

Abstract
We sought to identify fibroblast growth factor receptor 2 (FGFR2) kinase domain mutations that confer resistance to the pan-FGFR inhibitor, dovitinib, and explore the mechanism of action of the drug-resistant mutations. We cultured BaF3 cells overexpressing FGFR2 in high concentrations of dovitinib and identified 14 dovitinib-resistant mutations, including the N550K mutation observed in 25% of FGFR2mutant endometrial cancers (ECs). Structural and biochemical in vitro kinase analyses, together with BaF3 proliferation assays, showed that the resistance mutations elevate the intrinsic kinase activity of FGFR2. BaF3 lines were used to assess the ability of each mutation to confer cross-resistance to PD173074 and ponatinib. Unlike PD173074, ponatinib effectively inhibited all the dovitinib-resistant FGFR2 mutants except the V565I gatekeeper mutation, suggesting ponatinib but not dovitinib targets the active conformation of FGFR2 kinase. EC cell lines expressing wild-type FGFR2 were relatively resistant to all inhibitors, whereas EC cell lines expressing mutated FGFR2 showed differential sensitivity. Within the FGFR2mutant cell lines, three of seven showed marked resistance to PD173074 and relative resistance to dovitinib and ponatinib. This suggests that alternative mechanisms distinct from kinase domain mutations are responsible for intrinsic resistance in these three EC lines. Finally, overexpression of FGFR2N550K in JHUEM-2 cells (FGFR2C383R) conferred resistance (about five-fold) to PD173074, providing independent data that FGFR2N550K can be associated with drug resistance. Biochemical in vitro kinase analyses also show that ponatinib is more effective than dovitinib at inhibiting FGFR2N550K. We propose that tumors harboring mutationally activated FGFRs should be treated with FGFR inhibitors that specifically bind the active kinase.
Introduction
Constitutive fibroblast growth factor receptor (FGFR) signaling due to FGFR amplifications, chromosomal translocations, or gain-of-function mutations contributes to the development and progression of multiple cancers (reviewed in [1–3]). Tumor types associated with genetic aberrations in the FGF/FGFR family include lung and breast cancer (FGFR1), gastric cancer and endometrial cancer (EC; FGFR2), bladder cancer and multiple myeloma (FGFR3), and rhabdomyosarcoma (FGFR4). Preclinical in vitro and in vivo studies have indicated that FGFR kinase inhibition in FGFR-dependent tumors is a rational approach to target these cancers. While more selective anti-FGFR inhibitors are entering early clinical development, the most clinically advanced inhibitors are multi-kinase inhibitors, often developed as anti-angiogenic agents. Dovitinib is the multi-kinase inhibitor that has shown the most promising results in multiple FGFR-dependent cancers.
Dovitinib (TKI258, previously CHIR258) is an adenosine triphosphate (ATP)-competitive tyrosine kinase inhibitor (TKI) with activity against FGFR1–4, vascular endothelial growth factor receptors 1 to 3 (VEGFR1–3), PDGFRB, c-KIT, CSF1R, and FLT3 [4]. It has shown preclinical anti-tumor activity in a range of different cancers [5–8] including cancer models characterized by FGFR activation such as multiple myeloma, acute myelogenous leukemia, and prostate, bladder, and gastric cancers [4,9–14]. Dovitinib has demonstrated anti-tumor activity in several phase I clinical trials with partial responses and stable disease observed in several patients [15]. Dovitinib is currently in phase II clinical trials in renal cell carcinoma patients as an anti-angiogenic agent as well as in several malignancies associated with FGFR activation, e.g., multiple myeloma with t(4;14) translocation (activated FGFR3; Clinical Trials identifier: NCT01058434) and advanced urothelial carcinomas with and without mutations in FGFR3 (NCT00790426). It is also in a clinical phase II study in patients with advanced ECs expressing wild-type (WT) or mutant FGFR2 (NCT01379534).
Despite the initial clinical effectiveness of kinase inhibitors, the long-term efficacy of these agents is hampered by intrinsic resistance in a subset of patients and the development of acquired resistance in a proportion of responders. One resistance mechanism common to many kinase inhibitors is the acquisition of secondary mutations in the kinase domain. Mutations of the gatekeeper residue of the target kinase are the most frequently detected drug-resistant mutation in the clinic. Notably, mutation of the gatekeeper residue in Bcr-Abl (T315I) is detected with high frequency in chronic myelogenous leukemia patients with resistance against imatinib [16,17]. Likewise, mutation of the gatekeeper residue (T790M) in the epidermal growth factor receptor (EGFR) occurs in ∼50% of tumors with acquired erlotinib or gefitinib resistance and represents a major obstacle for treatment success with targeted EGFR inhibitors [18–20]. Substitutions of gatekeeper residues with larger hydrophobic residues have been shown to sterically interfere with access of drug to the hydrophobic pocket in the ATP-binding cleft. Bcr-Abl inhibitors have also been shown to form critical hydrogen bonds with the side chain hydroxyl group of T315 [21]. Moreover, the gatekeeper mutations appear to enhance tyrosine kinase activity by stabilizing a hydrophobic spine, a network of hydrophobic interactions characteristic of activated kinases [22]. In chronic myelogenous leukemia, the realization that patients acquire resistance after initial response led to the development of more potent second-generation inhibitors such as nilotinib and dasatinib [23]; however, like imatinib, these inhibitors do not have activity against the T315I gatekeeper mutation. This led to the structure-based design of ponatinib (AP24534), a third-generation inhibitor designed to have activity against WT Bcr-Abl as well as Bcr-Abl-T315I.
