

Abstract
Oncogenic receptor tyrosine kinase (RTK) signaling through the Ras-Raf-Mek-Erk (Ras-MAPK) pathway is implicated in a wide array of carcinomas, including those of the breast. The cyclin-dependent kinases (CDKs) are implicated in regulating proliferative and survival signaling downstream of this pathway. Here, we show that CDK inhibitors exhibit an order of magnitude greater cytotoxic potency than a suite of inhibitors targeting RTK and Ras-MAPK signaling in cell lines representative of clinically recognized breast cancer (BC) subtypes. Drug combination studies show that the pan-CDK inhibitor, flavopiridol (FPD), synergistically potentiated cytotoxicity induced by the Raf inhibitor, sorafenib (SFN). This synergy was most pronounced at sub-EC50 SFN concentrations in MDA-MB-231 (KRAS-G13D and BRAF-G464V mutations), MDA-MB-468 [epidermal growth factor receptor (EGFR) overexpression], and SKBR3 [ErbB2/EGFR2 (HER-2) overexpression] cells but not in hormone-dependent MCF-7 and T47D cells. Potentiation of SFN cytotoxicity by FPD correlated with enhanced apoptosis, suppression of retinoblastoma (Rb) signaling, and reduced Mcl-1 expression. SFN and FPD were also tested in an MDA-MB-231 mammary fat pad engraftment model of tumorigenesis. Mice treated with both drugs exhibited reduced primary tumor growth rates and metastatic tumor load in the lungs compared to treatment with either drug alone, and this correlated with greater reductions in Rb signaling and Mcl-1 expression in resected tumors. These findings support the development of CDK and Raf co-targeting strategies in EGFR/HER-2-overexpressing or RAS/RAF mutant BCs.
Introduction
Chemotherapy-related morbidity and drug-resistant recurrence have prompted intensive investigation of the molecular mechanisms underlying carcinogenesis. These efforts have led to the development of monotargeted therapies such as trastuzumab, a humanized monoclonal antibody, and lapatinib, a receptor tyrosine kinase (RTK) inhibitor to target ErbB2/epidermal growth factor receptor 2 (HER-2)-positive breast cancer (BC) [1–3]. Notwithstanding these early successes, the use of these monotargeted therapies alongside chemotherapy has been fraught with clinical dilemmas including intertumor and intratumor heterogeneity, intrinsic and adaptive drug resistance, and off-target toxicity [4,5].
The epidermal growth factor receptor (EGFR/HER-1/ErbB1) and HER-2 have been implicated in BC pathogenesis. HER-2 is over-expressed in approximately 20% of BC cases and is associated with poor prognosis [3], while EGFR has been shown to be overexpressed in 35% of recurrent cases [6]. Expression of EGFR in BC has been inversely correlated with relapse-free survival and is associated with a lack of response to hormone therapy [7]. Both EGFR and HER-2 regulate downstream tumor cell proliferation and survival through the Ras-Raf-Mek-Erk (Ras-MAPK) and phosphatidylinositol-3-kinase (PI3K)-Akt pathways. Mutations in RAS, RAF, PI3K, and PTEN are strongly associated with tumor initiation and progression and have been identified with high frequency in various malignancies including BC [8,9].
The Ras-MAPK pathway is a common downstream pathway for numerous RTK systems and is a major focal point in cancer therapy development [8]. Upstream RTK overexpression or activating mutations in RAS and RAF are strongly implicated in driving oncogenesis through constitutive activation of the MAPK pathway. The Ras-Raf oncogenic axis transduces survival and proliferative signals through both Mek-Erk-dependent and Mek-Erk-independent Raf signaling pathways [10]. Hence, Raf is a regulatory nexus in RTK-mediated proliferative and survival signaling and its inhibition has become a central strategy in the development of treatments for melanoma, kidney, liver, and breast carcinoma [11,12].
In addition to Raf, cyclin-dependent kinases (CDKs) have also emerged as attractive targets for cancer intervention [13]. CDKs are an important regulatory component of the retinoblastoma (Rb)–E2F signaling axis. The Rb-E2F axis can be activated by Mek-Erk-dependent or Mek-Erk-independent Raf signaling [10]. Activation through Mek-Erk-dependent signaling occurs through cyclin D1 induction and subsequent assembly of cyclin D1–CDK4/CDK6 and –CDK2 complexes. These complexes promote Rb phosphorylation, resulting in Rb-E2F dissociation and G1-S cell cycle transition through E2F-dependent transcriptional programs. Perturbation of the Rb-E2F signaling axis commonly occurs in cancer through gene amplifications, deletions, and functional alterations of its key signaling regulators. Perturbations frequently involve loss of Rb, cyclin D1 amplification, or CDK4 overexpression; these alterations have been associated with poor prognosis in many cancers including HER-2-positive BC [14–16].
Sorafenib (SFN; BAY 43-9006; Nexavar) is a broad-spectrum multi-kinase small molecule inhibitor. In vitro recombinant kinase assays have shown that SFN potently inhibits both the CRAF (Raf-1) and BRAF isoforms, including the highly oncogenic V600E variant [17]. SFN disrupts Ras-MAPK signaling in cell-based assays and this has been correlated with antitumor effects in colon, kidney, lung, and breast xenograft models [11,17]. SFN has been approved for treatment in advanced renal cell and hepatocellular carcinoma [18,19]. Most recently, SFN has been shown to increase progression-free survival in HER-2-negative metastatic BC when used in combination with capecitabine [12].
Flavopiridol (FPD; HMR-1275; Alvocidib) is a small molecule semisynthetic alkaloid that competes with ATP to inhibit CDK-1, -2, -4, -6, -7, and -9 [20]. This pan-CDK inhibitor exhibited considerable promise in phase I and II trials, showing partial or complete response as well as disease stabilization in advanced malignancies including ovarian, pancreas, gastric, and colon cancers and BC [21,22]. CDK inhibitors with increased selectivity (i.e., AZD5438 and PD0332991) have also been developed, which inhibit Rb-associated tumorigenesis in various preclinical human tumor xenograft models [23,24]. Cell-based studies have shown that FPD and other CDK inhibitors induce cell cycle arrests in G1-S or G2-M that are associated with antiproliferative and cytotoxic effects. CDK-induced cytotoxicity may underlie systemic toxicity issues of FPD and other CDK inhibitors in clinical trials [24–26].
In this study, we tested the cytotoxic potency and efficacy of a panel of RTK pathway (RTK-P) and CDK inhibitors across cell line models representative of clinically recognized BC subtypes. CDK inhibitors were found to exhibit significantly greater cytotoxic potency than RTK-P inhibitors in all tested lines. FPD most strongly potentiated cytotoxicity induced by SFN in vitro and in vivo. Potentiation occurred in cell lines harboring constitutive Ras-MAPK activation associated with EGFR/HER-2 overexpression or KRAS-BRAF mutations. Our data suggest that co-targeting CDKs and the Ras-MAPK pathway with FPD and SFN, respectively, enhances SFN killing efficacy at low doses. These data suggest a potential role for FPD in potentiating SFN therapeutic efficacy in the clinic.
Materials and Methods
Cell Lines and Reagents
MDA-MB-468, SK-BR3, MCF-7, and T47D BC lines were obtained from American Type Culture Collection (Manassas, VA). MDA-MB-231 cells were provided by Dr Peter Siegel (McGill University, Montreal, Quebec). SFN, FPD, lapatinib, and erlotinib were obtained from Toronto Research Chemicals (Toronto, Ontario). Fascaplysin, U0126, LY294002, and U73122 were purchased from Sigma-Aldrich (Oakville, Ontario). AZD5438 and PD0332991 were purchased from Selleckchem (Houston, TX). SFN and FPD used for xenograft studies were acquired from UHN Shanghai R&D Co (Shanghai, China).
