importance for immune function since they are longer-lived, self-renewing cells that maintain CD4+ T-cell homeostasis by replenishing the pool of shorter-lived, non-self-renewing CD4+ TEM cells (88). CD4+ TCM represent the largest reservoir of infected CD4+ T cells in HIV-1 infection (89-91). In SIV-infected MAC, progressive depletion of CD4+ TCM is the key factor dictating the tempo of progression to AIDS, both in the natural history of infection and in the context of vaccination (92-94). In vivo comparative studies in nonhuman primates recently showed that the infection frequencies of CD4+ TCM cells of SMs are remarkably lower as compared to both CD4+ TEM of SMs and CD4+ TCM of MACs in both blood and lymph nodes (95, 96). Consistent with this relative preservation from infection, in SIV-infected SMs CD4+ TCM cells are significantly more stable than in SIV-infected MAC (Paiardini M, unpublished data). Remarkably, recent data suggest that a phenotype of CD4+ TCM protection similar to those that we described in SMs is also present in nonprogressive HIV-infection. Specifically, low levels of CD4+ TCM infection have been described in (i) long-term non-progressors with protective HLA alleles, (ii) earlytreated patients that show a prolonged control of viremia and preserve CD4+ T cells after ARTinterruption, and (iii) VNPs, a rare subset of HIV-infected patients that preserve CD4+ T-cell counts despite high levels of viral load, thus resembling the SM phenotype (97, 98, Klatt N, personal communication).
Infection and depletion of CD4+ TCM cells is hypothesized to contribute to the establishment of chronic immune activation more significantly than does infection of CD4+ TEM cells, and by at least two different processes (11). First, since CD4+ TCM cells are critical to maintaining CD4+ T-cell homeostasis, their depletion may trigger T-cell homeostatic proliferation. Second, with CD4+ TCM cells concentrated in LN, their higher levels of infection may translate to higher viral replication, and thus activation, in the main anatomic sites in which immune responses are initiated (Fig. 2). In our opinion, it is likely that higher infection of CD4+ TCM cells and higher levels of immune activation feed into each other in a vicious cycle whose establishment is central to the immune deficiency that follows HIV/SIV infections of humans and RMs.
Fig. 2. Model of events in lymph nodes that could be associated with loss of CD4+ T cells.
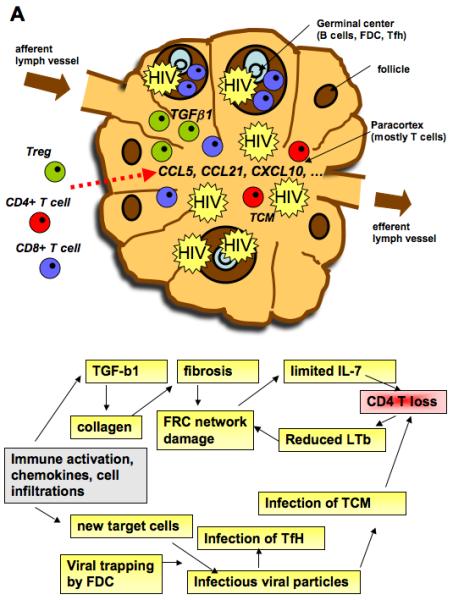
Immune activation is associated with a production of chemokines that allow immune cells to be held in secondary lymphoid organs. The number of T cells and B cells increases transiently and further contribute to secretion of chemokines. Morevoer, HIV infection is characterized by infiltrations of CD8+ T cells into germinal centers and accumulation of Treg in the T zone of lymph nodes. Treg associated TGFβ production induces collagen deposition and fibrosis. The latter causes damage of the FRC resulting in reduced access of naive CD4+ T cells to IL-7 and increased cell death by apoptosis. A consequence of CD4+ T-cell loss is a decreased production of LTb, which is necessary for the FRC network. The attraction of cells also leads to the arrival of new target cells for the virus that fuels the vicious cycle. Treg, regulatory T cell; GC, germinal center; FDC, follicular dendritic cell; Tfh, T-follicular helper cell, FRC, fibroblastic reticular cell
Regulatory T cells
An additional CD4+ T-cell subset whose function and/or homeostatic balance is perturbed by HIV/SIV infection is that of CD4+ regulatory T cells (Tregs). Classical Tregs, immunophenotypically defined as being CD25hiCD127low and by their intracellular expression of Forkhead Box P3 protein (FoxP3), are specialized in downmodulating the effector activity of other immune cell types via secretion of cytokines like IL-10 and TGF-β, as well as through a poorly-defined mechanism dependent on cell-to-cell contact. The importance of Tregs for maintaining immune homeostasis has been highlighted by signs of autoimmune pathology that occur when Treg activity is deficient. Tregs also facilitate early protective responses to local viral infection by allowing a timely entry of immune cells into infected tissue and diminishing pro-inflammatory chemokine levels in the LNs (99).
The data on the levels of Tregs during HIV-infection are conflicting, mainly due to the lack of Treg-specific markers (with different markers used in different studies), the multiplicity of distinct Treg subsets and to whether frequencies or absolute numbers were assessed. The majority of findings indicate that the relative frequency of CD25+ Tregs is increased in blood, while their absolute counts are reduced as a consequence of lower total CD4+ T-cell counts during progressive HIV-1 infection. In parallel, Foxp3+ Tregs are maintained or accumulate in the gut and lymph nodes according to some but not all studies (33, 100, 101). The ratio of Th17 to Treg cells is decreased in both blood and GI tract of HIV-infected individuals and correlates with increased plasma markers of microbial translocation and higher level of activated CD8+ T cells (102).
How Tregs impact HIV-associated chronic immune activation is still intensively debated. On one hand, Tregs seem to be ‘harmful’, since they are able to suppress HIV-specific immune responses (103-105). Studies of acute SIVmac infection have identified a distinct Treg subset (CD8+CD25+FoxP3+ T cells) that expands during the acute phase of infection and that also has a negative impact on the infection outcome (106). In this scenario, interventions aimed at decreasing Tregs numbers and functions may limit chronic immune activation. On the other hand, Tregs could be ‘beneficial’, since they might reduce the pro-inflammatory environment typical of HIV infection. In this case, interventions aimed at increasing their numbers and functions would limit chronic immune activation and thus ameliorate the course of HIV-disease. This latest hypothesis is supported by studies showing that (i) depletion of Treg numbers strongly correlate with both CD4+ and CD8+ T-cell activation in HIV-infected individuals; (ii) high levels of double negative Treg cells in acute HIV-1 infection are predictive of low T-cell activation at set-point, and (iii) the in vitro suppressive activity of blood and lymph node-derived Tregs correlates with high CD4+ T-cell counts and low levels of T-cell activation in SIV-infected cynomolgus macaques (107-110). Tregs, however, might be efficient in limiting residual immune activation in the context of controlled viral load but unable to suppress strong immune activation in viremic patients (111).
T-follicular helper cells
Tfh are a specialized subset of CD4+ T cells that reside within the germinal centers of secondary lymphoid tissues and play a critical role in the activation, differentiation, and survival of B cells. Expression of CXCR5, together with loss of the T-cell zone homing chemokine receptor CCR7, allows Tfh cells to relocate from the T-cell zone to the B-cell follicles, where they are positioned to directly support B cell expansion and differentiation. Moreover, Tfh cells express high levels of the surface receptors ICOS, CD40 ligand, PD-1, BTLA, as well as high levels of the cytokine IL-21 and the transcription factors Bcl-6 and c-Maf. Recent papers investigated the levels, function and susceptibility to infection of Tfh cells in HIV-infected humans and SIV-infected macaques (96, 112-115). These studies consistently showed that the frequency of Tfh cells within secondary lymphoid tissues is preserved or even increased in viremic HIV/SIV infected subjects. Expansion of Tfh cells associates with an increase in activated germinal center B cells and plasma cells (112, 113). Interestingly, expansion of Tfh cells does not result from reduced susceptibility to HIV/SIV infection, since Tfh cells were infected at frequencies similar to or higher than non-Tfh cells (96, 113, 115). Less is known on how HIV/SIV infection impacts the function of Tfh cells. One study showed that SIV infection altered the gene profile and decreased the survival, cycling, and trafficking of Tfh cells, thus suggesting a loss of function, while another study showed that Tfh cells remain capable of stimulating B cells’ Ig production (113, 115).
These findings suggest that Tfh cells are expanded and infected at high frequencies in chronic HIV/SIV infection and thus critically contribute to HIV replication and production. If and how the expansion of Tfh cells contributes to the maintenance of chronic immune activation still remains to be determined.
In summary, a large body of evidence generated in the last few years is consistent with a mechanism of HIV pathogenesis in which the changes in the balance of the different CD4+ T-cell subsets (Fig. 1) are more important than the total number of infected cells or the level of plasma viremia in causing the immune deficiency.
Type I interferons and pro-inflammatory mediators
Recognition of HIV products by pathogen-recognition receptors (PRRs) leads to the activation/maturation of innate immune cells and to the release of multiple inflammatory molecules, such as IFN-I. Recent findings generated in HIV-infected humans and SIV-infected non-human primates highlighted the potential role of type I interferon response as a contributor to HIV-associated chronic immune activation. Interferons, which include at least 14 different isoforms, induce the expression of hundreds of genes [interferon-stimulated genes (ISGs)], resulting in a number of clearly beneficial outcomes, including restriction of intracellular pathogens. However, in the context of chronic HIV/SIV infection, persistent upregulation of ISGs can have profound and possibly harmful effects, among others the chronic production of chemokines and the upregulation of IDO. Indeed, comparative studies of natural and non-natural hosts for SIV showed that the production of IFN-α and the expression of ISGs are remarkably lower during chronic SIV infection of SMs and AGMs than in MAC (35, 116, 117). Thus, chronic high expression of ISGs is a key distinguishing factor between pathogenic and nonpathogenic SIV infection. Interestingly, it also distinguishes long term non-progressors from progressors among HIV-1 infected individuals (15).
Based on these findings, several studies were recently designed to directly address in vivo the impact of type I interferons on viral load and immune activation. AIDS Clinical Trials Group (ACTG) study 5192 investigated the activity of 12 weekly administrations of IFNα to HIV-infected individuals (without HCV infection) not receiving ART (118). IFN-α was well tolerated, increased the expression of the activation markers CD38 and HLA-DR on CD8+ (but not CD4+) T cells and significantly reduced plasma viral load (118, 119). Of note, no signs of accelerated progression to AIDS have been observed in the thousands of HIV-infected individuals that were treated with IFN-α due to concomitant HCV infection. In two recent studies IFN-α was administered to SIV-infected natural hosts. When administered to chronically SIVinfected SMs, IFN-α induced a significant anti-viral effect (consistent with the data in human) but did not increase chronic immune activation (120). When administered to AGMs in the acute phase of infection, IFN-α did not lead to chronic ISG expression, nor impact the outcome of infection (Jacquelin B, personal communication). These latest findings are more consistent with a mechanism in which high IFN-α levels may be a consequence of high viral load and not sufficient alone to cause HIV-associated immune activation. However, further preclinical and clinical studies are warranted to better understand the cause-effect relationship between IFN-α and chronic immune activation in the context of HIV-infection.
Several processes have been suggested to contribute to the establishment and maintenance of HIV-associated immune activation. Their relative contribution may change in different subsets of HIV-infected patients and/or in different phases of infection. It will be necessary to evaluate the relevance of each alone and in combination as some of these processes might synergize in their promotion of immune activation.
Learn from the experts: how to maintain low chronic immune activation in the setting of HIV/SIV infections
One important approach to better identify the key mechanisms regulating chronic immune activation is to define the immunologic and genetic features distinguishing HIV/SIV-infected subjects who spontaneously limit chronic immune activation from those who fail to do so. HIV-infected humans who spontaneously limit chronic immune activation in the presence of viral replication (VNP) are extremely rare. In addition, most of the complex virus-host interactions take place in secondary lymphoid organs and mucosal tissues, and thus are difficult to study in humans. Finally, in humans, it is often impossible to study the early virus-host interactions. This is an important limitation, since studying these early interactions may allow for time sensitive identification of the factors that initiate and regulate T cell activation and also could provide key insights on what distinguishes harmful from desirable immune activation (that needed to induce antiviral responses) (121). Many of these limitations, including the possibility to study virushost interaction at early stages of infection and in multiple tissues, are overcome by using animal models of HIV infection.
The best animal model so far that fully recapitulates the physiopathology of HIV infection is that of monkeys infected with SIV. The first SIV was isolated from an Asian monkey (MAC). Their infection resulted from an accidental transmission of SIV from West-African monkeys (SMs). African NHPs represent the only reservoir of SIV in the wild. These lentiviruses are the ancestors of HIV-1 and HIV-2. The first species identified as natural carriers of SIVs were SMs, AGMs, mandrills and chimpanzees. In most cases, the SIV is specific for the primate species from which they have been isolated and they are designated as such (e.g., SIV of AGMs is named SIVagm; SIV of SMs is called SIVsm). The average SIV seroprevalence of AGMs, SMs and mandrills in the wild has been reported to be 40-50% (10). In this section, we mainly focus on SIV infection in AGMs and SMs, which have been studied the most with respect to correlates of protection against AIDS (11).