Cytotoxicity Assay
Cells (2 x 104 per well in 96-well plates) were treated with increasing concentrations of the indicated drugs for 72 hours in standard culture media. For drug combination experiments, cells were treated with varying concentrations of a primary drug in the presence of a fixed concentration (0.2 µM) of a secondary drug. Cell viability was assessed by metabolic activity using 3-(4,5-dimethylthiazol-2-yl)-2, 5-diphenyltetrazolium bromide assay (Sigma-Aldrich). Drug interactions were analyzed using CALCUSYN software by Chou and Talalay (Biosoft, Ferguson, MO) that quantitatively assesses the nature of drug interactions using multiple drug effect/combination index (MDE-CI) isobologram analysis. This analysis calculates a CI for drug interactions. CI values less than 0.9 indicate synergy; CI values between 0.9 and 1.1 indicate additivity, and CI values greater than 1.1 indicate antagonism [27]. Fa reflects the proportion of cells killed or no longer viable as a result of drug treatment and was calculated using the following equation: Fa = (A570 control - A570 treated)/A570 control [2,28].
Apoptosis Assay
Cells (1 x 106 cell per well in a six-well plate) were treated with the indicated drugs for 24 hours. After treatment, cells were incubated (30 minutes, 37°C) with propidium iodide (PI) and the annexin XII-based phosphatidylserine fluorescent sensor pSIVA-IANBD (Imgenex, San Diego, CA). Annexin XII+|PI- cells representative of apoptotic cells were counted on a Beckman Coulter flow cytometer (Mississauga, Ontario).
Signaling Experiments
Cells (1 x 107 per 10-cm plate) were treated with SFN or FPD alone or in combination at the indicated times and concentrations in standard culture media. After treatment, cells were lysed in radio-immunoprecipitation assay buffer supplemented with protease inhibitors and clarified by centrifugation (12,000g, 10 minutes). Equal quantities of protein were separated by sodium dodecyl sulfate-polyacrylamide gel electrophoresis and assayed by immunoblot (IB) analysis using the indicated antibodies. All antibodies were purchased from Cell Signaling Technology (Beverly, MA).
Preclinical In Vivo Drug Testing
Logarithmically growing MDA-MB-231 cells were resuspended in 1:1 phosphate-buffered saline (PBS)/Matrigel (Sigma-Aldrich) solution. Fifty microliters (2 x 106 cells) of the solution was injected into the right inguinal mammary gland of BalbC-RAG2-/-|IL2Rγc-/- mice (kindly provided by Dr M. Ito, Central Institute of Experimental Animals, Kawasaki, Japan). SFN was dissolved in H2O containing 12.5% Cremophor EL (Sigma-Aldrich) and 12.5% ethanol and administered by oral gavage [29]. FPD was dissolved in PBS and administered by intraperitoneal injection (i.p.). Mice were randomized into four cohorts (six/cohort). The control cohort received PBS (i.p.) and treatment cohorts received either SFN (30 mg/kg, oral gavage), FPD(3mg/kg, i.p.), or SFN-FPD (30 and 3 mg/kg, respectively) [17,30]. Treatment was initiated 7 days post-engraftment and continued every 2 days for 28 days. Tumor sizes were measured every 2 days using calipers. After 35 days, tumors were resected, bisected, and either lysed in radioimmunoprecipitation assay buffer for IB analysis or formalin-fixed and paraffin-embedded (FFPE) for histologic analysis. IBs were quantified using Image J software [31].
Fluorescence In Situ Hybridization Analysis
Fluorescence in situ hybridization (FISH)-based identification of lung-metastasized xenografted MDA-MB-231 cells was performed using the human Cep17 SpectrumGreen Probe (D17Z1, 17p11.1-q11.1 Alpha Satellite DNA; Abbott Molecular, Markham, Ontario) and counterstained for nuclei with 4′,6-diamidino-2-phenylindole (DAPI). Ten randomly selected regions representative of general whole lung morphology from each lung section were independently assessed for total FISH Cep17 signal and nuclei number using ePATHOLOGY IMAGESCOPE (Aperio, Vista, CA) and IMAGEPRO software (Media Cybernetics, Rockville, MD). The ratio of Cep17 probe signals to DAPI-stained nuclei was calculated to determine the relative burden of MDAMB-231 cells metastasized in murine lungs from each treatment cohort.
Results
CDK Inhibitors Are Significantly More Cytotoxic than RTK-P Inhibitors
The kill efficacy (KEff; maximal number of cells killed) and cytotoxic potency (EC50; dose required to induce half-maximal cell kill) of a panel of RTK-P inhibitors targeting EGFR/HER-2, Raf, Mek, PI3K, and phospholipase C (PLC) were compared to a panel of CDK inhibitors (Table 1) in five BC cell lines. In metastatic MDA-MB-231 cells, the KEff of CDK and RTK-P inhibitors spanned similar ranges (21–100%; Table 2). In contrast, the EC50 ranges for the CDK inhibitor panel were markedly lower than the EC50 ranges for the RTK-P panel (0.2–0.7 µM vs 8.6–36.4 µM, respectively; Table 2). These differences were clearly indicated by the cytotoxicity profiles of the CDK inhibitors, which clustered distinctly left of RTK-P profiles on the dose axis (Figure 1A). Subsequent estimation of the average EC50 of the two inhibitor panels showed that CDK inhibitors induce cell death with approximately 54-fold more potency than RTK-P inhibitors in MDA-MB-231 cells (Table 2). To investigate whether this potency enhancement was cell-line specific, EGFR-overexpressing (MDA-MB-468), HER-2-overexpressing (SKBR3), and hormone-dependent (MCF-7 and T47D) BC cell lines were also tested (Table 3). The KEff of CDK and RTK-P inhibitors in these lines spanned ranges comparable to those in MDA-MB-231 cells. More strikingly however, the CDK cytotoxicity profiles of these four lines also clustered distinctly left of the RTK-P profiles on the dose axis (Figure 1, B–E). Taken together, these results indicate that CDK inhibitors induce cytotoxicity with significantly greater potency than RTK-P inhibitors in BC lines.
Table 1.
CDK Inhibitors.
| Targets | FPD | AZD5483 | PD0332991 | Fascaplysin |
| CDK1-CycB | 0.027 | 0.016 | >10 | >100 |
| CDK2-CycA | 0.405 | 0.045 | >10 | >50 |
| CDK2-CycE | 0.282 | 0.006 | >10 | >50 |
| CDK4-CycD1 | 0.132 | 0.449 | 0.011 | 0.35 |
| CDK4-CycD2 | - | - | - | >100 |
| CDK4-CycD3 | - | - | 0.009 | >100 |
| CDK6-CycD1 | - | - | - | 3.4 |
| CDK6-CycD2 | - | - | 0.015 | >50 |
| CDK6-CycD3 | 0.395 | 0.021 | - | - |
| CDK7-CycH | 0.514 | 0.821 | - | - |
| CDK9-CycT | 0.011 | 0.020 | - | - |
Comparison of IC50 (µM) of FPD, AZD5483, PD0332991, and fascaplysin against various CDK-cyclin complexes [23,47,48]; -, undetermined.
Table 2.
Potency and Efficacy of Inhibitor Panels.
| Cell Line | Drug Panel | Potency Range, EC50 (µM) | Mean Potency, EC50 (µM) (± SEM) | Maximal Kill Efficacy Range, KEff (%) |
| MDA-MB-231 | Anti-CDK | 0.2–0.7 | 0.43 ± 0.7 | 23–75 |
| Anti-RTK-P | 8.6–36.4 | 23.4 ± 0.6 | 21–100 | |
| MDA-MB-468 | Anti-CDK | 0.1–1.1* | 0.36 ± 0.5 | 23–97 |
| Anti-RTK-P | 7.7–40.3 | 16 ± 0.7 | 44–100 | |
| SKBR3 | Anti-CDK | 0.1–0.6 | 0.36 ± 0.9 | 18–100 |
| Anti-RTK-P | 3.9–25.7 | 17.9 ± 0.5 | 41–100 | |
| MCF-7 | Anti-CDK | 0.2–0.6 | 0.45 ± 0.8 | 37–95 |
| Anti-RTK-P | 13.3–67.2 | 29.3 ± 0.5 | 29–100 | |
| T47D | Anti-CDK | 0.3–0.9 | 0.66 ± 0.6 | 37–90 |
| Anti-RTK-P | 5.8–61.7** | 21.9 ± 0.4 | 43–100 |
Potency (EC50) and maximal kill efficacy (KEff) of each inhibitor panel in BC lines. KEff and EC50 were estimated by fitting inhibition data to a four-parameter sigmoidal function. In cases where cell kill did not reach saturation, KEff was taken to be the value at the highest drug dose tested and this value was used to constrain fitting. EC50 values in these cases would be underestimated. Estimates for PD0332991 (*) and U0126 (**) were indeterminate.
Figure 1.

Comparison of cytotoxicity induced by CDK and RTK-P inhibitors. (A) MDA-MB-231, (B) MDA-MB-468, (C) SKBR3, (D) MCF-7, and (E) T47D cells were treated with increasing concentrations of the indicated inhibitors. Cell viability was assayed after 72 hours. The fraction of cells killed (Fa ± SEM) by treatment with a panel of CDK inhibitors (solid markers) and RTK-P inhibitors (open markers) are shown. The RTK-P inhibitors U0126, erlotinib, lapatinib, SFN, U73122, and LY294002 were used to inhibit Mek, EGFR, EGFR/HER-2, Raf, PLC, and PI3K, respectively. Dose-effect profiles are representative of two to three independent experiments.
Table 3.
Human Breast Cancer Cell Line Models [55,56].
| Cell Line | Clinical Markers | Mutational Status* | Tumor Type | Tumor Classification |
| MDA-MB-231 | TN-Claudin low | TP53 | Adenocarcinoma† | Basal B |
| KRAS‡ | ||||
| BRAF§ | ||||
| CDKN2A | ||||
| MDA-MB-468 | EGFR positive¶ | PTEN | Adenocarcinoma† | Basal A |
| TP53 | ||||
| MADH4 | ||||
| RB1 | ||||
| SKBR3 | HER-2 positive# | (NA)** | Adenocarcinoma† | Luminal |
| T47D | ER/PR positive | PIK3CA | Invasive ductal carcinoma† | Luminal |
| TP53 | ||||
| MCF-7 | ER/PR positive | PIK3CA | Invasive ductal carcinoma† | Luminal |
| CDKN2A |
Cancer Genome Project, Sanger Institute (http://www.sanger.ac.uk/genetics/CGP/CellLines).
Isolated from metastatic pleural effusion.
KRAS G13D mutation.
BRAF G464V mutation.
EGFR overexpression (gene amplification).
HER-2 overexpression (gene amplification).
Not available.
FPD and Fascaplysin Potentiate SFN Cytotoxicity in MDA-MB-231 Cells
The marked increases in potency observed with the CDK inhibitor panel underscored their potential use as cytotoxicity-enhancing agents for therapies targeting the Ras-MAPK pathway. This prompted us to explore potential synergistic cytotoxic effects of CDK inhibitors with the Raf inhibitor SFN in MDA-MB-231 cells. These cells harbor oncogenic KRAS and BRAF mutations associated with constitutive Ras-MAPK signaling (Table 3). When CDK inhibitors were added at fixed concentrations to varying concentrations of SFN, cytotoxicity was potentiated in this line (Figure 2, A and B). Potentiation was substantially more pronounced with FPD and fascaplysin than with either PD0332991 or AZD5438 at SFN concentrations below its EC50 (≤5 µM; compare Figure 2, A and B to C and D). The fraction of cells killed by 0.2 µM of either FPD or fascaplysin alone was significantly less than that observed in combination with SFN, indicating that these combinations can potentiate cytotoxicity to levels greater than the summed effects of individual drugs (compare shaded line with drug response curves in Figure 2, A and B). We further assessed the nature of these drug interactions using MDE-CI analysis (see Materials and Methods section) [2,27]. This analysis yielded CI values indicative of synergy (<0.9) and additivity (0.9–1.1) at the higher range of tested ratio combinations (Figure 2, A and B, lower panels, black and gray bars, respectively). Synergy was associated with FPD/SFN and fascaplysin/SFN combination ratios ranging from 1:6 to 1:25. MDE-CI analysis did not indicate synergistic or additive interactions between PD0332991 or AZD5438 and SFN (Figure 2, C and D, lower panels). Taken together, these results show that specific ratio combinations of SFN and the CDK inhibitor FPD or fascaplysin synergistically potentiate cytotoxicity in MDA-MB-231 cells.
Figure 2.

Potentiation of SFN-induced cytotoxicity by a panel of CDK inhibitors in MDA-MB-231 cells. (A–D, upper panels) The fraction of cells killed (Fa ± SEM) by SFN in the presence of DMSO (vehicle) or a fixed concentration (0.2 µM) of FPD (A), fascaplysin (B), PD0332991 (C), and AZD5438 (D). The fraction of cells killed by CDK inhibitors at a fixed concentration of 0.2 µM is shown for comparison [dashed lines; gray shading (±SEM)]. (A–D; lower panels) MDE-CI analysis of drug interactions in the upper panels. CIs as a function of SFN concentration are shown. Black, gray, and white bars denote synergistic (CI < 0.9), additive (CI = 0.9–1.1), or antagonistic interactions (CI > 1.1), respectively. Synergistic ratios and SFN-EC50 are indicated. Data are representative of two to three independent experiments.
FPD Potentiates SFN Cytotoxicity in MDA-MB-468 and SKBR3 Cells
We extended our analyses to MDA-MB-468, SKBR3, MCF-7, and T47D cells and focused efforts on FPD, which has undergone extensive testing in clinical trials [22,25,26]. Interestingly, FPD failed to potentiate cytotoxicity in hormone-dependent MCF-7 and T47D cells (Figure 3, A and B, upper panels). Consistent with this, synergistic interactions between these drugs were not detected in these lines (Figure 3, A and B, lower panels). We next tested for potentiating interactions between these drugs in MDA-MB-468 and SKBR3 cells, which harbor constitutive Ras-MAPK signaling associated with EGFR and HER-2 overexpression, respectively (Table 3). Exploration of drug ratio combinations in MDA-MB-468 cells indicated synergy between FPD and SFN ratios ranging from 1:6 to 1:25; these interactions became additive as ratios decreased (Figure 3C). Synergy was only indicated at the highest FPD to SFN ratio tested in SKBR3 cells (1:6; Figure 3D). As observed in MDA-MB-231 cells, synergy was only evident at SFN concentrations below the EC50 of the tested lines. Together with the observations made in MDA-MB-231 cells, these data suggest that specific ratio combinations of FPD and SFN synergistically potentiate cytotoxicity in BC cells harboring constitutive Ras-MAPK signaling.
Figure 3.

Potentiation of SFN-induced cytotoxicity by FPD in MCF-7, T47D, MDA-MB-468, and SKBR3 cells. (A–D, upper panels) The fraction of cells killed (Fa ± SEM) by SFN in the presence of DMSO (vehicle) or a fixed concentration (0.2 µM) of FPD in MCF-7 (A), T47D (B), MDA-MB-468 (C), and SKBR3 (D) cells. The fraction of cells killed by CDK inhibitors at a fixed concentration of 0.2 µM is shown for comparison [dashed lines; gray shading (±SEM)]. (A–D, lower panels) MDE-CI analysis of drug interactions in the upper panels. CIs as a function of SFN concentration are shown. Black, gray, and white bars denote synergistic (CI < 0.9), additive (CI = 0.9–1.1), or antagonistic interactions (CI > 1.1), respectively. Synergistic ratios and SFN-EC50 are indicated. Data are representative of two to three independent experiments.
FPD Potentiates SFN-Induced Apoptosis in MDA-MB-231 and MDA-MB-468 Cells
Combination studies showed that FPD-SFN used at a ratio of 1:25 induced cytotoxic synergy in MDA-MB-231 and MDA-MB468 cells (Figures 2A and 3C). Focusing on these two lines, we tested whether the same FPD-SFN combination ratio could also potentiate apoptosis. Annexin XII-based phosphatidylserine-binding assays showed that FPD and SFN alone induced levels of apoptosis that were significantly less than that induced by FPD-SFN combinations used at 1:25 ratios in MDA-MB-231 (Figure 4A) and MDA-MB-468 (Figure 4B) cells. The enhancement produced by the combination was greater than theoretically expected additive effects, suggesting that these drugs synergistically interact to induce programmed cell death in both lines (Figure 4, A and B). These data suggest that a 1:25 ratio of FPD-SFN supra-additively potentiates apoptosis in MDA-MB-231 and MDA-MB-468 cells.
Figure 4.