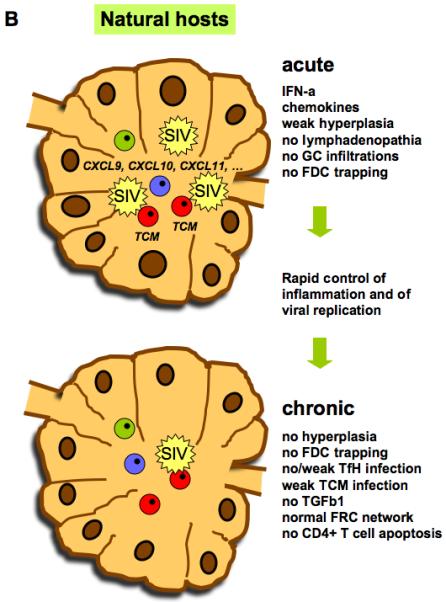
Natural history of SIV infection in natural hosts: early transient T-cell activation In SIV-infected natural hosts, the levels of plasma viremia are similar to those associated with disease progression in HIV/SIV-infected humans and macaques. In addition, similar to pathogenic HIV/SIV-infection in humans and macaques, SIV also replicates at high levels in the gut of natural hosts, both in the acute and the chronic phase of infection (42, 122, 142). A key feature of SIV infection in natural hosts is the absence of aberrant chronic immune activation, as manifested by many parameters. Chronically infected natural hosts display normal lymphocyte turnover and no increases of pro- or anti-inflammatory proteins in the plasma and tissues (123). Their lymph nodes show (i) a normal architecture; (ii) absence of marked lymphadenopathy or extensive follicular hyperplasia; (iii) no infiltration of CD8+ T cells into germinal centers; and (iv) a normal fibroblastic reticular cell (FRC) network (124) (Müller-Trutwin, unpublished data). Another distinguishing feature of non-progressive SIV infection is the maintenance of mucosal barrier integrity and the absence of microbial translocation (42, 70, 71). Therefore, fundamental clues regarding the role of immune activation in AIDS and the mechanisms that protect (or do not) against it lie in the natural hosts of SIV.
It is clear that the lack of chronic immune activation is not simply due to an ignorance of the virus by the host. For instance, natural hosts mount SIV-specific T and B-cell responses (125-127). Binding anti-SIV antibodies start to be detectable by two weeks post-infection, and seroconversion is completed within the same time frame as in MAC (124, 128). In line with the lack of efficient viral control, the levels of neutralizing antibody and SIV-specific T cells in blood do not exceed those in humans and macaques (129). Longitudinal studies in natural hosts clearly show an increase in T-cell activation around the time of viral peak, which then returns to baseline levels by the end of the acute infection and remains stable thereafter (123, 130). Thus, in sharp contrast to progressive HIV and SIV infections, SIV-infection of natural hosts is characterized by an early resolution of acute T cell activation.
Differences at the level of target cells
Recent studies have highlighted several differences in the nature of cells targeted by the virus in natural versus non-natural hosts, which could contribute to their better capacity to control immune activation. In both natural hosts and pathogenic HIV/SIV infections, the main cell type supporting virus replication is a population of short-lived, activated CD4+ T cells (131, 132). Interestingly, CD4+ T cells of natural hosts show lower frequencies of CCR5 expression (133, 134). However, SIVsmm and SIVagm, which largely use CCR5 as a co-receptor, still induce high viremia in the context of these low CCR5 levels (135, 136). Thus, it may be possible that SIV infects distinct target cells in natural hosts, or that the levels of CCR5 expression on cells may still be sufficient to allow infection, or that the SIVs use additional co-receptors in vivo. Indeed, SIVagm and SIVsm can, at least in vitro, also use the co-receptors Bonzo and Bob and more rarely CXCR4 (134-137). A recent report showed that SMs lacking a functional CCR5 on their cell surface still had robust plasma viral loads and was capable of using CXCR4 (134). The low level of CCR5 expression is more pronounced in TCM than other CD4+ T cells. We recently showed that SM CD4+ T cells fail to upregulate CCR5 after in vitro stimulation (95). CD4+ Tcell activation was similarly uncoupled from CCR5 expression during acute SIVsm infection and as a result of the homeostatic proliferation that follows antibody-mediated CD4+ T-cell depletion. Remarkably, SM CD4+ TCM cells showed reduced susceptibility to SIV infection both in vivo and in vitro when compared to CD4+ TCM cells of rhesus MAC. These data suggest that SM are protecting the ‘precious’ CD4+ TCM cell subset from direct virus infection. AGMs have also developed a mechanism to protect memory CD4+ T cells. Indeed, their CD4 molecules get downregulated from the surface of the CD4+ T cells as these cells get activated (138, 139). The functional properties usually associated with memory CD4+ T cells seem to be maintained and/or assumed by these CD4− T cells (140). This has also been reported in a subset of SMs in which a significant loss of CD4+ T cells is partially compensated by a population of double-negative T cells, which perform CD4+ T-cell function (141).
In addition to those for TCM, major differences in natural and non-natural hosts for HIV/SIV have been reported for three other CD4+ T-cell subsets. Specifically, natural hosts are characterized by a low infection rate of Tfh cells, a maintenance of intestinal Th17 cells and a conservation of intestinal IL-21 producing CD4+ T cells (66, 81, 96) (see ‘Proposed mechanisms inducing chronic immune activation’ section above).
While SIVs also have the potential to replicate in macrophages, such as those in the lung and brain, few studies have been performed on other target cells in natural hosts (142). The susceptibility of dendritic cells to SIV infection in natural hosts is not known, but it has been shown that these cells have the ability to efficiently attach and transmit the virus through DCSIGN (143).
These findings suggest that during their fight to cope with SIV infection, natural hosts have acquired mechanisms of protection for specific, and probably the most critical, CD4+ T-cell subsets such as CD4+ TCM, Tfh, and Th17 cells. Better preservation of these cells may be critical for the maintenance of CD4+ T-cell homeostasis, the lack of chronic immune activation, and the preservation of normal immune competence.
Low viral load in lymph nodes during the chronic phase of SIV infection in natural hosts Similar to progressive HIV/SIV infections in humans and macaques, SIV can be found in many tissues in the early and late infection of natural hosts (e.g. lymph nodes, intestine, thymus, cerebrospinal fluid, lung) (122, 142, 144). Additionally, the major site of viral replication in both natural and unnatural hosts is the gut (42, 122, 142).
While the viral load is similar in the blood and gut between natural and unnatural hosts, viral replication is significantly lower in the lymph nodes of the former (Fig. 2B). Thus, only a few productively infected cells can be detected in the lymph nodes during chronic SIVagm infection (145-147). The proviral load in AGM lymph nodes is also very low during the chronic phase (124, 144, 146). This contrasts with humans infected with HIV-1 and macaques infected with SIVmac, which both show a five to ten times more extensive proviral burden in the LNs than in blood cells (148). Recent studies reveal that this is also the case in SMs, thus pointing towards a common feature of natural hosts (96, 149). The mechanism responsible for the lower viral load in the LN of natural hosts is not known but likely to be multifactorial. The lack of viral trapping by follicular dendritic cells (FDC) contributes substantially to the lower total viral burden. Moreover, while Tfh cells are a major cellular target for the virus in pathogenic HIV/SIV infections, this is not the case in natural hosts (96). In addition, central memory CD4+ T cells, whose frequency is superior in LNs than in blood or mucosa, are less susceptible to SIV infection compared to RM (95). Finally, the lower viral load in lymph nodes may also be related to a more rapid immune clearance of virus from lymphoid tissue in natural hosts (149).
The lower viral load could be responsible for a lower immune activation in this tissue. However, one cannot exclude that the lower viral load in LNs is not the cause but instead the consequence of better control of immune activation in this tissue. Indeed, less activated T cells would deliver a lower number of targets cells to viral replication. In line with this, the expressions of CXCL9, CXCL10 (IP-10), and CXCL11, the ligands of CXCR3, are not upregulated in chronic infection in natural hosts, in contrast to macaques, in which their levels associate with disease progression (32, 116). CXCR3-ligands are responsible for the recruitment of circulating activated immune cells (such as Th1 cells). Although essential for effective host defense against infection, the sustained recruitment of activated cells can fuel inflammation and replication. Altogether, natural hosts have found a way to allow for preferential immune preservation of secondary lymphoid organs, i.e. those organs that have the most crucial role in the induction of immune responses.
Natural hosts are able to resolve inflammation
The study of early events following experimental SIV infection of SM and AGM have conclusively shown that the absence of chronic immune activation is not a failure of the innate immune system to respond to SIV infection but rather the result of a rapid downmodulation of inflammation. Indeed, natural hosts mount an innate immune response post-SIV-infection manifested by rapid and robust increases in pro-inflammatory cytokines, such as IFN-α, IP-10, and MCP-1, during the first two weeks that follow SIV infection (35, 116, 150, 151). In all cases, the elevation was only transient and there was no persistent increase of inflammatory cytokines during the chronic phase in natural hosts. The early but transient inflammation could also be measured at the level of gene expression. Many of the genes which were up-regulated during acute infection in natural hosts were associated with inflammation, such as the genes known to be inducible by IFN (ISGs). The gene expressions returned to normal levels by the end of the acute infection (116, 117 ). Similarly, LTNP and even viremic LTNP (VNP) display significantly lower levels of ISG expressions than HIV-infected individuals progressing to AIDS (15). Studies on IFN and ISGs in natural hosts contributed to the change in view on IFN. While a defect in IFN-I production had been considered to be associated with disease progression, studies in natural hosts emphasized the potential harm of chronic IFN-associated inflammation. The controversial results both in SIV and HIV infections encouraged the deeper studies of IFN-I regulation and function in HIV infection. The processes inducing the down-modulation of inflammation in natural hosts, which is lacking in pathogenic SIV infection, are currently a major field of investigation. Of note, some other pro-inflammatory cytokines, such as TNF-α, IL-6 and IL-8, are only weakly elevated in natural hosts (123, 152, 153). The lack of sustained inflammation in natural hosts might play a key role in the preservation of epithelial barrier integrity (Fig. 1).
The rapid control of inflammation in natural hosts had led us to raise the hypothesis that HIV-infected individuals with lower levels of inflammation by the end of acute HIV-1 infection would have a higher probability to become long term non progressors. We have quantified 28 plasma proteins in HIV-1 infected individuals with a known disease progression profile (31). Already during primary infection, rapid progressors showed a higher number of inflammatory cytokines than progressors or slow progressors. The plasma levels of TGF-β1 and IL-18 in acute HIV-1 infection were able to predict 74% of the T-cell activation variation at set-point. Plasma IP-10 was positively and negatively associated with, respectively, T cell activation and CD4+ Tcell counts at set-point and capable of predicting 30% of the CD4+ T-cell count variation. Moreover, plasma IP-10 levels during acute infection were predictive of rapid progression. This study showed for the first time that the inflammatory profile in acute HIV-1 infection is predictive of subsequent T-cell activation levels (31). It is urgent to identify the mechanism responsible for the resolution of inflammation in natural hosts and to understand how this impacts the maintenance of normal tissue structure and function.
Proposed mechanisms for the pathogenic role of HIV-associated chronic immune activation
Although the mechanisms inducing chronic immune activation are still not completely defined, the paradoxical hypothesis that aberrant immune activation is an essential contributor to the immunosuppressive state associated with pathogenic HIV/SIV infection is widely recognized and supported by a growing number of observations. When foreign antigens are recognized, the cells of the immune system proliferate and produce molecules that will eliminate the pathogen as well as the cells that harbor these pathogens. While crucial for host defense, this process can become very destructive to the host itself if protracted for long periods of time, as in the case of chronic infection with HIV and SIV. As for the mechanisms that may trigger immune activation, the hypotheses which suggest how chronic immune activation may cause damage to the immune system are multiple and may vary in different classes of patients.
One mechanism by which generalized immune activation may damage the immune system is by providing available targets for HIV replication. Indeed, activation, proliferation and differentiation of naïve and memory CD4+ T cells lead to increased CCR5 expression that renders these cells more susceptible to infection. The concomitant presence of high levels of CD4+ T-cell activation and a virus infecting and killing activated CD4+ T cells may help to sustain a vicious cycle, in which infection stimulates activation and activation stimulates infection, which may lead to a severe loss of CD4+ T cells. Due to the critical role of CD4+ T cells for the host immune response, the increased virus-mediated killing of activated CD4+ T cells may eventually translate to the inability to successfully control a wide range of potential pathogens. This phenomenon is even more pronounced for HIV-specific CD4+ T cells that are indeed particularly susceptible to HIV infection (154).