Potentiation of SFN-induced apoptosis by FPD. Cells were treated with either DMSO, 0.2 µM FPD, 5 µM SFN, or 1:25 ratio of FPD-SFN (0.2 µMand 5 µM, respectively) for 24 hours. The percentages (mean ± SEM) of early apoptotic (annexin XII+), non-necrotic (PI-) MDA-MB-231 (A) and MDA-MB-468 (B) cells detected after drug treatment (gray bars) are shown. The white bar indicates the fraction of apoptotic cells expected by simple addition of the percentage of apoptotic cells induced by SFN or FPD alone. Significant P values are indicated (t test; n = 3–4).
FPD and SFN Cooperatively Dysregulate Rb and Mcl-1
The Ras-MAPK and Rb-E2F signaling axes play critical roles in cancer cell proliferation and survival [8,14]. Assessment of Ras-MAPK activity using an active-state antibody against Erk (pErk) showed that SFN alone strongly suppressed pErk in MDA-MB-231 cells for up to 2 hours before undergoing periodic oscillations in activity at 4 and 24 hours (Figure 5A). This periodic Erk signal recovery could represent normal oscillatory behavior of Erk activity recently described in asynchronously growing cells [32]. Expectedly, FPD did not affect pErk signaling in this line (Figure 5B), and consistent with this, it did not significantly alter Erk inhibition when added to SFN (Figure 5C). In MDA-MB-468 cells, FPD and SFN alone or in combination failed to inhibit pErk signaling (Figure 5, D–F). Signaling through the Rb-E2F pathway was also assessed by Rb phosphorylation (pRb) at S807/811 [33]. pRb was partially suppressed by SFN and only intermittently by FPD in MDA-MB-231 cells (Figure 5, A and B). However, when FPD was combined with SFN, maximal suppression was observed for the full length of the treatment period (Figure 5C). In contrast, FPD or SFN alone or in combination failed to suppress pRb in MDA-MB-468 cells (Figure 5, D–F). Lastly, SFN and FPD in combination reduced expression of Mcl-1 to greater extents than either drug alone in both MDA-MB-231 and MDA-MB-468 cells (Figure 5, A–F). These data suggest that pRb suppression and reduced Mcl-1 expression underlie enhanced cytotoxicity associated with FPD-SFN combinations.
Figure 5.

Inhibition of Ras-MAPK and Rb-E2F signaling by SFN-FPD combinations. MDA-MB-231 (A–C) and MDA-MB-468 (D–F) cells were treated with SFN (5 µM; A, D), FPD (0.2 µM; B, E), or a 1:25 combination ratio of FPD-SFN (0.2 µMand 5 µM, respectively; C, F). Erk phosphorylation at T202/Y204 indicative of its active state. Rb phosphorylation at S807/811 indicative of inactivation catalyzed by CDKs [33]. Loading was assessed by levels of the Ras GTPase-activating protein, RasGAP. Blots are representative of two to three independent experiments.
Maximal Tumor Growth Suppression In Vivo with FPD and SFN Coadministration
We next tested whether FPD-SFN combinations could potentiate antitumor activity in vivo. MDA-MB-231 cells were orthotopically engrafted in mammary fat pads of BalbC-RAG2-/-|IL2Rγc-/- mice. FPD and SFN alone or in combination at a 1:10 ratio—which falls within the 1:6 to 1:25 range observed in vitro—was administered to assess relative effects on antitumor activity. SFN alone significantly reduced the rate of tumor development relative to control mice (Figure 6A) [17]. FPD alone also significantly suppressed tumor development rates compared to controls, although not to the same extent observed with SFN. FPD and SFN in combination, however, produced the greatest level of tumor growth suppression (Figure 6A). Importantly, all six mice of each treatment cohort survived with no adverse toxicity reactions of weight loss. Thus, FPD-SFN combinations associated with enhanced cytotoxicity and apoptosis in vitro also promote antitumor effects when administered at a similar ratio in vivo.
Figure 6.

Combined treatment of SFN and FPD results in enhanced tumor suppression in vivo. (A) Tumor development profiles of drug-treated cohorts (six mice/cohort). Estimates of mean tumor volume ± SEM are shown. Control versus SFN, FPD, or SFN-FPD (P < .0001; two-way analysis of variance). SFN-FPD versus SFN or FPD (P < .0001; two-way analysis of variance). (B) Pulmonary metastatic tumor burden assessed by quantification of the ratio (±SEM) of human Cep17 signal and DAPI-stained nuclei in each cohort (n = 24; see also Figure W2). Significant P values (t test) areindicated.
Cooperative Inhibition of Pulmonary Metastatic Tumor Load by Combined FPD and SFN Treatment
Whole lungs resected after 35 days of treatment were examined for histologic evidence of metastases (Figures W1–W3). Pathologic assessment suggested that metastatic tumor burden in the lung was significantly less in the SFN-treated group compared to the FPD- or control-treated group (Figure W1). FISH analyses quantifying the ratio of human-specific Cep17 signal to the number of DAPI-stained nuclei corroborated this pathologic assessment (Figure W2) and further verified that the number of metastatic BC cells in the lung was suppressed by either FPD or SFN treatment alone. The most pronounced suppression, however, was observed with FPD and SFN co-treatment (Figure 6B), which paralleled the suppressive effects of this combination on tumor growth at the orthotopic site.
FPD and SFN Combinations Maximally Inhibited Erk, Rb, and Mcl-1 In Vivo
The molecular mechanisms underlying tumor suppression were assessed by measuring pErk, pRb, and Mcl-1 expression in resected tumors (Figure 7A). In comparison to the control cohort, the FPD-treated group did not show significant differences in pErk inhibition (Figure 7B), Rb phosphorylation (Figure 7C), or Mcl-1 expression (Figure 7D). SFN also failed to suppress Rb phosphorylation and paradoxically increased pErk and Mcl-1 levels. However, FPD and SFN in combination affected all three signaling proteins as indicated by significant reductions in pErk, pRb, and Mcl-1 levels relative to controls (Figure 7, B–D). These data show that combined FPD and SFN treatment maximally suppress levels of pErk, pRb, and Mcl-1, and this correlates with the enhanced antitumor effects produced by this combination.
Figure 7.
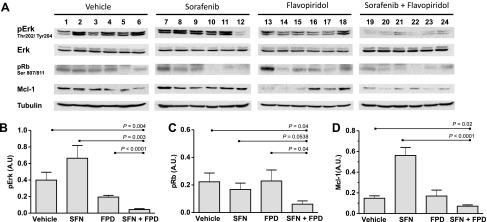
SFN and FPD cooperatively inhibit Erk, Mcl-1, and Rb signaling in tumors. (A) Immunoblots of lysates prepared from resected tumors assessing pErk, pRb, and Mcl-1 levels in the indicated cohorts (n = 24). Loading was assessed by tubulin levels. (B–D) Densitometry analyses of the average intensity of pErk, pRb, and Mcl-1. The mean ± SEM intensity expressed as a ratio of tubulin intensity across treatment cohorts (six mice/cohort) is shown. Significant P values (t test) are indicated. A.U., arbitrary units.
Discussion
Overexpression of RTKs or activating mutations residing within nodes of the Ras-Raf-MAPK and PI3K-Akt pathways are associated with poor prognosis and have become focal points of targeted therapy strategies. We evaluated the general merit of such strategies in BC by exploring the cytotoxic effects of a panel of RTK-P inhibitors targeting EGFR, HER-2, PLC, Raf, Mek, and PI3K in cell lines representative of EGFR/HER-2-overexpressing, hormone-dependent, and triple-negative (TN) BC subtypes. Our findings showed that the mean cytotoxic potency (EC50) of the RTK-P inhibitor panel was narrow, ranging from 16 to 29 µM across all lines (Table 2). In comparison, a panel of four CDK inhibitors with highly varied substrate specificities exhibited a very tight mean potency range of 0.4 to 0.7 µM across the same five lines (Table 2). These relatively narrow ranges suggest that improvements in potency achievable within anti-RTK-P or anti-CDK treatment modalities are limited. Across these modalities however, we see compelling improvements in cytotoxic potential. Our data show that the average EC50 of the CDK inhibitor panel is well over an order of magnitude less than the average EC50 of the RTK-P inhibitor panel (0.45 µM vs 21.7 µM, respectively) reflecting a 48-fold improvement in potency. Perhaps more compelling is the ubiquity of this improvement across a morphologically and clinically diverse panel of BC lines (Table 3). These data clearly indicate that CDK targeting is a substantially more potent alternative to inducing cell death than RTK-P targeting. The reasons for this are presently unclear but may be due to the fact that CDKs lie downstream of RTK-P targets and constitute a major regulatory point of mitogenic and survival signal convergence.
The potency with which CDK inhibitors induce cell death indicates that BC cell survival may be highly sensitive to disruption in CDK activity. Consistent with this, FPD and fascaplysin strongly potentiated SFN cytotoxicity in MDA-MB-231 cells (Figure 2). MDE-CI analysis indicated that FPD or fascaplysin synergistically interacts with SFN. Focusing on the clinically tested drug FPD, we also observed synergy with SFN in MDA-MB-468 and SKBR3 cells but not in hormone-dependent MCF-7 and T47D cells (Figure 3). Cytotoxic synergy in all lines occurred at low (sub-EC50) SFN concentrations and at FPD-SFN ratios ranging from 1:6 to 1:25. We also observed that cytotoxic synergy correlated with supra-additive effects on apoptosis at a 1:25 combination ratio. This ratio falls within the 1:6 to 1:25 range associated with cytotoxic synergy, suggesting a link between cell death and the induction of apoptotic mechanisms by these drugs (Figure 4). Taken together, our data show that FPD synergizes with SFN at specific combination ratios to induce cell death in EGFR/HER-2 and mutant RAS/RAF BC model systems. We hypothesize that the ability of FPD to synergize with SFN in these models may be associated with dependency on constitutively elevated EGFR/HER2 and Ras-MAPK activity. Interestingly, FPD has previously been shown to synergize with trastuzumab in inducing cytotoxicity in HER-2 BC lines through apoptotic mechanisms that involved suppression of EGFR expression [34]. In line with our observations, synergy between FPD and trastuzumab was not observed in hormone-dependent cells [34]. FPD has also been shown to promote lethality of the dual EGFR/HER-2 inhibitor lapatinib in a greater than additive manner [35]. The latter studies support our observations and, together, highlight the importance of co-targeting strategies centered on inhibiting Ras-MAPK and CDK signaling concurrently.
Significant toxicity issues in phase I FPD trials have been observed with maximal tolerable doses (MTDs) of FPD including leukopenia, nausea, vomiting, diarrhea, hypotension, and pro-inflammatory syndrome [36]. In this study, animal cohorts were treated with sub-MTDs of FPD, SFN, or FPD-SFN. In addition, Cremophor EL, a polyethoxylated castor oil commonly used to improve water solubility of SFN and other drugs, was used at levels below reported toxicity thresholds in mice [29,37]. Importantly, adverse toxicity issues such as lethality or severe weight loss were not observed in any of the individual cohorts over the course of the treatment period (vehicle, 22.8 ± 0.09 g; SFN, 25.5 ± 0.04 g; FPD, 24.6 ± 0.08 g; SFN + FPD, 26.7 ± 0.06 g; mean ± SD). At these nontoxic sub-MTDs, FPD potentiated the antitumor effects of SFN in a mammary xenograft tumor model highlighting its potential use at low-toxicity doses in FPD-SFN combination modalities [30,38,39]. Cytotoxic potentiation by FPD, however, was associated with FPD-SFN ratios ranging from 1:6 to 1:25 in cell lines. This would limit the maximal SFN concentration in such combinations to about 5 µM that is 25-fold more than FPD (0.2 µM) and is below the EC50 of SFN. At sub-EC50 SFN concentrations, the maximal kill efficacy (KEff) of the cocktail is in the order of 50% to 60% (Figure 2A). This suggests that reduced systemic toxicity associated with the use of synergistic sub-MTD FPD-sub-EC50 SFN ratios come at the expense of a 40% to 50% reduction in KEff. Assuming these ratios are also synergistic at higher FPD concentrations (i.e., 0.8 µM), the maximal SFN concentration allowed at a 1:25 ratio would be 20 µM. At this concentration, the KEff of the cocktail would be approximately 90%, which is 15% greater than the KEff of SFN alone. Under this assumption, synergy (and its associated benefit of reduced systemic toxicity) would not come at the expense of a loss in KEff. Importantly, we used a 1:10 FPD/SFN dosing ratio in mice that falls within ratio boundaries for cytotoxic potentiation determined in cell lines (1:6-1:25). Thus, drug ratio effects observed in vitro may correlate with effects in vivo, supporting the use of cell-based drug ratio studies for guiding initial dosing experiments in preclinical mouse models. However, it remains unclear what the true impact of changes in FPD/SFN ratios and/or doses would be on systemic toxicity and tumor response in the clinic. It also remains unclear whether potential reductions in toxicity associated with FPD-SFN combinations come at the expense of an overall loss in tumor response. The latter questions, including possible trade-offs between toxicity and tumor response, still remain to be addressed in the clinic.
Erk inhibition by SFN in MDA-MB-231 cells correlated with reductions in Mcl-1 expression levels and suppression of pRb, suggesting that Mcl-1 and Rb are regulated by Mek-Erk-dependent signaling. Reduced Mcl-1 expression and Rb phosphorylation have been previously correlated with antiapoptotic and proliferative effects associated with inhibition of the Ras-MAPK pathway [40]. For example, Mcl-1 induction downstream of the Ras-MAPK signaling has been shown to promote BC cell survival [41]. In addition, phosphorylation-dependent inactivation of Rb through cyclin D1 induction downstream of the Ras-MAPK pathway was shown to lock cells in G1 and induce apoptosis [14,24]. Our results therefore are consistent with a model in which Mek-Erk-dependent Raf signal suppression by SFN inhibits downstream Mcl-1 and pRb-dependent survival signaling in MDA-MB-231 cells (Figure 5). SFN also inhibited Mcl-1 levels in MDA-MB-468 cells but paradoxically increased Erk and Rb signaling. Failure to suppress Erk signaling suggested that Mek-Erk-independent Raf signaling mechanisms may contribute to survival in this line, possibly through inhibition of direct Rb-Raf interactions or Rb phosphorylation by Raf [13,42]. Consistent with this, SFN has previously been shown to promote apoptosis with Mcl-1 suppression through Mek-Erk-independent mechanisms [11,43–45]. However, the mechanistic basis for the paradoxical increases in pErk and pRb in these cells is unclear. SFN has been shown to dose-dependently activate CRAF through BRAF-CRAF dimerization [46,47]. SFN was also shown to induce CRAF (and downstream Erk activation) in epithelial cells isolated from polycystic livers through a protein kinase A-dependent BRAF-CRAF dimerization mechanism [47]. It is hence plausible that in MDA-MB-468 cells, SFN induces CRAF activation through BRAF heterodimerization leading to enhanced pErk and pRb signaling through Mek-Erk-dependent and –independent pathways, respectively.
FPD, which has been shown to induce apoptosis through Mcl-1 [35,48], enhanced the extent to which Mcl-1 levels were suppressed by SFN in both MDA-MB-231 and MDA-MB-468 cells. FPD also enhanced the durability of suppression of Rb phosphorylation by SFN in MDA-MB-231 cells. The failure of FPD to inhibit Erk suggests that the latter FPD effects are Mek-Erk independent, likely involving direct inhibition of CDKs. Thus, FPD-SFN-potentiating effects on pRb suppression and reduction of Mcl-1 expression correlate with cytotoxic synergy exhibited by this combination at the cellular level. We speculate that concomitant suppression of MekErk-dependent and -independent pathways could underlie cytotoxic synergy exhibited by SFN-FPD combinations. Similar molecular mechanisms may also operate in vivo, where the enhanced antitumor growth effects and reduced pulmonary metastatic burden of this combination correlated with maximal repression of pErk, pRb, and Mcl-1 expression (Figures 6 and 7). These observations are not without precedent. Lapatinib and CDK inhibitors (including FPD) synergized to kill BC cells in vivo and in vitro, and this correlated with inhibition of Mcl-1 expression [35]. In addition, trastuzumab and FPD synergistically inhibited proliferation of HER-2-overexpressing SKBR3 and BT474 cells and inhibited Erk signaling, cyclin D1 expression, and Rb phosphorylation more potently in combination [28]. Importantly, we cannot exclude the involvement of the antiangiogenic properties that have been associated with both SFN and FPD. SFN has been shown to directly target the RTK activity of vascular endothelial growth factor receptors 1 to 3 and PDGFR-β, while FPD has been shown to downregulate vascular endothelial growth factor receptor mRNA and protein levels induced by hypoxia [17,49,50]. Consistent with these observations, we have observed that FPD- and SFN-treated tumors exhibit reduced expression of the angiogenic marker CD31 (PECAM-1). More strikingly, combined FPD and SFN treatment correlated with lower CD31 expression levels than that observed in the presence of either drug alone (Figure W4). Taken together, these data suggest that cooperative inhibition of angiogenesis by FPD and SFN may also contribute to cytotoxic synergy exhibited by this combination.