Furthermore, the depletion of CD4+ T cells may trigger a homeostatic response of the immune system that induces activation and proliferation of the remaining cells, particularly those with a naïve and central memory phenotype. While triggered in an attempt to replenish the depleted compartment, this homeostasis-driven CD4+ T-cell proliferation may paradoxically have harmful effects during pathogenic HIV/SIV infection by increasing the number of cells targeted by the virus. In addition, it is widely admitted that the chronic stimulation of these regenerative compartments that occurs during HIV infection translates into a premature aging of the immune system (155). Of note, the use of homeostatic cytokines, particularly interleukin-7 (IL-7), is very promising when HIV replication is inhibited, a context in which this cytokine may exert its beneficial effect (proliferation with reconstitution of the compartment) without providing more target to the virus (see ‘Targeting immune activation’ section).
An additional key mechanism by which HIV-associated chronic immune activation negatively impacts the immune system is via the destruction and/or dysregulation of the architecture of tissues that are crucial for T-cell homeostasis and function, such as the bone marrow, thymus, and lymph node (156, 157). Immune activation in HIV/SIV infections is associated with chronic TGF-β1 production in secondary lymphoid tissue leading to collagen deposition which damages the fibroblastic reticular cell (FRC) network (156) (Fig. 2A). Loss of the FRC network results in reduced availability of IL-7, impairing the survival of naive T cells. This leads to a decreased availability of CD4+ T-cell-produced lymphotoxin-β by the FRC and to a vicious cycle of further destruction of the FRC network. The FRC damage limits immune reconstitution after antiretroviral therapy in HIV infection (156).
Another way in which chronic immune activation can exert suppressive effects on the immune system is by direct inhibition of the normal functions of B cells, NK cells, dendritic cells, and monocytes, leading to less efficient viral control, more virus replication, and thus further immune activation (158, 159). Furthermore, the proliferation and activation of immune cells (i.e. monocyte/macrophages) together with increased levels and a prolonged presence of many proteins (IL-6, sCD14, C-reactive protein, D-dimer, TGF-β1 etc.) can induce damage in the vasculature, as well as lipid abnormalities, and result in cardiovascular diseases (160, 161). They also facilitate the proliferation of pre-malignant cells and malignant cells, thus predicting development of non-AIDS associated cancers. A mechanistic link between HIV-associated microbial translocation and cardiovascular disease has been recently proposed (161). LPS induces the expression of tissue factor on monocytes, which in turn activates the coagulation cascade. Consistent with this finding, an association between the levels of LPS and those of Ddimer has been described in HIV infection and may potentially contribute to the increased risk of cardiovascular disease (162, 163). Strong support for the link between microbial translocation, chronic immune activation and vascular disease was generated from a recent study in non-human primates. As observed in HIV-infected individuals, chronically SIV-infected pigtail macaques develop atherosclerosis and thrombotic disease, whose severity associates with increased levels of monocyte/macrophage activation and coagulation markers (164). Remarkably, vascular disease was not observed in SIV-infected natural hosts that lack microbial translocation, and administration of LPS to SIV-infected AGMs increased both monocyte/macrophage activation and D-dimer levels, providing strong direct evidence that chronic immune activation, and particularly microbial translocation, can cause cardiovascular disease in the setting of HIV/SIV infection (164).
Chronic immune activation established during pathogenic HIV and SIV infections in humans and RMs appears to undermine the integrity and functionality of the host immune system by utilizing at least three interrelated mechanisms: (i) generation of activated CCR5+ cells that are more susceptible to infection by HIV/SIV; (ii) disruption of secondary lymphoid tissue niches required for the long-term homeostasis of the naive and memory CD4+ T-cell compartments, which then severely undermines the capacity of the host to mount effective adaptive immune responses; and (iii) contributing to premature aging and non-AIDS related events. The surest way to prove that activation/inflammation is driving HIV morbidity and mortality is by blocking them and testing whether that is beneficial.
Targeting chronic immune activation: where are we, and where are we going?
We hope that we have convinced the reader that targeting chronic immune activation and thus reducing its negative impact on overall immune function is of the highest importance to optimize the treatment of HIV-infection in humans. Importantly, it is now clear that HIV-associated chronic immune activation is often not fully resolved even in the setting of long-term, successful HAART and that this residual immune activation may be largely responsible for the increased ‘non-AIDS’ morbidity and mortality observed in treated HIV-infected individuals (165). Based on this fact, a role for immune suppressive interventions in the clinical management of HIV-infected individuals is increasingly being considered. Believing on the paradox of treating an immune deficiency with immune suppressive drugs, from the early 1990s several investigators indeed started to test immunomodulatory compounds having the potential, at least theoretically, to reduce chronic immune activation during HIV infection (166). Overall, the vast majority of these original immunomodulatory interventions had limited and/or temporary effects on disease progression. The main reason for these limited effects, in our opinion, was that "generic" immune suppressive drugs were originally used, due to our incomplete understanding of the molecular mechanisms responsible for the HIV-associated chronic immune activation. In recent years, however, more specific immunomodulatory approaches have been designed and, although still at the early steps of investigation, more encouraging results have been generated. These immunomodulatory interventions are discussed in this section.
Chloroquine and Hydroxychloroquine
Chloroquine (CQ) and hydroxychloroquine (HCQ), two antimalarial drugs that have also been used to reduce inflammation in the treatment of systemic lupus erythematosus and rheumatoid arthritis, have recently captured the attention of HIV researchers. Interest for CQ in the setting of HIV-infection arose when two of its properties were better elucidated. First, CQ may interfere with the intracellular processing of antigens and cytokines and inhibits Toll-like receptor signaling, thus reducing T cell proliferation (167). Second, through modification of the gp120 glycosylation patterns, CQ stimulates the production of neutralizing antibodies and inhibits in vitro viral replication (168). Several recent studies investigated the ability of CQ/HCQ to reduce immune activation in HIV infection. In one study, CQ was administered for two months to nine HIV-infected patients not receiving antiretroviral therapy (169). CQ-treated patients showed reduced levels of activated (CD38+HLA-DR+) CD8+ T cells during the complete course of treatment, as well as decreased levels of proliferating (Ki-67+) T cells and of plasma LPS in the first month of treatment (169). In a second study, HCQ was administered to 20 HIV-infected immunologic non-responders for 6 months (170). This treatment resulted in the significant reduction of several markers of immune activation and inflammation, including plasma LPS, percentages of CD4+Ki-67+ T cells, IL-6, and TNF-α. These HCQ-induced effects on immune activation were stable (maintained up to two months after treatment interruption) and increased the percentages but not the absolute numbers of CD4+ T cells (170). These encouraging results were recently contrasted by a larger double-blind, placebo-controlled trial in which HIV-infected individuals not receiving ART were treatment for 48 weeks with HCQ. Not only did HCQ not reduce the levels of immune activation, but treatment also resulted in a greater decline of CD4+ T-cell counts and increased HIV replication (171). Thus, the effects of CQ/HCQ may be very different based on the time (early vs. chronic infection) and category of patients (not in ART vs. ART-treated) in which it is used, as well as based on dose and length of treatment. In our opinion, this is a crucial message that may apply to many of the proposed immune modulatory intervention.
Cyclosporine A
Cyclosporine A (CsA) is a cyclic undecapeptide that has been used as an immunosuppressive agent in transplant recipients as well as in the treatment of autoimmune disorders. CsA inhibits T-cell activation, proliferation, and effector functions by inhibiting nuclear factor of activated T cells (NFAT) and the transactivation of NFAT-dependent genes. As a result, CsA reduces the gene expression of proinflammatory cytokines such as IL-2, IL-4, TNF-α, and upregulates the expression of the immune-suppressive cytokine TGF-β (172). Encouraging results were generated in early studies of CsA treatment in HIV-infected individuals. In one of such study, the mean CD4+ T-cell counts remained remarkably stable while patients were on CsA monotherapy (median duration of CsA treatment was 11 months), while it significantly decreased after CsA interruption (173). In a subsequent study, CsA was administered in the first 8 weeks of ART in nine patients with primary HIV infection (174). Although no significant differences were found in viral replication between the CsA+ART and the ART alone cohorts, CsA treatment was associated with a persistent increase in the percentages and absolute numbers of CD4+ T cells within the first week of treatment (174). Further support for a potentially beneficial role of CsA in the treatment of HIV infection was provided in a review of 53 transplant patients who acquired HIV (either from an infected transplant or a blood transfusion) shortly after transplant (175). This study showed that the 5-year cumulative risk of developing AIDS was remarkably lower (31%) in patients receiving CsA as part of their immunosuppression regimen than in those not on CsA therapy (90%). On the basis of these promising results, ACTG conducted a randomized study (ACTG 5138) on treatment-naïve, chronic HIV-infected patients starting ART with or without CsA (176). Disappointingly, CsA did not provide any sustained immunologic benefit. Similarly, CsA did not change levels of proviral DNA or CD4+ T-cell count when used with ART in acute and early HIV infection (177). Thus, despite early promising results, the enthusiasm for CsA treatment of HIV infection has declined.
Mycophenolic acid
Mycopenolic acid (MPA) and its esther derivative mycophenolic mofetil (MMF) have been successfully used in the last two decades for prevention of acute allograft rejection. MPA inhibits T-cell proliferation by blocking the synthesis of guanosine nucleotides through a reversible inhibition of the enzyme inosine monophosphate dehydrogenase (IMPDH). As such, MPA antiproliferative effects are specific to lymphocytes, since these cells are dependent on the de novo synthesis of purine nucleotides and preferentially targets activated T cells, since the isoform (type II) of IMPDH expressed in these cells is more sensitive than the type I found in resting T cells. Significant enthusiasm was generated following the first pilot study in which MMF was administered daily for a period of 24 weeks in eight ART-treated HIV-infected individuals with undetectable viral loads. The addition of MMF resulted in a significant decrease in the fraction of proliferating (i.e. Ki-67+) CD4+ and CD8+ T cells and in reduced virus titer in CD4+ T cells isolated from these patients (178). Based on this result, additional clinical studies were designed. Unfortunately, the results of these studies, which used different MPA/MMF and ART regimens as well as HIV-infected patients at different stages of disease, have been variable and inconsistent. Thus, it remains unclear if MPA/MMF is of any benefit for HIV-infected individuals, particularly for those in which viral load is controlled by ART (179, 180).
Rapamycin
Rapamycin (Rapa) is a macrocyclic lactone antibiotic, currently approved to prevent transplant rejection and as an anti-tumor agent, that exerts its immunosuppressive function by blocking mTOR, an intracellular Ser/Thr kinase that play an important role in regulating T-cell activation and proliferation. By this mechanism, Rapa reduces T-cell proliferation, inhibits the expression of CCR5 and up-regulates that of CCR5 ligands, thus it is of potential therapeutic interest for the treatment of HIV infection (181, 182). Indeed, in vitro and animal studies confirmed the inhibitory effects of Rapa on HIV replication and the synergistic effects of Rapa on the antiviral activities of CCR5 antagonists (182-185). A first prospective, nonrandomized trial of HIV-infected patients receiving Rapa monotherapy after liver transplantation (LT) has been reported (186). While primary immunosuppression was based on calcineurin inhibitors (CI), 6 out of 14 HIV-infected patients switched to Rapa. Although the treatment did not impact CD4+ T-cell counts, significantly better control of HIV and HCV replication was found among liver transplant recipients treated with Rapa vs. those on CI (186). This first clinical report of significant benefits in long-term HIV-1 control generated some hope for the utility of Rapa as an additional drug in the arsenal for HIV infection. Of note, in a previous study in 7 ART-treated HIV-infected individuals with Kaposi’s sarcoma, Rapa treatment was safe and well tolerated, but did not significantly change viral loads or CD4+ T cell counts (187).
TNF inhibitors
As previously discussed, in HIV-infected individuals, TNF levels are abnormally high and may significantly contribute to induction and/or maintenance of chronic immune activation (24, 67). As such, it is not surprising that TNF inhibitors have been recently proposed as a therapy for HIV-infected individuals. While TNF inhibitors are widely used to treat numerous autoimmune diseases including rheumatoid arthritis, psoriatic arthritis, ankylosing spondylitis, and plaque psoriasis, few data are available on their effectiveness and safety in subjects with chronic HIV infection. In one study, eight HIV-1-positive patients with rheumatic diseases refractory to standard therapy were treated with different TNF inhibitors (etanercept, infliximab or adalimumab) (188). Although no significant differences were found for CD4+ T-cell counts and HIV viral loads in any of the patients, six of them experienced sustained clinical improvement in their rheumatic diseases. Of note, in a different study, infliximab did not cause any notable adverse effects in 4 years of follow-up when administered to HIV-infected individuals with high viral load and very severe CD4+ T-cell depletion (189). Two additional studies reported on etanercept use in two ART-treated HIV-infected patients with psoriasis and psoriatic arthritis (190, 191). In both patients, etanercept treatment was associated with an excellent clinical response for psoriasis, with no change in CD4+ T cell counts and the viral load remaining undetectable throughout treatment. Very recently, adalimumab, a human anti-TNF monoclonal antibody, was tested in a pilot study in the acute phase of SIV infection in RMs (41). While Adalimumab did not affect plasma SIV RNA or T-cell immune activation in peripheral blood or lymph nodes, it decreased infiltration of polymorphonuclear cells into the T-cell zone of lymphoid tissues, reduced lymphoid tissue fibrosis and improved preservation of CD4+ T cells (41). This preclinical study suggests that TNF inhibitors may favor the maintenance of lymphoid tissue structure necessary for CD4+ T-cell homeostasis and reconstitution, and thus may benefit HIV-infected individuals. Since lymph nodes were never assessed in the clinical studies of TNF inhibitors in HIV-infected individuals, this study perfectly illustrates the critical information which can be gained from preclinical studies in the nonhuman primate model for HIV. Because this study administered adalimumab in the acute phase of SIV infection, it will be very important to perform the same treatment during the chronic phase of infection in ART-treated SIV-infected RMs, thus in a setting very relevant for HIV-infected individuals, to better appreciate its clinical potential.
Cytokine administration and cytokine blockade
As previously described, progressive HIV infection is characterized by loss of CD4+ T cells from many anatomical locations, with a resulting increase in T-cell homeostatic proliferation of the remaining cells. It has been proposed that this proliferation may significantly contribute to the establishment and/or maintenance of chronic immune activation. Thus, it is not surprising that homeostatic cytokines are under extensive consideration in combination with ART for the treatment of HIV-infected individuals. Among these, significant interest has been generated by IL-7, a γ-chain cytokine that is critical in regulating the proliferation and survival of CD4+ and CD8+ T cells. Two main findings provided rationale for the use of IL-7 in the setting of HIV-infection. First, naïve and memory CD4+ T cells, the subsets more responsive to IL-7 due to their high expression of IL-7 receptor, are gradually depleted during progressive HIV infection; second, the IL-7/IL-7R signaling is dysfunctional in both CD4+ and CD8+ T cells from HIV-infected individuals (192). Exogenous administration of IL-7 has been recently tested in three clinical trials in ART-treated, HIV-infected individuals as a way to improve CD4+ T-cell levels and overall T-cell homeostasis. In all three studies, administration of IL-7 was well tolerated and resulted in a dose-dependent increase of CD4+ and CD8+ T cells, particularly those with a naive and central memory phenotype (193). IL-7 therapy has been tested in SIV-infected MAC to improve T-cell renewal as well as a means to overcome IFN-α-induced lymphopenia (194, 195). Although low-dose IFN-α induced strong lymphopenia, high-dose IFN-α treatment stimulated IL-7 production, leading to increased circulating T-cell counts. In addition, IL-7 therapy more than abrogated the lymphopenic effect of low-dose IFN-α, with the administration of both cytokines resulting in an expansion of naive T cells (195).
In summary, the ability of IL-7 to improve T-cell homeostasis may counteract the establishment of chronic immune activation by preserving a more intact immune system as well as by limiting the homeostasis-dependent T-cell proliferation. Finally, IL-7 treatment may also help in targeting viral reservoirs by promoting activation and reactivation of virus replication in latently infected CD4+ T cells with a transitional and central memory phenotype, which at present represent the best characterized cellular reservoir of HIV (90).
Another γ-chain cytokine that is catching the attention of HIV-investigators is IL-21, a pleiotropic cytokine mainly produced by activated CD4+ T cells, including Tfh, and NKT cells. IL-21 is a key factor in regulating central processes that are defective and/or compromised in HIV/SIV infected humans and RMs, including differentiation of Th17 cells, long-term maintenance of functional CD8+ T cells, differentiation of memory B cells and antibody-secreting plasma cells (196). Confirming the molecular link between availability of IL-21 and Th17 cell homeostasis, we recently described a significant association between the levels of IL-21-producing CD4+ T cells and those of Th17 cells in chronically SIV-infected RMs (81). The ability of IL-21 to stimulate differentiation and maintenance of functional Th17 cells is of particular importance in the context of HIV infection due to the contribution of Th17 cell loss to chronic immune activation. Based on these data, we hypothesized that IL-21 administration is beneficial during HIV/SIV infection, and recently tested this hypothesis in acutely SIV-infected rhMAC. IL-21-treatment was safe and did not increase plasma viral load or systemic immune activation. Consistent with the main functions of IL-21, IL-21 treated animals had higher expressions of perforin and granzyme B in total and SIV-specific CD8+ T cells and higher frequencies of intestinal Th17 cells as compared to untreated controls. Remarkably, increased levels of Th17 cells during early infection was associated with reduced levels of intestinal T cell proliferation and limited microbial translocation in the chronic infection (Paiardini M, unpublished observations). Thus, our results suggest that, by preservation of intestinal Th17 cells, IL-21 treatment may counteract HIV-induced chronic immune activation at mucosal sites. To better appreciate the potential of IL-21 in the clinical management of HIV infection, further studies are investigating the effects of IL-21 in ART-treated chronically SIV-infected RMs alone or in combination with other immune modulatory intervention such as a probiotics administration.
Probiotic supplementation
As discussed, compromised mucosal integrity with increased translocation of bacterial and fungal products is considered a key contributor to the persistent status of activation/inflammation both in the natural history of infection and during ART-treatment. Thus, supplementation of ART with therapeutic interventions aimed at decreasing GI tract inflammation and microbial translocation has been actively investigated in the past few years. In particular, several studies tested the hypothesis that, by decreasing activation/inflammation at mucosal level and competing with pathogenic bacteria, dietary supplementation of probiotic bacteria would be beneficial to HIV-infected individuals on ART. The first results were not very encouraging, with three studies in which supplementation of probotics to ART-treated HIV-infected individuals induced only a modest improvement in circulating CD4+ T-cell reconstitution (197, 198). However, in more recent studies combination of two or four different probiotics resulted in moderately decreased microbial translocation and immune activation in ART-treated HIV-infected individuals (199, 200). Of note, supplementation of different strains of Lactobacilli, Bifidobacteria, and Streptococcus reduced fibrosis and increased reconstitution of CD4+ T cells in the GI tract of ART-treated SIV-infected rhMAC (201). This very interesting study suggests that the beneficial effects of probiotics may be particularly prominent at the mucosal level and, as with TNF inhibitors, is another example on how informative and critical are pre-clinical studies in nonhuman primates, in particular when the main effects of an intervention are at anatomical locations difficult to be longitudinally accessed in humans.
Other therapeutic approaches involving administration or inhibition of cytokines, inhibition of fibrosis, and inhibition of cell trafficking that may be critical in modulating the extent of chronic immune activation are currently under active investigation in HIV-infected or SIV-infected subjects. These approaches, which include blockade of IFN-α, IFNα-receptor, IL-6 and statins, are not discussed in this review, since limited data are available at present.
Final remarks
Four critical messages arise from the enormous amount of evidence generated on the link between HIV-infection and chronic immune activation. First, effective inhibition of viral replication alone is not sufficient to restore a fully functional immune system and to prevent its premature aging. Effective immunomodulatory interventions are critical for the clinical management of HIV-infected individuals. Second, the complex nature of the mechanisms causing the establishment and maintenance of HIV-associated chronic immune activation has limited the impact of the interventions tested up to date. These interventions were too general, thus not specifically directed at the causes of immune activation, or too narrow, i.e. able to block only one of the main mechanisms driving immune activation or targeting a factor that is not an initial component of the activation chain. Third, a deeper understanding of the specific molecular, cellular, and pathophysiological mechanisms causing chronic T-cell immune activation and of the interaction between these mechanisms is crucial to targeting immune activation in HIV-infected individuals in a specific and effective way. Fourth, it is very likely that ART therapies will need to be complemented by multiple, combined immunomodulatory approaches, each specific for a particular cause of chronic immune activation. As such, studies of these combined immunomodulatory strategies should be prioritized in ART-treated SIV-infected nonhuman primates, thus allowing their quick translation to HIV-infected humans. Even if we will be unsuccessful in eradicating or substantially reducing HIV reservoirs, the design of novel therapeutic interventions that might improve the recovery of a competent immune system and reduce/eliminate activation/inflammation-induced cardiovascular disease, neurocognitive dysfunction, cancer, osteoporosis, among others, would be of major benefit to people living with HIV. A status of persistent activation/inflammation is also more and more observed in other diseases. Quoting Dr. Couzin-Frankel in the Science 2010 issue ‘Top inside of the decade’, ‘Over the past decade, it has become widely accepted that inflammation is a driving force behind chronic diseases that will kill nearly all of us’ (202). Thus, if achieved, the ability to specifically modulate chronic immune activation and inflammation will be a major advance for human health.
Table 2. Therapeutic interventions targeting HIV-associated chronic immune activation.
This table summarizesimmune-modulatory approacheswhich ability to modulate the extent of chronic immune activation has been investigated so far the most in HIV infected individuals. Detailed description of the studies and references are included in section 6 “Targeting chronic immune activation: Where are we and where are we going”.
| Intervention | Target | Rationale | Main result |
|---|---|---|---|
| Chloroquine Hydroxychloroquine |
Interfere with antigen processing; inhibit TLR signaling; inhibit viral replication |
Reduces innate responses | Tested in the presence and absence of ART - Discordant |
| Cyclosporin | Inhibits NFAT activity | Reduces the gene expression of proinflammatory cytokines |
Did not provide any sustained immunologic benefit when used with ART |
| Mycopenolic acid | Blocks the synthesis of guanosine nucleotides through a reversible inhibition of IMPDH. |
Reduces T cell proliferation; preferentially targets activated T cells |
Discordant |
| Rapamycin | Blocks mTor | Reduces T cell proliferation; inhibits CCR5 expression; up- regulates CCR5 ligands |
Few available data |
| TNF Inhibitors | TNF | TNF levels are abnormally high and contribute to chronic immune activation |
Few available data |
| IL-7 | IL-7R expressing cells | Contributes to regulate proliferation and survival of T cells; limits lymphopenia-induced T cell proliferation |
Dose-dependent increase of naive and central memory CD4 and CD8 T cells |
| Probiotic supplementation | Decreases activation/inflammation at mucosal level; competes with pathogenic bacteria |
Few available data; moderately decreased microbial translocation and immune activation during ART-treatment in recent studies |
TLR= Toll-like receptor; NFAT= Nuclear factor of activated T cells; IMPDH= inosine monophosphate dehydrogenase; mTor= mammalian target of rapamycin; TNF= Tumor Necrosis Factor; IL-7= Interleukin-7
Acknowledgments
We thank Drs. Steven Deeks, Steven Bosinger, and the Cleveland Immunopathogenesis Consortium (BBC/CLIC) for helpful discussions, as well as Colleen McGary and Emily Ryan for critical reading of the manuscript. This work was supported by NIH grants R01-AI084836 and R56-AI087186 (to MP), by P51OD011132 (to Yerkes National Primate Research Center), by the ANRS (to MM), the AREVA foundation (to MM) and Sidaction (to MM). The content of this publication is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Footnotes
There is no conflict of interest.