SFN alone did not inhibit Erk activity in tumors, although marked inhibition was observed in MDA-MB-231 cells in vitro. This is in agreement with a number of studies showing that inhibition of tumor growth by SFN is not always associated with Erk inhibition [51]. For example, SFN inhibited tumor growth in renal and colorectal tumor models but failed to inhibit Erk activity [17,50]. Unaffected pErk levels in response to SFN could reflect failure to detect Erk inhibition due to inappropriate sampling time or to failure to achieve sufficient inhibitor levels in microenvironmental and cell compartments. Alternatively, Erk-independent antitumor effects may be attributable to SFN's antiangiogenic function [51] or to Mek-Erk-independent antiproliferative/apoptotic Rb signaling [13,52]; this is supported by reduced pRb levels in SFN-treated MDA-MB-231 cells and tumors (Figure 7C). Lastly, FPD (and SFN) alone suppressed Mcl-1 expression in MDA-MB-231 cells but failed to inhibit its expression in tumors. We propose that similar effects as those proposed for pErk apply to failed Mcl-1 suppression in tumors; these include pharmacodynamic factors, as well as the antiangiogenic function ascribed to FPD.
In summary, our data show that FPD-SFN combinations exhibit cytotoxic synergy in EGFR/HER-2 and mutant RAS/RAF BC model systems. This correlates with enhanced tumor growth inhibition at the orthotopic site and with reduced pulmonary metastatic burden. These preclinical data support possible antitumor applications of FPD-SFN modalities in adjuvant and advanced settings of BC therapy. The potential therapeutic benefits of adding FPD to current SFN treatment modalities in BC are suggested by recent successes of SFN in BC trials. SFN has been shown to prolong progression-free survival of advanced or metastatic HER-2-negative BC patients previously treated with anthracyclines and/or taxanes (SOLTI-0701; [12]). SFN has also been shown to increase survival of HER-2-negative patients with disease progression during or after bevacizumab therapy [53]. Trastuzumab is currently the main targeted treatment for HER-2-positive cases and lapatinib for advanced/metastatic cases. We have observed that FPDSFN combinations exhibit synergy in the HER-2–overexpressing cell line SKBR3, supporting the use of SFN as a potential alternative to trastuzumab and lapatinib in combination with FPD (discussed above). In this context, SFN may provide potential improvements in therapeutic efficacy over HER-2 inhibitors because it directly targets RAF, a central regulatory nexus in RTK-mediated proliferative and survival signaling. Unlike HER-2 inhibitors, SFN provides the added benefit of enhancing FPD-associated antiangiogenic effects (Figure W4). Alternatively, FPD-SFN combination therapy could also be employed as antiresistance strategies in cases refractory to trastuzumab or lapatinib. Lastly, TN BC, the most aggressive BC subtype, is characterized by a lack of estrogen, progesterone, and HER-2 receptor expression and with EGFR overexpression [7,54]. The observed synergy in EGFR-overexpressing MDA-MB-468 and in TN MDA-MB-231 cell/tumor models supports the use of this combination in TN BC. Thus, FPD-SFN combination modalities may be suitable for treating HER-2-positive BC, as well as aggressive HER-2-negative BCs exhibiting EGFR or TN molecular signatures.
Supplementary Material
Acknowledgments
We thank Christine Hall and Changnian Shi for their technical assistance.
Abbreviations
- BC
breast cancer
- CDKs
cyclin-dependent kinases
- CI
combination index
- EGFR
epidermal growth factor receptor
- FFPE
formalin fixed and paraffin embedded
- FISH
fluorescence in situ hybridization
- FPD
flavopiridol
- HER-2
ErbB2/epidermal growth factor receptor 2
- IB
immunoblot
- MDE
multiple drug effect
- MTD
maximal tolerable dose
- PI3K
phosphatidylinositol-3-kinase
- PLC
phospholipase C
- Rb
retinoblastoma
- RTK
receptor tyrosine kinase
- RTK-P
receptor tyrosine kinase pathway
- SFN
sorafenib
- TN
triple-negative
Footnotes
This work was supported by operating grants from the Canadian Institutes of Health Research (219806 and 219762), Canadian Cancer Society Research Institute (700866), and Canadian Breast Cancer Foundation (369404). T.S.N. received scholarship from the Terry Fox Foundation training program in transdisciplinary cancer research in partnership with Canadian Institutes of Health Research. The authors declare no conflict of interest.
This article refers to supplementary materials, which are designated by Figures W1 to W4 and are available online at www.neoplasia.com.
References
- 1.Rusnak DW, Alligood KJ, Mullin RJ, Spehar GM, Arenas-Elliott C, Martin AM, Degenhardt Y, Rudolph SK, Haws TF, Jr, Hudson-Curtis BL, et al. Assessment of epidermal growth factor receptor (EGFR, ErbB1) and HER2 (ErbB2) protein expression levels and response to lapatinib (Tykerb, GW572016) in an expanded panel of human normal and tumour cell lines. Cell Prolif. 2007;40:580–594. doi: 10.1111/j.1365-2184.2007.00455.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Pegram MD, Konecny GE, O'Callaghan C, Beryt M, Pietras R, Slamon DJ. Rational combinations of trastuzumab with chemotherapeutic drugs used in the treatment of breast cancer. J Natl Cancer Inst. 2004;96:739–749. doi: 10.1093/jnci/djh131. [DOI] [PubMed] [Google Scholar]
- 3.Slamon DJ, Godolphin W, Jones LA, Holt JA, Wong SG, Keith DE, Levin WJ, Stuart SG, Udove J, Ullrich A, et al. Studies of the HER-2/neu protooncogene in human breast and ovarian cancer. Science. 1989;244:707–712. doi: 10.1126/science.2470152. [DOI] [PubMed] [Google Scholar]
- 4.Janne PA, Gray N, Settleman J. Factors underlying sensitivity of cancers to small-molecule kinase inhibitors. Nat Rev Drug Discov. 2009;8:709–723. doi: 10.1038/nrd2871. [DOI] [PubMed] [Google Scholar]
- 5.Gerlinger M, Rowan AJ, Horswell S, Larkin J, Endesfelder D, Gronroos E, Martinez P, Matthews N, Stewart A, Tarpey P, et al. Intratumor heterogeneity and branched evolution revealed by multiregion sequencing. NEngl J Med. 2012;366:883–892. doi: 10.1056/NEJMoa1113205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Tsutsui S, Kataoka A, Ohno S, Murakami S, Kinoshita J, Hachitanda Y. Prognostic and predictive value of epidermal growth factor receptor in recurrent breast cancer. Clin Cancer Res. 2002;8:3454–3460. [PubMed] [Google Scholar]
- 7.Ferraro DA, Gaborit N, Maron R, Cohen-Dvashi H, Porat Z, Pareja F, Lavi S, Lindzen M, Ben-Chetrit N, Sela M, et al. Inhibition of triple-negative breast cancer models by combinations of antibodies to EGFR. Proc Natl Acad Sci USA. 2013;110:1815–1820. doi: 10.1073/pnas.1220763110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Roberts PJ, Der CJ. Targeting the Raf-MEK-ERK mitogen-activated protein kinase cascade for the treatment of cancer. Oncogene. 2007;26:3291–3310. doi: 10.1038/sj.onc.1210422. [DOI] [PubMed] [Google Scholar]