REFERENCES
- 1.Barré-Sinoussi F, et al. Isolation of a T-lymphotropic retrovirus from a patient at risk of acquired immunodeficiency syndrome. Science. 1983;220:868–871. doi: 10.1126/science.6189183. [DOI] [PubMed] [Google Scholar]
- 2.Ascher MS, Sheppard HW. AIDS as immune system activation: a model for pathogenesis. Clinical and experimental immunology. 1988;73:165–167. [PMC free article] [PubMed] [Google Scholar]
- 3.Fuchs D, Hausen A, Reibnegger G, Werner ER, Dierich MP, Wachter H. Neopterin as a marker for activated cell-mediated immunity: application in HIV infection. Immunology today. 1988;9:150–155. doi: 10.1016/0167-5699(88)91203-0. [DOI] [PubMed] [Google Scholar]
- 4.Sheppard HW, Ascher MS, McRae B, Anderson RE, Lang W, Allain JP. The initial immune response to HIV and immune system activation determine the outcome of HIV disease. Journal of acquired immune deficiency syndromes. 1991;4:704–712. [PubMed] [Google Scholar]
- 5.Giorgi JV, et al. The Multicenter AIDS Cohort Study Group CD8+ lymphocyte activation at human immunodeficiency virus type 1 seroconversion: development of HLA-DR+ CD38-CD8+ cells is associated with subsequent stable CD4+ cell levels. J Infect Dis. 1994;170:775–781. doi: 10.1093/infdis/170.4.775. [DOI] [PubMed] [Google Scholar]
- 6.Ho DD, Neumann AU, Perelson AS, Chen W, Leonard JM, Markowitz M. Rapid turnover of plasma virions and CD4 lymphocytes in HIV-1 infection. Nature. 1995;373:123–126. doi: 10.1038/373123a0. [DOI] [PubMed] [Google Scholar]
- 7.Wei X, et al. Viral dynamics in human immunodeficiency virus type 1 infection. Nature. 1995;373:117–122. doi: 10.1038/373117a0. [DOI] [PubMed] [Google Scholar]
- 8.Schneider T, Jahn HU, Schmidt W, Riecken EO, Zeitz M, Ullrich R, Berlin Diarrhea/Wasting Syndrome Study Group Loss of CD4 T lymphocytes in patients infected with human immunodeficiency virus type 1 is more pronounced in the duodenal mucosa than in the peripheral blood. Gut. 1995;37:524–529. doi: 10.1136/gut.37.4.524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Veazey RS, et al. Gastrointestinal tract as a major site of CD4+ T cell depletion and viral replication in SIV infection. Science. 1998;280:427–431. doi: 10.1126/science.280.5362.427. [DOI] [PubMed] [Google Scholar]
- 10.Liovat AS, Jacquelin B, Ploquin MJ, Barre-Sinoussi F, Muller-Trutwin MC. African non human primates infected by SIV - why don’t they get sick? Lessons from studies on the early phase of non-pathogenic SIV infection. Current HIV research. 2009;7:39–50. doi: 10.2174/157016209787048546. [DOI] [PubMed] [Google Scholar]
- 11.Chahroudi A, Bosinger SE, Vanderford TH, Paiardini M, Silvestri G. Natural SIV hosts: showing AIDS the door. Science. 2012;335:1188–1193. doi: 10.1126/science.1217550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Giorgi JV, et al. Shorter survival in advanced human immunodeficiency virus type 1 infection is more closely associated with T lymphocyte activation than with plasma virus burden or virus chemokine coreceptor usage. J Infect Dis. 1999;179:859–870. doi: 10.1086/314660. [DOI] [PubMed] [Google Scholar]
- 13.Sousa AE, Carneiro J, Meier-Schellersheim M, Grossman Z, Victorino RM. CD4 T cell depletion is linked directly to immune activation in the pathogenesis of HIV-1 and HIV-2 but only indirectly to the viral load. J Immunol. 2002;169:3400–3406. doi: 10.4049/jimmunol.169.6.3400. [DOI] [PubMed] [Google Scholar]
- 14.Choudhary SK, et al. Low immune activation despite high levels of pathogenic human immunodeficiency virus type 1 results in long-term asymptomatic disease. J Virol. 2007;81:8838–8842. doi: 10.1128/JVI.02663-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Rotger M, et al. Comparative transcriptomics of extreme phenotypes of human HIV-1 infection and SIV infection in sooty mangabey and rhesus macaque. The Journal of clinical investigation. 2011;121:2391–2400. doi: 10.1172/JCI45235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sauce D, et al. HIV disease progression despite suppression of viral replication is associated with exhaustion of lymphopoiesis. Blood. 2011;117:5142–5151. doi: 10.1182/blood-2011-01-331306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hunt PW, et al. Relationship between T cell activation and CD4+ T cell count in HIV-seropositive individuals with undetectable plasma HIV RNA levels in the absence of therapy. The Journal of infectious diseases. 2008;197:126–133. doi: 10.1086/524143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Giorgi JV, Detels R. T-cell subset alterations in HIV-infected homosexual men: NIAID Multicenter AIDS cohort study. Clinical immunology and immunopathology. 1989;52:10–18. doi: 10.1016/0090-1229(89)90188-8. [DOI] [PubMed] [Google Scholar]
- 19.Deeks SG, et al. Immune activation set point during early HIV infection predicts subsequent CD4+ T-cell changes independent of viral load. Blood. 2004;104:942–947. doi: 10.1182/blood-2003-09-3333. [DOI] [PubMed] [Google Scholar]
- 20.van Asten L, et al. Pre-seroconversion immune status predicts the rate of CD4 T cell decline following HIV infection. Aids. 2004;18:1885–1893. doi: 10.1097/00002030-200409240-00004. [DOI] [PubMed] [Google Scholar]
- 21.Doisne JM, et al. CD8+ T cells specific for EBV, cytomegalovirus, and influenza virus are activated during primary HIV infection. Journal of immunology. 2004;173:2410–2418. doi: 10.4049/jimmunol.173.4.2410. [DOI] [PubMed] [Google Scholar]
- 22.Aziz N, et al. Stability of plasma levels of cytokines and soluble activation markers in patients with human immunodeficiency virus infection. The Journal of infectious diseases. 1999;179:843–848. doi: 10.1086/314673. [DOI] [PubMed] [Google Scholar]
- 23.Birx DL, Redfield RR, Tencer K, Fowler A, Burke DS, Tosato G. Induction of interleukin-6 during human immunodeficiency virus infection. Blood. 1990;76:2303–2310. [PubMed] [Google Scholar]
- 24.Molina JM, Scadden DT, Byrn R, Dinarello CA, Groopman JE. Production of tumor necrosis factor alpha and interleukin 1 beta by monocytic cells infected with human immunodeficiency virus. The Journal of clinical investigation. 1989;84:733–737. doi: 10.1172/JCI114230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wiercinska-Drapalo A, Flisiak R, Jaroszewicz J, Prokopowicz D. Increased plasma transforming growth factor-beta1 is associated with disease progression in HIV-1-infected patients. Viral Immunol. 2004;17:109–113. doi: 10.1089/088282404322875502. [DOI] [PubMed] [Google Scholar]
- 26.Thieblemont N, Weiss L, Sadeghi HM, Estcourt C, Haeffner-Cavaillon N. CD14lowCD16high: a cytokine-producing monocyte subset which expands during human immunodeficiency virus infection. European journal of immunology. 1995;25:3418–3424. doi: 10.1002/eji.1830251232. [DOI] [PubMed] [Google Scholar]
- 27.Dutertre CA, et al. Pivotal role of M-DC8(+) monocytes from viremic HIV-infected patients in TNFalpha overproduction in response to microbial products. Blood. 2012;120:2259–2268. doi: 10.1182/blood-2012-03-418681. [DOI] [PubMed] [Google Scholar]
- 28.Hasegawa A, et al. The level of monocyte turnover predicts disease progression in the macaque model of AIDS. Blood. 2009;114:2917–2925. doi: 10.1182/blood-2009-02-204263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Thiebaut R, et al. Association of soluble CD14 and inflammatory biomarkers with HIV-2 disease progression. Clinical infectious diseases: an official publication of the Infectious Diseases Society of America. 2012;55:1417–1425. doi: 10.1093/cid/cis708. [DOI] [PubMed] [Google Scholar]
- 30.Stacey AR, et al. Induction of a striking systemic cytokine cascade prior to peak viremia in acute human immunodeficiency virus type 1 infection, in contrast to more modest and delayed responses in acute hepatitis B and C virus infections. Journal of virology. 2009;83:3719–3733. doi: 10.1128/JVI.01844-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Liovat AS, et al. Acute plasma biomarkers of T cell activation set-point levels and of disease progression in HIV-1 infection. PLoS ONE. 2012;7:e46143. doi: 10.1371/journal.pone.0046143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Durudas A, et al. Differential innate immune responses to low or high dose oral SIV challenge in Rhesus macaques. Current HIV research. 2011;9:276–288. doi: 10.2174/157016211797635928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Estes JD, et al. Premature induction of an immunosuppressive regulatory T cell response during acute simian immunodeficiency virus infection. J Infect Dis. 2006;193:703–712. doi: 10.1086/500368. [DOI] [PubMed] [Google Scholar]
- 34.Nascimbeni M, et al. Plasmacytoid dendritic cells accumulate in spleens from chronically HIV-infected patients but barely participate in interferon-alpha expression. Blood. 2009;113:6112–6119. doi: 10.1182/blood-2008-07-170803. [DOI] [PubMed] [Google Scholar]
- 35.Harris LD, et al. Downregulation of robust acute type I interferon responses distinguishes nonpathogenic simian immunodeficiency virus (SIV) infection of natural hosts from pathogenic SIV infection of rhesus macaques. Journal of virology. 2010;84:7886–7891. doi: 10.1128/JVI.02612-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Berlier W, et al. Seminal plasma promotes the attraction of Langerhans cells via the secretion of CCL20 by vaginal epithelial cells: involvement in the sexual transmission of HIV. Hum Reprod. 2006;21:1135–1142. doi: 10.1093/humrep/dei496. [DOI] [PubMed] [Google Scholar]
- 37.Li Q, et al. Glycerol monolaurate prevents mucosal SIV transmission. Nature. 2009;458:1034–1038. doi: 10.1038/nature07831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Emilie D, et al. Production of interleukins in human immunodeficiency virus-1-replicating lymph nodes. The Journal of clinical investigation. 1990;86:148–159. doi: 10.1172/JCI114678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Pantaleo G, Graziosi C, Fauci AS. The role of lymphoid organs in the pathogenesis of HIV infection. Seminars in Immunology. 1993;5:157–163. doi: 10.1006/smim.1993.1019. [DOI] [PubMed] [Google Scholar]
- 40.Racz P, et al. Lymphatic tissue changes in AIDS and other retrovirus infections: tools and insights. Lymphology. 1990;23:85–91. [PubMed] [Google Scholar]
- 41.Tabb B, et al. Reduced inflammation and lymphoid tissue immunopathology in rhesus macaques receiving anti-tumor necrosis factor treatment during primary simian immunodeficiency virus infection. The Journal of infectious diseases. 2013;207:880–892. doi: 10.1093/infdis/jis643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Pandrea IV, et al. Acute loss of intestinal CD4+ T cells is not predictive of simian immunodeficiency virus virulence. J Immunol. 2007;179:3035–3046. doi: 10.4049/jimmunol.179.5.3035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kwa S, et al. Plasmacytoid dendritic cells are recruited to the colorectum and contribute to immune activation during pathogenic SIV infection in rhesus macaques. Blood. 2011;118:2763–2773. doi: 10.1182/blood-2011-02-339515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Reeves RK, et al. SIV infection induces accumulation of plasmacytoid dendritic cells in the gut mucosa. The Journal of infectious diseases. 2012;206:1462–1468. doi: 10.1093/infdis/jis408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Gray F, et al. Encephalitis with infiltration by CD8+ Lymphocytes in HIV Patients receiving Combination Antiretroviral Treatment. Brain Pathol. 2013 doi: 10.1111/bpa.12038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Beignon AS, et al. Endocytosis of HIV-1 activates plasmacytoid dendritic cells via Toll-like receptor-viral RNA interactions. J Clin Invest. 2005;115:3265–3275. doi: 10.1172/JCI26032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Heil F, et al. Species-specific recognition of single-stranded RNA via toll-like receptor 7 and 8. Science. 2004;303:1526–1529. doi: 10.1126/science.1093620. [DOI] [PubMed] [Google Scholar]
- 48.Meier A, et al. MyD88-dependent immune activation mediated by human immunodeficiency virus type 1-encoded Toll-like receptor ligands. J Virol. 2007;81:8180–8191. doi: 10.1128/JVI.00421-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Vabret N, Bailly-Bechet M, Najburg V, Muller-Trutwin M, Verrier B, Tangy F. The biased nucleotide composition of HIV-1 triggers type I interferon response and correlates with subtype D increased pathogenicity. PLoS ONE. 2012;7:e33502. doi: 10.1371/journal.pone.0033502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Finkel TH, et al. Apoptosis occurs predominantly in bystander cells and not in productively infected cells of HIV- and SIV-infected lymph nodes. Nature medicine. 1995;1:129–134. doi: 10.1038/nm0295-129. [DOI] [PubMed] [Google Scholar]
- 51.Hellerstein MK, et al. Subpopulations of long-lived and short-lived T cells in advanced HIV-1 infection. The Journal of clinical investigation. 2003;112:956–966. doi: 10.1172/JCI17533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Burdo TH, et al. Soluble CD163 made by monocyte/macrophages is a novel marker of HIV activity in early and chronic infection prior to and after anti-retroviral therapy. The Journal of infectious diseases. 2011;204:154–163. doi: 10.1093/infdis/jir214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Brown KN, Trichel A, Barratt-Boyes SM. Parallel loss of myeloid and plasmacytoid dendritic cells from blood and lymphoid tissue in simian AIDS. J Immunol. 2007;178:6958–6967. doi: 10.4049/jimmunol.178.11.6958. [DOI] [PubMed] [Google Scholar]
- 54.Hunt PW, et al. T cell activation is associated with lower CD4+ T cell gains in human immunodeficiency virus-infected patients with sustained viral suppression during antiretroviral therapy. The Journal of infectious diseases. 2003;187:1534–1543. doi: 10.1086/374786. [DOI] [PubMed] [Google Scholar]
- 55.Rodriguez B, et al. Predictive value of plasma HIV RNA level on rate of CD4 T-cell decline in untreated HIV infection. JAMA: the journal of the American Medical Association. 2006;296:1498–1506. doi: 10.1001/jama.296.12.1498. [DOI] [PubMed] [Google Scholar]
- 56.Hazenberg MD, et al. Persistent immune activation in HIV-1 infection is associated with progression to AIDS. Aids. 2003;17:1881–1888. doi: 10.1097/00002030-200309050-00006. [DOI] [PubMed] [Google Scholar]
- 57.Jiang W, et al. Plasma levels of bacterial DNA correlate with immune activation and the magnitude of immune restoration in persons with antiretroviral-treated HIV infection. The Journal of infectious diseases. 2009;199:1177–1185. doi: 10.1086/597476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Schindler M, et al. Nef-mediated suppression of T cell activation was lost in a lentiviral lineage that gave rise to HIV-1. Cell. 2006;125:1055–1067. doi: 10.1016/j.cell.2006.04.033. [DOI] [PubMed] [Google Scholar]
- 59.Munch J, et al. Nef-mediated enhancement of virion infectivity and stimulation of viral replication are fundamental properties of primate lentiviruses. Journal of virology. 2007;81:13852–13864. doi: 10.1128/JVI.00904-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Sacre K, et al. A role for cytomegalovirus-specific CD4+CX3CR1+ T cells and cytomegalovirus-induced T-cell immunopathology in HIV-associated atherosclerosis. Aids. 2012;26:805–814. doi: 10.1097/QAD.0b013e328351f780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Parrinello CM, et al. Cytomegalovirus immunoglobulin G antibody is associated with subclinical carotid artery disease among HIV-infected women. The Journal of infectious diseases. 2012;205:1788–1796. doi: 10.1093/infdis/jis276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Wittkop L, et al. Effect of Cytomegalovirus-Induced Immune Response, Self Antigen-Induced Immune Response, and Microbial Translocation on Chronic Immune Activation in Successfully Treated HIV Type 1-Infected Patients: The ANRS CO3 Aquitaine Cohort. The Journal of infectious diseases. 2013;207:622–627. doi: 10.1093/infdis/jis732. [DOI] [PubMed] [Google Scholar]
- 63.Hunt PW, et al. Valganciclovir reduces T cell activation in HIV-infected individuals with incomplete CD4+ T cell recovery on antiretroviral therapy. The Journal of infectious diseases. 2011;203:1474–1483. doi: 10.1093/infdis/jir060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Brenchley JM, et al. CD4+ T cell depletion during all stages of HIV disease occurs predominantly in the gastrointestinal tract. The Journal of experimental medicine. 2004;200:749–759. doi: 10.1084/jem.20040874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Mattapallil JJ, Douek DC, Hill B, Nishimura Y, Martin M, Roederer M. Massive infection and loss of memory CD4+ T cells in multiple tissues during acute SIV infection. Nature. 2005;434:1093–1097. doi: 10.1038/nature03501. [DOI] [PubMed] [Google Scholar]
- 66.Brenchley JM, et al. Differential Th17 CD4 T-cell depletion in pathogenic and nonpathogenic lentiviral infections. Blood. 2008;112:2826–2835. doi: 10.1182/blood-2008-05-159301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Ruemmele FM, et al. Lipopolysaccharide modulation of normal enterocyte turnover by toll-like receptors is mediated by endogenously produced tumour necrosis factor alpha. Gut. 2002;51:842–848. doi: 10.1136/gut.51.6.842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Kaser A, Tilg H. Inflammatory bowel diseases: highlights from the United European Gastroenterology Week 2008. Expert Opin Ther Targets. 2009;13:259–263. doi: 10.1517/14728220802682325. [DOI] [PubMed] [Google Scholar]
- 69.Sandler NG, Douek DC. Microbial translocation in HIV infection: causes, consequences and treatment opportunities. Nat Rev Microbiol. 2012;10:655–666. doi: 10.1038/nrmicro2848. [DOI] [PubMed] [Google Scholar]
- 70.Brenchley JM, et al. Microbial translocation is a cause of systemic immune activation in chronic HIV infection. Nat Med. 2006;12:1365–1371. doi: 10.1038/nm1511. [DOI] [PubMed] [Google Scholar]
- 71.Estes JD, et al. Damaged intestinal epithelial integrity linked to microbial translocation in pathogenic simian immunodeficiency virus infections. PLoS pathogens. 2010;6:e1001052. doi: 10.1371/journal.ppat.1001052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Pandrea I, et al. Cutting edge: Experimentally induced immune activation in natural hosts of simian immunodeficiency virus induces significant increases in viral replication and CD4+ T cell depletion. Journal of immunology. 2008;181:6687–6691. doi: 10.4049/jimmunol.181.10.6687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Pandrea I. XIX International AIDS Conference; Washington. 2012. [Google Scholar]
- 74.Cecchinato V, et al. Altered balance between Th17 and Th1 cells at mucosal sites predicts AIDS progression in simian immunodeficiency virus-infected macaques. Mucosal Immunol. 2008;1:279–288. doi: 10.1038/mi.2008.14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Klatt NR, et al. Loss of mucosal CD103+ DCs and IL-17+ and IL-22+ lymphocytes is associated with mucosal damage in SIV infection. Mucosal Immunol. 2012;5:646–657. doi: 10.1038/mi.2012.38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Raffatellu M, et al. Simian immunodeficiency virus-induced mucosal interleukin-17 deficiency promotes Salmonella dissemination from the gut. Nat Med. 2008;14:421–428. doi: 10.1038/nm1743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Favre D, et al. Critical loss of the balance between Th17 and T regulatory cell populations in pathogenic SIV infection. PLoS Pathog. 2009;5:e1000295. doi: 10.1371/journal.ppat.1000295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Salgado M, Rallon NI, Rodes B, Lopez M, Soriano V, Benito JM. Long-term non-progressors display a greater number of Th17 cells than HIV-infected typical progressors. Clinical Immunology. 2011;139:110–114. doi: 10.1016/j.clim.2011.02.008. [DOI] [PubMed] [Google Scholar]
- 79.Brandt L, et al. Low level of regulatory T cells and maintenance of balance between regulatory T cells and TH17 cells in HIV-1-infected elite controllers. Journal of acquired immune deficiency syndromes. 2011;57:101–108. doi: 10.1097/QAI.0b013e318215a991. [DOI] [PubMed] [Google Scholar]
- 80.Ciccone EJ, et al. CD4+ T cells, including Th17 and cycling subsets, are intact in the gut mucosa of HIV-1-infected long-term nonprogressors. Journal of virology. 2011;85:5880–5888. doi: 10.1128/JVI.02643-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Micci L, et al. Paucity of IL-21-producing CD4(+) T cells is associated with Th17 cell depletion in SIV infection of rhesus macaques. Blood. 2012;120:3925–3935. doi: 10.1182/blood-2012-04-420240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Gordon SN, et al. Disruption of intestinal CD4+ T cell homeostasis is a key marker of systemic CD4+ T cell activation in HIV-infected individuals. Journal of immunology. 2010;185:5169–5179. doi: 10.4049/jimmunol.1001801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Macal M, et al. Effective CD4+ T-cell restoration in gut-associated lymphoid tissue of HIV-infected patients is associated with enhanced Th17 cells and polyfunctional HIV-specific T-cell responses. Mucosal Immunol. 2008;1:475–488. doi: 10.1038/mi.2008.35. [DOI] [PubMed] [Google Scholar]
- 84.Hartigan-O’Connor DJ, Abel K, Van Rompay KK, Kanwar B, McCune JM. SIV replication in the infected rhesus macaque is limited by the size of the preexisting TH17 cell compartment. Sci Transl Med. 2012;4:136ra169. doi: 10.1126/scitranslmed.3003941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Campillo-Gimenez L, et al. AIDS progression is associated with the emergence of IL-17-producing cells early after simian immunodeficiency virus infection. Journal of immunology. 2010;184:984–992. doi: 10.4049/jimmunol.0902316. [DOI] [PubMed] [Google Scholar]
- 86.Reeves RK, et al. Gut inflammation and indoleamine deoxygenase inhibit IL-17 production and promote cytotoxic potential in NKp44+ mucosal NK cells during SIV infection. Blood. 2011;118:3321–3330. doi: 10.1182/blood-2011-04-347260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Xu H, Wang X, Liu DX, Moroney-Rasmussen T, Lackner AA, Veazey RS. IL-17-producing innate lymphoid cells are restricted to mucosal tissues and are depleted in SIV-infected macaques. Mucosal Immunol. 2012;5:658–669. doi: 10.1038/mi.2012.39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Brenchley JM, Silvestri G, Douek DC. Nonprogressive and progressive primate immunodeficiency lentivirus infections. Immunity. 2010;32:737–742. doi: 10.1016/j.immuni.2010.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Brenchley JM, et al. T-cell subsets that harbor human immunodeficiency virus (HIV) in vivo: implications for HIV pathogenesis. Journal of virology. 2004;78:1160–1168. doi: 10.1128/JVI.78.3.1160-1168.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Chomont N, et al. HIV reservoir size and persistence are driven by T cell survival and homeostatic proliferation. Nature medicine. 2009;15:893–900. doi: 10.1038/nm.1972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Pfaff JM, et al. HIV-1 resistance to CCR5 antagonists associated with highly efficient use of CCR5 and altered tropism on primary CD4+ T cells. Journal of virology. 2010;84:6505–6514. doi: 10.1128/JVI.00374-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Okoye A, et al. Progressive CD4+ central memory T cell decline results in CD4+ effector memory insufficiency and overt disease in chronic SIV infection. The Journal of experimental medicine. 2007;204:2171–2185. doi: 10.1084/jem.20070567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Mattapallil JJ, et al. Vaccination preserves CD4 memory T cells during acute simian immunodeficiency virus challenge. The Journal of experimental medicine. 2006;203:1533–1541. doi: 10.1084/jem.20060657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Letvin NL, et al. Preserved CD4+ central memory T cells and survival in vaccinated SIV-challenged monkeys. Science. 2006;312:1530–1533. doi: 10.1126/science.1124226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Paiardini M, et al. Low levels of SIV infection in sooty mangabey central memory CD(4)(+) T cells are associated with limited CCR5 expression. Nature medicine. 2011;17:830–836. doi: 10.1038/nm.2395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Brenchley JM, et al. Differential infection patterns of CD4+ T cells and lymphoid tissue viral burden distinguish progressive and nonprogressive lentiviral infections. Blood. 2012;120:4172–4181. doi: 10.1182/blood-2012-06-437608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Descours B, et al. Immune responses driven by protective human leukocyte antigen alleles from long-term nonprogressors are associated with low HIV reservoir in central memory CD4 T cells. Clinical infectious diseases. 2012;54:1495–1503. doi: 10.1093/cid/cis188. [DOI] [PubMed] [Google Scholar]
- 98.Bacchus C. International AIDS Society’s Pre-Conference Symposium “Towards and HIV Cure”; Washington, DC. 2012. [Google Scholar]
- 99.Lund JM, Hsing L, Pham TT, Rudensky AY. Coordination of early protective immunity to viral infection by regulatory T cells. Science. 2008;320:1220–1224. doi: 10.1126/science.1155209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Epple HJ, et al. Mucosal but not peripheral FOXP3+ regulatory T cells are highly increased in untreated HIV infection and normalize after suppressive HAART. Blood. 2006;108:3072–3078. doi: 10.1182/blood-2006-04-016923. [DOI] [PubMed] [Google Scholar]