- 9.Berns K, Horlings HM, Hennessy BT, Madiredjo M, Hijmans EM, Beelen K, Linn SC, Gonzalez-Angulo AM, Stemke-Hale K, Hauptmann M, et al. A functional genetic approach identifies the PI3K pathway as a major determinant of trastuzumab resistance in breast cancer. Cancer Cell. 2007;12:395–402. doi: 10.1016/j.ccr.2007.08.030. [DOI] [PubMed] [Google Scholar]
- 10.Hindley A, Kolch W. Extracellular signal regulated kinase (ERK)/mitogen activated protein kinase (MAPK)-independent functions of Raf kinases. J Cell Sci. 2002;115:1575–1581. doi: 10.1242/jcs.115.8.1575. [DOI] [PubMed] [Google Scholar]
- 11.Wilhelm SM, Adnane L, Newell P, Villanueva A, Llovet JM, Lynch M. Preclinical overview of sorafenib, a multikinase inhibitor that targets both Raf and VEGF and PDGF receptor tyrosine kinase signaling. Mol Cancer Ther. 2008;7:3129–3140. doi: 10.1158/1535-7163.MCT-08-0013. [DOI] [PubMed] [Google Scholar]
- 12.Baselga J, Segalla JG, Roche H, Del Giglio A, Pinczowski H, Ciruelos EM, Filho SC, Gomez P, Van Eyll B, Bermejo B, et al. Sorafenib in combination with capecitabine: an oral regimen for patients with HER2-negative locally advanced or metastatic breast cancer. J Clin Oncol. 2012;30:1484–1491. doi: 10.1200/JCO.2011.36.7771. [DOI] [PubMed] [Google Scholar]
- 13.Singh S, Johnson J, Chellappan S. Small molecule regulators of RbE2F pathway as modulators of transcription. Biochim Biophys Acta. 2010;1799:788–794. doi: 10.1016/j.bbagrm.2010.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sherr CJ, McCormick F. The RB and p53 pathways in cancer. Cancer Cell. 2002;2:103–112. doi: 10.1016/s1535-6108(02)00102-2. [DOI] [PubMed] [Google Scholar]
- 15.Davis R, Chellappan S. Disrupting the Rb-Raf-1 interaction: a potential therapeutic target for cancer. Drug News Perspect. 2008;21:331–335. doi: 10.1358/dnp.2008.21.6.1246832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sutherland R, Musgrove E. CDK inhibitors as potential breast cancer therapeutics: new evidence for enhanced efficacy in ER+ disease. Breast Cancer Res. 2009;11:112. doi: 10.1186/bcr2454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wilhelm SM, Carter C, Tang L, Wilkie D, McNabola A, Rong H, Chen C, Zhang X, Vincent P, McHugh M, et al. BAY 43-9006 exhibits broad spectrum oral antitumor activity and targets the RAF/MEK/ERK pathway and receptor tyrosine kinases involved in tumor progression and angiogenesis. Cancer Res. 2004;64:7099–7109. doi: 10.1158/0008-5472.CAN-04-1443. [DOI] [PubMed] [Google Scholar]
- 18.Escudier B, Eisen T, Stadler WM, Szczylik C, Oudard S, Staehler M, Negrier S, Chevreau C, Desai AA, Rolland F, et al. Sorafenib for treatment of renal cell carcinoma: final efficacy and safety results of the phase III treatment approaches in renal cancer global evaluation trial. J Clin Oncol. 2009;27:3312–3318. doi: 10.1200/JCO.2008.19.5511. [DOI] [PubMed] [Google Scholar]
- 19.Llovet JM, Ricci S, Mazzaferro V, Hilgard P, Gane E, Blanc JF, de Oliveira AC, Santoro A, Raoul JL, Forner A, et al. Sorafenib in advanced hepatocellular carcinoma. N Engl J Med. 2008;359:378–390. doi: 10.1056/NEJMoa0708857. [DOI] [PubMed] [Google Scholar]
- 20.Senderowicz AM. Flavopiridol: the first cyclin-dependent kinase inhibitor in human clinical trials. Invest New Drugs. 1999;17:313–320. doi: 10.1023/a:1006353008903. [DOI] [PubMed] [Google Scholar]
- 21.Fornier MN, Rathkopf D, Shah M, Patil S, O'Reilly E, Tse AN, Hudis C, Lefkowitz R, Kelsen DP, Schwartz GK. Phase I dose-finding study of weekly docetaxel followed by flavopiridol for patients with advanced solid tumors. Clin Cancer Res. 2007;13:5841–5846. doi: 10.1158/1078-0432.CCR-07-1218. [DOI] [PubMed] [Google Scholar]
- 22.Bible KC, Peethambaram PP, Oberg AL, Maples W, Groteluschen DL, Boente M, Burton JK, Gomez Dahl LC, Tibodeau JD, Isham CR, et al. A phase 2 trial of flavopiridol (Alvocidib) and cisplatin in platin-resistant ovarian and primary peritoneal carcinoma: MC0261. Gynecol Oncol. 2012;127:55–62. doi: 10.1016/j.ygyno.2012.05.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Byth KF, Thomas A, Hughes G, Forder C, McGregor A, Geh C, Oakes S, Green C, Walker M, Newcombe N, et al. AZD5438, a potent oral inhibitor of cyclin-dependent kinases 1, 2, and 9, leads to pharmacodynamic changes and potent antitumor effects in human tumor xenografts. Mol Cancer Ther. 2009;8:1856–1866. doi: 10.1158/1535-7163.MCT-08-0836. [DOI] [PubMed] [Google Scholar]
- 24.Dickson MA, Schwartz GK. Development of cell-cycle inhibitors for cancer therapy. Curr Oncol. 2009;16:36–43. doi: 10.3747/co.v16i2.428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Senderowicz AM. The cell cycle as a target for cancer therapy: basic and clinical findings with the small molecule inhibitors flavopiridol and UCN-01. Oncologist. 2002;7(suppl 3):12–19. doi: 10.1634/theoncologist.7-suppl_3-12. [DOI] [PubMed] [Google Scholar]
- 26.Musgrove EA, Caldon CE, Barraclough J, Stone A, Sutherland RL. Cyclin D as a therapeutic target in cancer. Nat Rev Cancer. 2011;11:558–572. doi: 10.1038/nrc3090. [DOI] [PubMed] [Google Scholar]
- 27.Chou TC. Theoretical basis, experimental design, and computerized simulation of synergism and antagonism in drug combination studies. Pharmacol Rev. 2006;58:621–681. doi: 10.1124/pr.58.3.10. [DOI] [PubMed] [Google Scholar]
- 28.Wu K, Wang C, D'Amico M, Lee RJ, Albanese C, Pestell RG, Mani S. Flavopiridol and trastuzumab synergistically inhibit proliferation of breast cancer cells: association with selective cooperative inhibition of cyclin D1-dependent kinase and Akt signaling pathways. Mol Cancer Ther. 2002;1:695–706. [PubMed] [Google Scholar]
- 29.Shimizu S, Takehara T, Hikita H, Kodama T, Tsunematsu H, Miyagi T, Hosui A, Ishida H, Tatsumi T, Kanto T, et al. Inhibition of autophagy potentiates the antitumor effect of the multikinase inhibitor sorafenib in hepatocellular carcinoma. Int J Cancer. 2012;131:548–557. doi: 10.1002/ijc.26374. [DOI] [PubMed] [Google Scholar]
- 30.Motwani M, Rizzo C, Sirotnak F, She Y, Schwartz G. Flavopiridol enhances the effect of docetaxel in vitro and in vivo in human gastric cancer cells. Mol Cancer Ther. 2003;2:549–555. [PubMed] [Google Scholar]
- 31.Collins T. Image J for microscopy. Biotechniques. 2007;43:25–30. doi: 10.2144/000112517. [DOI] [PubMed] [Google Scholar]
- 32.Albeck J, Mills G, Brugge J. Frequency-modulated pulses of ERK activity transmit quantitative proliferation signals. Mol Cell. 2013;49:249–261. doi: 10.1016/j.molcel.2012.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Knudsen ES, Wang JY. Dual mechanisms for the inhibition of E2F binding to RB by cyclin-dependent kinase-mediated RB phosphorylation. Mol Cell Biol. 1997;17:5771–5783. doi: 10.1128/mcb.17.10.5771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Nahta R, Iglehart JD, Kempkes B, Schmidt EV. Rate-limiting effects of Cyclin D1 in transformation by ErbB2 predicts synergy between herceptin and flavopiridol. Cancer Res. 2002;62:2267–2271. [PubMed] [Google Scholar]