- 101.Chase AJ, et al. Severe depletion of CD4+ CD25+ regulatory T cells from the intestinal lamina propria but not peripheral blood or lymph nodes during acute simian immunodeficiency virus infection. Journal of virology. 2007;81:12748–12757. doi: 10.1128/JVI.00841-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Favre D, et al. Tryptophan catabolism by indoleamine 2,3-dioxygenase 1 alters the balance of TH17 to regulatory T cells in HIV disease. Sci Transl Med. 2010;2:32ra36. doi: 10.1126/scitranslmed.3000632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Kinter AL, et al. CD25(+)CD4(+) regulatory T cells from the peripheral blood of asymptomatic HIV-infected individuals regulate CD4(+) and CD8(+) HIV-specific T cell immune responses in vitro and are associated with favorable clinical markers of disease status. J Exp Med. 2004;200:331–343. doi: 10.1084/jem.20032069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Aandahl EM, Michaelsson J, Moretto WJ, Hecht FM, Nixon DF. Human CD4+ CD25+ regulatory T cells control T-cell responses to human immunodeficiency virus and cytomegalovirus antigens. J Virol. 2004;78:2454–2459. doi: 10.1128/JVI.78.5.2454-2459.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Weiss L, Donkova-Petrini V, Caccavelli L, Balbo M, Carbonneil C, Levy Y. Human immunodeficiency virus-driven expansion of CD4+CD25+ Regulatory T cells Which Suppress HIV-specific CD4 T-cell Responses in HIV-infected Patients. Blood. 2004;104(10):3249–3256. doi: 10.1182/blood-2004-01-0365. [DOI] [PubMed] [Google Scholar]
- 106.Karlsson I, et al. FoxP3+ CD25+ CD8+ T-cell induction during primary simian immunodeficiency virus infection in cynomolgus macaques correlates with low CD4+ T-cell activation and high viral load. J Virol. 2007;81:13444–13455. doi: 10.1128/JVI.01466-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Eggena MP, et al. Depletion of regulatory T cells in HIV infection is associated with immune activation. J Immunol. 2005;174:4407–4414. doi: 10.4049/jimmunol.174.7.4407. [DOI] [PubMed] [Google Scholar]
- 108.Petitjean G, et al. Level of double negative T cells, which produce TGF-beta and IL-10, predicts CD8 T-cell activation in primary HIV-1 infection. Aids. 2012;26:139–148. [Google Scholar]
- 109.Chase AJ, Yang HC, Zhang H, Blankson JN, Siliciano RF. Preservation of FoxP3+ regulatory T cells in the peripheral blood of human immunodeficiency virus type 1-infected elite suppressors correlates with low CD4+ T-cell activation. Journal of virology. 2008;82:8307–8315. doi: 10.1128/JVI.00520-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Karlsson I, et al. Suppressive activity of regulatory T cells correlates with high CD4(+) T-cell counts and low T-cell activation during chronic simian immunodeficiency virus infection. Aids. 2011;25:585–593. doi: 10.1097/QAD.0b013e3283437c7b. [DOI] [PubMed] [Google Scholar]
- 111.Weiss L, et al. Relationship between regulatory T cells and immune activation in human immunodeficiency virus-infected patients interrupting antiretroviral therapy. PLoS ONE. 2010;5:e11659. doi: 10.1371/journal.pone.0011659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Hong JJ, Amancha PK, Rogers K, Ansari AA, Villinger F. Spatial alterations between CD4(+) T follicular helper, B, and CD8(+) T cells during simian immunodeficiency virus infection: T/B cell homeostasis, activation, and potential mechanism for viral escape. Journal of immunology. 2012;188:3247–3256. doi: 10.4049/jimmunol.1103138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Petrovas C, et al. CD4 T follicular helper cell dynamics during SIV infection. The Journal of clinical investigation. 2012;122:3281–3294. doi: 10.1172/JCI63039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Lindqvist M, et al. Expansion of HIV-specific T follicular helper cells in chronic HIV infection. The Journal of clinical investigation. 2012;122:3271–3280. doi: 10.1172/JCI64314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Perreau M, et al. Follicular helper T cells serve as the major CD4 T cell compartment for HIV-1 infection, replication, and production. The Journal of experimental medicine. 2013;210:143–156. doi: 10.1084/jem.20121932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Jacquelin B, et al. Nonpathogenic SIV infection of African green monkeys induces a strong but rapidly controlled type I IFN response. The Journal of clinical investigation. 2009;119:3544–3555. doi: 10.1172/JCI40093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Bosinger SE, et al. Global genomic analysis reveals rapid control of a robust innate response in SIV-infected sooty mangabeys. The Journal of clinical investigation. 2009;119:3556–3572. doi: 10.1172/JCI40115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Asmuth DM, et al. Safety, tolerability, and mechanisms of antiretroviral activity of pegylated interferon Alfa-2a in HIV-1-monoinfected participants: a phase II clinical trial. The Journal of infectious diseases. 2010;201:1686–1696. doi: 10.1086/652420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Manion M, et al. Interferon-alpha administration enhances CD8+ T cell activation in HIV infection. PLoS ONE. 2012;7:e30306. doi: 10.1371/journal.pone.0030306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Vanderford TH, et al. Treatment of SIV-infected sooty mangabeys with a type-I IFN agonist results in decreased virus replication without inducing hyperimmune activation. Blood. 2012;119:5750–5757. doi: 10.1182/blood-2012-02-411496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Ploquin MJ, Jacquelin B, Jochems SP, Barre-Sinoussi F, Muller-Trutwin MC. Innate immunity in the control of HIV/AIDS: recent advances and open questions. Aids. 2012;26:1269–1279. doi: 10.1097/QAD.0b013e328353e46b. [DOI] [PubMed] [Google Scholar]
- 122.Gueye A, et al. Viral load in tissues during the early and chronic phase of non-pathogenic SIVagm infection. J Med Primatol. 2004;33:83–97. doi: 10.1111/j.1600-0684.2004.00057.x. [DOI] [PubMed] [Google Scholar]
- 123.Kornfeld C, et al. Antiinflammatory profiles during primary SIV infection in African green monkeys are associated with protection against AIDS. J Clin Invest. 2005;115:1082–1091. doi: 10.1172/JCI23006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Diop OM, et al. High levels of viral replication during primary simian immunodeficiency virus SIVagm infection are rapidly and strongly controlled in African green monkeys. J Virol. 2000;74:7538–7547. doi: 10.1128/jvi.74.16.7538-7547.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Zahn RC, et al. Simian Immunodeficiency Virus (SIV)-Specific CD8+ T-Cell Responses in Vervet African Green Monkeys Chronically Infected with SIVagm. J Virol. 2008;82:11577–11588. doi: 10.1128/JVI.01779-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Wang Z, Metcalf B, Ribeiro RM, McClure H, Kaur A. Th-1-type cytotoxic CD8+ T-lymphocyte responses to simian immunodeficiency virus (SIV) are a consistent feature of natural SIV infection in sooty mangabeys. J Virol. 2006;80:2771–2783. doi: 10.1128/JVI.80.6.2771-2783.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Lozano Reina JM, et al. Gag p27-specific B- and T-cell responses in Simian immunodeficiency virus SIVagm-infected African green monkeys. Journal of Virology. 2009;83:2770–2777. doi: 10.1128/JVI.01841-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Onanga R, et al. High levels of viral replication contrast with only transient changes in CD4+ and CD8+ cell numbers during the early phase of experimental infection with SIVmnd-1 in Mandrillus sphinx. J Virol. 2002;76:10256–10263. doi: 10.1128/JVI.76.20.10256-10263.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Li B, et al. Nonpathogenic simian immunodeficiency virus infection of sooty mangabeys is not associated with high levels of autologous neutralizing antibodies. Journal of virology. 2010;84:6248–6253. doi: 10.1128/JVI.00295-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Silvestri G, et al. Divergent Host Responses during Primary Simian Immunodeficiency Virus SIVsm Infection of Natural Sooty Mangabey and Nonnatural Rhesus Macaque Hosts. J Virol. 2005;79:4043–4054. doi: 10.1128/JVI.79.7.4043-4054.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Klatt NR, et al. Availability of activated CD4+ T cells dictates the level of viremia in naturally SIV-infected sooty mangabeys. J Clin Invest. 2008;118:2039–2049. doi: 10.1172/JCI33814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Gordon SN, et al. Short-lived infected cells support virus replication in sooty mangabeys naturally infected with simian immunodeficiency virus: implications for AIDS pathogenesis. Journal of virology. 2008;82:3725–3735. doi: 10.1128/JVI.02408-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Pandrea I, et al. Paucity of CD4+CCR5+ T cells is a typical feature of natural SIV hosts. Blood. 2007;109:1069–1076. doi: 10.1182/blood-2006-05-024364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Riddick NE, et al. A novel CCR5 mutation common in sooty mangabeys reveals SIVsmm infection of CCR5-null natural hosts and efficient alternative coreceptor use in vivo. PLoS pathogens. 2010;6:e1001064. doi: 10.1371/journal.ppat.1001064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Kuhmann SE, et al. Frequent substitution polymorphisms in African green monkey CCR5 cluster at critical sites for infections by simian immunodeficiency virus SIVagm, implying ancient virus-host coevolution. J Virol. 2001;75:8449–8460. doi: 10.1128/JVI.75.18.8449-8460.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Chen Z, et al. Natural infection of a homozygous delta24 CCR5 red-capped mangabey with an R2b-tropic simian immunodeficiency virus. The Journal of experimental medicine. 1998;188:2057–2065. doi: 10.1084/jem.188.11.2057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Elliott ST, et al. Cloning and analysis of sooty mangabey alternative coreceptors that support simian immunodeficiency virus SIVsmm entry independently of CCR5. Journal of virology. 2012;86:898–908. doi: 10.1128/JVI.06415-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Beaumier CM, et al. CD4 downregulation by memory CD4+ T cells in vivo renders African green monkeys resistant to progressive SIVagm infection. Nature medicine. 2009;15:879–885. doi: 10.1038/nm.1970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Murayama Y, et al. CD4 and CD8 expressions in African green monkey helper T lymphocytes: implication for resistance to SIV infection. Int Immunol. 1997;9:843–851. doi: 10.1093/intimm/9.6.843. [DOI] [PubMed] [Google Scholar]
- 140.Murayama Y, Mukai R, Inoue-Murayama M, Yoshikawa Y. An African green monkey lacking peripheral CD4 lymphocytes that retains helper T cell activity and coexists with SIVagm. Clin Exp Immunol. 1999;117:504–512. doi: 10.1046/j.1365-2249.1999.00999.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Milush JM, et al. Lack of clinical AIDS in SIV-infected sooty mangabeys with significant CD4+ T cell loss is associated with double-negative T cells. The Journal of clinical investigation. 2011;121:1102–1110. doi: 10.1172/JCI44876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Goldstein S, et al. Wide range of viral load in healthy african green monkeys naturally infected with simian immunodeficiency virus. J Virol. 2000;74:11744–11753. doi: 10.1128/jvi.74.24.11744-11753.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Ploquin MJ, et al. DC-SIGN from African green monkeys is expressed in lymph nodes and mediates infection in trans of simian immunodeficiency virus SIVagm. J Virol. 2004;78:798–810. doi: 10.1128/JVI.78.2.798-810.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Broussard SR, Staprans SI, White R, Whitehead EM, Feinberg MB, Allan JS. Simian immunodeficiency virus replicates to high levels in naturally infected African green monkeys without inducing immunologic or neurologic disease. J Virol. 2001;75:2262–2275. doi: 10.1128/JVI.75.5.2262-2275.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Goldstein S, et al. Plateau levels of viremia correlate with the degree of CD4+-T-cell loss in simian immunodeficiency virus SIVagm-infected pigtailed macaques: variable pathogenicity of natural SIVagm isolates. J Virol. 2005;79:5153–5162. doi: 10.1128/JVI.79.8.5153-5162.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Beer B, Scherer J, Megede JZ, Norley S, Baier M, Kurth R. Lack of Dichotomy between Virus Load of Peripheral Blood and Lymph Nodes during Long-Term Simian Immunodeficiency Virus Infection of African Green Monkeys. Virology. 1996;219:367. doi: 10.1006/viro.1996.0262. [DOI] [PubMed] [Google Scholar]
- 147.Cumont MC, et al. Early Divergence in Lymphoid Tissue Apoptosis between Pathogenic and Nonpathogenic Simian Immunodeficiency Virus Infections of Nonhuman Primates. J Virol. 2008;82:1175–1184. doi: 10.1128/JVI.00450-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Pantaleo G, et al. HIV infection is active and progressive in lymphoid tissue during the clinically latent stage of disease. Nature. 1993;362:355–358. doi: 10.1038/362355a0. [DOI] [PubMed] [Google Scholar]