- 35.Mitchell C, Yacoub A, Hossein H, Martin A, Bareford M, Eulitt P, Yang C, Nephew K, Dent P. Inhibition of MCL-1 in breast cancer cells promotes cell death in vitro and in vivo. Cancer Biol Ther. 2010;10:903–917. doi: 10.4161/cbt.10.9.13273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Senderowicz A, Headlee D, Stinson S, Lush R, Kalil N, Villalba L, Hill K, Steinberg S, Figg W, Tompkins A, et al. Phase I trial of continuous infusion flavopiridol, a novel cyclin-dependent kinase inhibitor, in patients with refractory neoplasms. J Clin Oncol. 1998;16:2986–2999. doi: 10.1200/JCO.1998.16.9.2986. [DOI] [PubMed] [Google Scholar]
- 37.Sparreboom A, van Tellingen O, Nooijen W, Beijnen J. Nonlinear pharmacokinetics of paclitaxel in mice results from the pharmaceutical vehicle Cremophor EL. Cancer Res. 1996;56:2112–2115. [PubMed] [Google Scholar]
- 38.Arguello F, Alexander M, Sterry J, Tudor G, Smith E, Kalavar N, Greene J, Koss W, Morgan C, Stinson S, et al. Flavopiridol induces apoptosis of normal lymphoid cells, causes immunosuppression, and has potent antitumor activity in vivo against human leukemia and lymphoma xenografts. Blood. 1998;91:2482–2490. [PubMed] [Google Scholar]
- 39.Drees M, Dengler W, Roth T, Labonte H, Mayo J, Malspeis L, Grever M, Sausville E, Fiebig H. Flavopiridol (L86-8275): selective antitumor activity in vitro and activity in vivo for prostate carcinoma cells. Clin Cancer Res. 1997;3:273–279. [PubMed] [Google Scholar]
- 40.Peng CL, Guo W, Ji T, Ren T, Yang Y, Li DS, Qu HY, Li X, Tang S, Yan TQ, et al. Sorafenib induces growth inhibition and apoptosis in human synovial sarcoma cells via inhibiting the RAF/MEK/ERK signaling pathway. Cancer Biol Ther. 2009;8:1729–1736. doi: 10.4161/cbt.8.18.9208. [DOI] [PubMed] [Google Scholar]
- 41.Booy E, Henson E, Gibson S. Epidermal growth factor regulates Mcl-1 expression through the MAPK-Elk-1 signalling pathway contributing to cell survival in breast cancer. Oncogene. 2011;30:2367–2378. doi: 10.1038/onc.2010.616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Dasgupta P, Sun J, Wang S, Fusaro G, Betts V, Padmanabhan J, Sebti SM, Chellappan SP. Disruption of the Rb-Raf-1 interaction inhibits tumor growth and angiogenesis. Mol Cell Biol. 2004;24:9527–9541. doi: 10.1128/MCB.24.21.9527-9541.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Liu L, Cao Y, Chen C, Zhang X, McNabola A, Wilkie D, Wilhelm S, Lynch M, Carter C. Sorafenib blocks the RAF/MEK/ERK pathway, inhibits tumor angiogenesis, and induces tumor cell apoptosis in hepatocellular carcinoma model PLC/PRF/5. Cancer Res. 2006;66:11851–11858. doi: 10.1158/0008-5472.CAN-06-1377. [DOI] [PubMed] [Google Scholar]
- 44.Ulivi P, Arienti C, Amadori D, Fabbri F, Carloni S, Tesei A, Vannini I, Silvestrini R, Zoli W. Role of RAF/MEK/ERK pathway, p-STAT-3 and Mcl-1 in sorafenib activity in human pancreatic cancer cell lines. J Cell Physiol. 2009;220:214–221. doi: 10.1002/jcp.21753. [DOI] [PubMed] [Google Scholar]
- 45.Augustine CK, Toshimitsu H, Jung SH, Zipfel PA, Yoo JS, Yoshimoto Y, Selim MA, Burchette J, Beasley GM, McMahon N, et al. Sorafenib, a multi-kinase inhibitor, enhances the response of melanoma to regional chemotherapy. Mol Cancer Ther. 2010;9:2090–2101. doi: 10.1158/1535-7163.MCT-10-0073. [DOI] [PubMed] [Google Scholar]
- 46.Arnault JP, Mateus C, Escudier B, Tomasic G, Wechsler J, Hollville E, Soria JC, Malka D, Sarasin A, Larcher M, et al. Skin tumors induced by sorafenib; paradoxic RAS-RAF pathway activation and oncogenic mutations of HRAS, TP53, and TGFBR1. Clin Cancer Res. 2012;18:263–272. doi: 10.1158/1078-0432.CCR-11-1344. [DOI] [PubMed] [Google Scholar]
- 47.Spirli C, Morell CM, Locatelli L, Okolicsanyi S, Ferrero C, Kim AK, Fabris L, Fiorotto R, Strazzabosco M. Cyclic AMP/PKA-dependent paradoxical activation of Raf/MEK/ERK signaling in polycystin-2 defective mice treated with sorafenib. Hepatology. 2012;56:2363–2374. doi: 10.1002/hep.25872. [DOI] [PMC free article] [PubMed] [Google Scholar] [Research Misconduct Found]
- 48.Ma Y, Cress W, Haura E. Flavopiridol-induced apoptosis is mediated through up-regulation of E2F1 and repression of Mcl-1. Mol Cancer Ther. 2003;2:73–81. [PubMed] [Google Scholar]
- 49.Melillo G, Sausville EA, Cloud K, Lahusen T, Varesio L, Senderowicz AM. Flavopiridol, a protein kinase inhibitor, down-regulates hypoxic induction of vascular endothelial growth factor expression in human monocytes. Cancer Res. 1999;59:5433–5437. [PubMed] [Google Scholar]
- 50.Chang YS, Adnane J, Trail PA, Levy J, Henderson A, Xue D, Bortolon E, Ichetovkin M, Chen C, McNabola A, et al. Sorafenib (BAY 43-9006) inhibits tumor growth and vascularization and induces tumor apoptosis and hypoxia in RCC xenograft models. Cancer Chemother Pharmacol. 2007;59:561–574. doi: 10.1007/s00280-006-0393-4. [DOI] [PubMed] [Google Scholar]
- 51.Semrad T, Gandara D, Lara P. Enhancing the clinical activity of sorafenib through dose escalation: rationale and current experience. Ther Adv Med Oncol. 2011;3:95–100. doi: 10.1177/1758834010396117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Nath N, Wang S, Betts V, Knudsen E, Chellappan S. Apoptotic and mitogenic stimuli inactivate Rb by differential utilization of p38 and cyclin-dependent kinases. Oncogene. 2003;22:5986–5994. doi: 10.1038/sj.onc.1206843. [DOI] [PubMed] [Google Scholar]
- 53.Schwartzberg LS, Tauer KW, Hermann RC, Makari-Judson G, Isaacs C, Beck JT, Kaklamani V, Stepanski EJ, Rugo HS, Wang W, et al. Sorafenib or placebo with either gemcitabine or capecitabine in patients with HER-2-negative advanced breast cancer that progressed during or after bevacizumab. Clin Cancer Res. 2013;19:2745–2754. doi: 10.1158/1078-0432.CCR-12-3177. [DOI] [PubMed] [Google Scholar]
- 54.Reis-Filho JS, Tutt AN. Triple negative tumours: a critical review. Histopathology. 2008;52:108–118. doi: 10.1111/j.1365-2559.2007.02889.x. [DOI] [PubMed] [Google Scholar]
- 55.Filmus J, Pollak MN, Cailleau R, Buick RN. MDA-468, a human breast cancer cell line with a high number of epidermal growth factor (EGF) receptors, has an amplified EGF receptor gene and is growth inhibited by EGF. Biochem Biophys Res Commun. 1985;128:898–905. doi: 10.1016/0006-291x(85)90131-7. [DOI] [PubMed] [Google Scholar]
- 56.Kenny PA, Lee GY, Myers CA, Neve RM, Semeiks JR, Spellman PT, Lorenz K, Lee EH, Barcellos-Hoff MH, Petersen OW, et al. The morphologies of breast cancer cell lines in three-dimensional assays correlate with their profiles of gene expression. Mol Oncol. 2007;1:84–96. doi: 10.1016/j.molonc.2007.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.