- 149.Meythaler M, et al. Early induction of polyfunctional simian immunodeficiency virus (SIV)-specific T lymphocytes and rapid disappearance of SIV from lymph nodes of sooty mangabeys during primary infection. Journal of immunology. 2011;186:5151–5161. doi: 10.4049/jimmunol.1004110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Diop OM, et al. Plasmacytoid dendritic cell dynamics and alpha interferon production during Simian immunodeficiency virus infection with a nonpathogenic outcome. J Virol. 2008;82:5145–5152. doi: 10.1128/JVI.02433-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Meythaler M, et al. Differential CD4+ T-Lymphocyte Apoptosis and Bystander T-Cell Activation in Rhesus Macaques and Sooty Mangabeys during Acute Simian Immunodeficiency Virus Infection. J Virol. 2009;83:572–583. doi: 10.1128/JVI.01715-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Ploquin MJ, et al. Distinct expression profiles of TGF-beta1 signaling mediators in pathogenic SIVmac and non-pathogenic SIVagm infections. Retrovirology. 2006;3:37. doi: 10.1186/1742-4690-3-37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Campillo-Gimenez L, et al. Nonpathogenesis of simian immunodeficiency virus infection is associated with reduced inflammation and recruitment of plasmacytoid dendritic cells to lymph nodes, not to lack of an interferon type I response, during the acute phase. Journal of virology. 2010;84:1838–1846. doi: 10.1128/JVI.01496-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Douek DC, et al. HIV preferentially infects HIV-specific CD4+ T cells. Nature. 2002;417:95–98. doi: 10.1038/417095a. [DOI] [PubMed] [Google Scholar]
- 155.Papagno L, et al. Immune activation and CD8+ T-cell differentiation towards senescence in HIV-1 infection. PLoS biology. 2004;2:E20. doi: 10.1371/journal.pbio.0020020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Zeng M, Haase AT, Schacker TW. Lymphoid tissue structure and HIV-1 infection: life or death for T cells. Trends in immunology. 2012;33:306–314. doi: 10.1016/j.it.2012.04.002. [DOI] [PubMed] [Google Scholar]
- 157.Dion ML, et al. Slow disease progression and robust therapy-mediated CD4+ T-cell recovery are associated with efficient thymopoiesis during HIV-1 infection. Blood. 2007;109:2912–2920. doi: 10.1182/blood-2006-09-047308. [DOI] [PubMed] [Google Scholar]
- 158.Moir S, Fauci AS. B cells in HIV infection and disease. Nat Rev Immunol. 2009;9:235–245. doi: 10.1038/nri2524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Muller-Trutwin M, Hosmalin A. Role for plasmacytoid dendritic cells in anti-HIV innate immunity. Immunology and Cell Biology. 2005;83:578–585. doi: 10.1111/j.1440-1711.2005.01394.x. [DOI] [PubMed] [Google Scholar]
- 160.Kuller LH, et al. Inflammatory and coagulation biomarkers and mortality in patients with HIV infection. PLoS Med. 2008;5:e203. doi: 10.1371/journal.pmed.0050203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Hsue PY, Deeks SG, Hunt PW. Immunologic basis of cardiovascular disease in HIV-infected adults. The Journal of infectious diseases. 2012;205(Suppl 3):S375–382. doi: 10.1093/infdis/jis200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Sandler NG, et al. Plasma levels of soluble CD14 independently predict mortality in HIV infection. The Journal of infectious diseases. 2011;203:780–790. doi: 10.1093/infdis/jiq118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Funderburg NT, et al. Increased tissue factor expression on circulating monocytes in chronic HIV infection: relationship to in vivo coagulation and immune activation. Blood. 2010;115:161–167. doi: 10.1182/blood-2009-03-210179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Pandrea I, et al. Coagulation biomarkers predict disease progression in SIV-infected nonhuman primates. Blood. 2012;120:1357–1366. doi: 10.1182/blood-2012-03-414706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Grund B, Neuhaus J, Phillips A. Relative risk of death in the SMART study. The Lancet infectious diseases. 2009;9:724–725. doi: 10.1016/S1473-3099(09)70302-0. [DOI] [PubMed] [Google Scholar]
- 166.Loftus R. Immunosuppressive drugs and corticosteroids for HIV infection. GMHC Treat Issues. 1995;9:5–7. [PubMed] [Google Scholar]
- 167.Kyburz D, Brentano F, Gay S. Mode of action of hydroxychloroquine in RA-evidence of an inhibitory effect on toll-like receptor signaling. Nat Clin Pract Rheumatol. 2006;2:458–459. doi: 10.1038/ncprheum0292. [DOI] [PubMed] [Google Scholar]
- 168.Naarding MA, Baan E, Pollakis G, Paxton WA. Effect of chloroquine on reducing HIV-1 replication in vitro and the DC-SIGN mediated transfer of virus to CD4+ T-lymphocytes. Retrovirology. 2007;4:6. doi: 10.1186/1742-4690-4-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Murray SM, et al. Reduction of immune activation with chloroquine therapy during chronic HIV infection. Journal of virology. 2010;84:12082–12086. doi: 10.1128/JVI.01466-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Piconi S, et al. Hydroxychloroquine drastically reduces immune activation in HIV-infected, antiretroviral therapy-treated immunologic nonresponders. Blood. 2011;118:3263–3272. doi: 10.1182/blood-2011-01-329060. [DOI] [PubMed] [Google Scholar]
- 171.Paton NI, et al. Effects of hydroxychloroquine on immune activation and disease progression among HIV-infected patients not receiving antiretroviral therapy: a randomized controlled trial. JAMA: the journal of the American Medical Association. 2012;308:353–361. doi: 10.1001/jama.2012.6936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Emmel EA, Verweij CL, Durand DB, Higgins KM, Lacy E, Crabtree GR. Cyclosporin A specifically inhibits function of nuclear proteins involved in T cell activation. Science. 1989;246:1617–1620. doi: 10.1126/science.2595372. [DOI] [PubMed] [Google Scholar]
- 173.Levy R, Jais JP, Tourani JM, Even P, Andrieu JM. Long-term follow-up of HIV positive asymptomatic patients having received cyclosporin A. Advances in experimental medicine and biology. 1995;374:229–234. doi: 10.1007/978-1-4615-1995-9_20. [DOI] [PubMed] [Google Scholar]
- 174.Rizzardi GP, et al. Treatment of primary HIV-1 infection with cyclosporin A coupled with highly active antiretroviral therapy. The Journal of clinical investigation. 2002;109:681–688. doi: 10.1172/JCI14522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Schwarz A, et al. The effect of cyclosporine on the progression of human immunodeficiency virus type 1 infection transmitted by transplantation--data on four cases and review of the literature. Transplantation. 1993;55:95–103. doi: 10.1097/00007890-199301000-00019. [DOI] [PubMed] [Google Scholar]
- 176.Lederman MM, et al. Cyclosporin A provides no sustained immunologic benefit to persons with chronic HIV-1 infection starting suppressive antiretroviral therapy: results of a randomized, controlled trial of the AIDS Clinical Trials Group A5138. The Journal of infectious diseases. 2006;194:1677–1685. doi: 10.1086/509261. [DOI] [PubMed] [Google Scholar]
- 177.Markowitz M, et al. The virologic and immunologic effects of cyclosporine as an adjunct to antiretroviral therapy in patients treated during acute and early HIV-1 infection. The Journal of infectious diseases. 2010;201:1298–1302. doi: 10.1086/651664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Chapuis AG, et al. Effects of mycophenolic acid on human immunodeficiency virus infection in vitro and in vivo. Nature medicine. 2000;6:762–768. doi: 10.1038/77489. [DOI] [PubMed] [Google Scholar]
- 179.Coull JJ, Turner D, Melby T, Betts MR, Lanier R, Margolis DM. A pilot study of the use of mycophenolate mofetil as a component of therapy for multidrug-resistant HIV-1 infection. Journal of acquired immune deficiency syndromes. 2001;26:423–434. doi: 10.1097/00126334-200104150-00004. [DOI] [PubMed] [Google Scholar]
- 180.Millan O, et al. Pharmacokinetics and pharmacodynamics of low dose mycophenolate mofetil in HIV-infected patients treated with abacavir, efavirenz and nelfinavir. Clin Pharmacokinet. 2005;44:525–538. doi: 10.2165/00003088-200544050-00006. [DOI] [PubMed] [Google Scholar]
- 181.Sehgal SN. Rapamune (RAPA, rapamycin, sirolimus): mechanism of action immunosuppressive effect results from blockade of signal transduction and inhibition of cell cycle progression. Clin Biochem. 1998;31:335–340. doi: 10.1016/s0009-9120(98)00045-9. [DOI] [PubMed] [Google Scholar]
- 182.Heredia A, et al. Rapamycin causes down-regulation of CCR5 and accumulation of anti-HIV beta-chemokines: an approach to suppress R5 strains of HIV-1. Proceedings of the National Academy of Sciences of the United States of America. 2003;100:10411–10416. doi: 10.1073/pnas.1834278100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Heredia A, et al. Reduction of CCR5 with low-dose rapamycin enhances the antiviral activity of vicriviroc against both sensitive and drug-resistant HIV-1. Proceedings of the National Academy of Sciences of the United States of America. 2008;105:20476–20481. doi: 10.1073/pnas.0810843106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Latinovic O, Heredia A, Gallo RC, Reitz M, Le N, Redfield RR. Rapamycin enhances aplaviroc anti-HIV activity: implications for the clinical development of novel CCR5 antagonists. Antiviral research. 2009;83:86–89. doi: 10.1016/j.antiviral.2009.02.199. [DOI] [PubMed] [Google Scholar]
- 185.Nicoletti F, et al. Inhibition of human immunodeficiency virus (HIV-1) infection in human peripheral blood leucocytes-SCID reconstituted mice by rapamycin. Clinical and experimental immunology. 2009;155:28–34. doi: 10.1111/j.1365-2249.2008.03780.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Di Benedetto F, et al. First report on a series of HIV patients undergoing rapamycin monotherapy after liver transplantation. Transplantation. 2010;89:733–738. doi: 10.1097/TP.0b013e3181c7dcc0. [DOI] [PubMed] [Google Scholar]
- 187.Krown SE, et al. Rapamycin with antiretroviral therapy in AIDS-associated Kaposi sarcoma: an AIDS Malignancy Consortium study. Journal of acquired immune deficiency syndromes. 2012;59:447–454. doi: 10.1097/QAI.0b013e31823e7884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Cepeda EJ, Williams FM, Ishimori ML, Weisman MH, Reveille JD. The use of anti-tumour necrosis factor therapy in HIV-positive individuals with rheumatic disease. Ann Rheum Dis. 2008;67:710–712. doi: 10.1136/ard.2007.081513. [DOI] [PubMed] [Google Scholar]
- 189.Sellam J, et al. Use of infliximab to treat psoriatic arthritis in HIV-positive patients. Joint Bone Spine. 2007;74:197–200. doi: 10.1016/j.jbspin.2006.05.012. [DOI] [PubMed] [Google Scholar]
- 190.Linardaki G, Katsarou O, Ioannidou P, Karafoulidou A, Boki K. Effective etanercept treatment for psoriatic arthritis complicating concomitant human immunodeficiency virus and hepatitis C virus infection. J Rheumatol. 2007;34:1353–1355. [PubMed] [Google Scholar]
- 191.Mikhail M, Weinberg JM, Smith BL. Successful treatment with etanercept of von Zumbusch pustular psoriasis in a patient with human immunodeficiency virus. Arch Dermatol. 2008;144:453–456. doi: 10.1001/archderm.144.4.453. [DOI] [PubMed] [Google Scholar]
- 192.Paiardini M. ditorial: Hijacking the IL-7/IL-7R system in HIV infection. Journal of leukocyte biology. 2011;89:491–493. doi: 10.1189/jlb.1110614. [DOI] [PubMed] [Google Scholar]
- 193.Levy Y, et al. Effects of recombinant human interleukin 7 on T-cell recovery and thymic output in HIV-infected patients receiving antiretroviral therapy: results of a phase I/IIa randomized, placebo-controlled, multicenter study. Clinical infectious diseases: an official publication of the Infectious Diseases Society of America. 2012;55:291–300. doi: 10.1093/cid/cis383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Beq S, et al. IL-7 induces immunological improvement in SIV-infected rhesus macaques under antiviral therapy. Journal of immunology. 2006;176:914–922. doi: 10.4049/jimmunol.176.2.914. [DOI] [PubMed] [Google Scholar]
- 195.Parker R, et al. Interleukin-7 treatment counteracts IFN-alpha therapy-induced lymphopenia and stimulates SIV-specific cytotoxic T lymphocyte responses in SIV-infected rhesus macaques. Blood. 2010;116:5589–5599. doi: 10.1182/blood-2010-03-276261. [DOI] [PubMed] [Google Scholar]
- 196.Pallikkuth S, Parmigiani A, Pahwa S. The role of interleukin-21 in HIV infection. Cytokine Growth Factor Rev. 2012;23:173–180. doi: 10.1016/j.cytogfr.2012.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Hummelen R, et al. Effect of 25 weeks probiotic supplementation on immune function of HIV patients. Gut Microbes. 2011;2:80–85. doi: 10.4161/gmic.2.2.15787. [DOI] [PubMed] [Google Scholar]
- 198.Hummelen R, et al. Effect of micronutrient and probiotic fortified yogurt on immune-function of anti-retroviral therapy naive HIV patients. Nutrients. 2011;3:897–909. doi: 10.3390/nu3100897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Gonzalez-Hernandez LA, et al. Synbiotic therapy decreases microbial translocation and inflammation and improves immunological status in HIV-infected patients: a double-blind randomized controlled pilot trial. Nutr J. 2012;11:90. doi: 10.1186/1475-2891-11-90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Schunter M, et al. Randomized pilot trial of a synbiotic dietary supplement in chronic HIV-1 infection. BMC Complement Altern Med. 2012;12:84. doi: 10.1186/1472-6882-12-84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Klatt NR, et al. Probiotic/prebiotic supplementation of antiretrovirals improves gastrointestinal immunity in SIV-infected macaques. The Journal of clinical investigation. doi: 10.1172/JCI66227. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Couzin-Frankel J. Inflammation bares a dark side. Science. 2010;330:1621. doi: 10.1126/science.330.6011.1621. [DOI] [PubMed] [Google Scholar]