Despite the importance of FGFRs as cancer drug targets, little is known about the repertoire of mutations in FGFRs that confer resistance to current FGFR inhibitors. Mutations of the gatekeeper residues in FGFR1 (V561M) and FGFR3 (V555M) have been shown to result in in vitro resistance to the multi-kinase inhibitor PP58 and the FGFR inhibitor AZ12908010, respectively [24], thus indicating that mutation of the gatekeeper residue may be a general mechanism of resistance to receptor tyrosine kinase (RTK) inhibitors.
The BaF3 screening strategy developed by von Bubnoff et al. is considered the “gold standard” method to identify drug-resistant mutations in a variety of RTKs and non-receptor kinases [25,26]. In this method, BaF3 cells are made dependent on the desired RTK, cultured in the presence of an inhibitor against that RTK, and resistant colonies that emerge are screened for drug-resistant mutations. This approach has been successfully used to identify TKI-resistant mutations in Bcr-Abl, FLT3, PDGFRA, MET, EGFR, and JAK2 [27–32] and has effectively reproduced the pattern and relative abundance of Bcr-Abl mutations seen clinically in imatinib-resistant patients [27]. In this study, we used the BaF3 screening strategy to identify FGFR2 mutations that impart resistance to dovitinib and examined the effect of these mutations on FGFR2 kinase activity in vitro and in stable FGFR2-expressing BaF3 cells. We show that the dovitinib-resistant FGFR2 mutations act by stabilizing the active conformation of the kinase. We also examined the ability of these dovitinib-resistant mutations to confer cross-resistance to other FGFR inhibitors including PD173074 and ponatinib. Importantly, we discovered that ponatinib is capable of inhibiting dovitinibresistant gain-of-function mutations indicating that ponatinib may be more effective as a first-line therapy as well as in the second-line setting to target tumors with resistance to dovitinib. Treatment of a panel of FGFR2 mutant EC cell lines with dovitinib and ponatinib revealed different levels of drug sensitivity within cell lines expressing the same FGFR2 mutation, suggesting that other intrinsic mechanisms of resistance may also be present in patient tumors.
Materials and Methods
Cell Lines and Reagents
BaF3 cells were cultured as previously described [33]. The BaF3 cells used in this study were obtained directly from ATCC (Manassas, VA) and were passaged for fewer than 6 months after their receipt, and as such, reauthentication was not performed. The JHUEM-2, MFE280, and MFE296 cell lines were purchased from the RIKEN Cell Bank (Tsukuba, Japan), the DSMZ (Berlin, Germany), and the European Collection of Cell Cultures (Salisbury, United Kingdom), respectively. AN3CA, HEC1A, Ishikawa, and KLE were provided by Dr Paul Goodfellow (Washington University, St Louis, MO). EI, EJ, and EN1078D were provided by Dr Gordon Mills (MD Anderson Cancer Center, Houston, TX). Recombinant murine interleukin-3 (IL-3) and human FGF10 were purchased from R&D Systems (Minneapolis, MN). Dovitinib and ponatinib were purchased from Selleck Chemicals (Houston, TX), and PD173074 was purchased from EMD Chemicals (Gibbstown, NJ). Phospho-FGFR (P-FGFR) antibody was purchased from Cell Signaling Technology (Genesearch Pty Ltd, Arundel, Australia), total FGFR2 (T-FGFR) antibody was purchased from Santa Cruz Biotechnology (Thermo Fisher Scientific Pty Ltd, Scoresby, Australia), α-tubulin antibody was purchased from Sigma-Aldrich (Castle Hill, Australia), and IRDye 800 and IRDye 680LT secondary antibodies were purchased from Rockland (Jomar Biosciences Pty Ltd, Kensington, Australia).
BaF3 Screen for Dovitinib-Resistant FGFR2 Mutations
BaF3 cells were stably transduced with pEF1a.FGFR2b.IRES.neo, pEF1a.FGFR2b.S252W.IRES.neo, or pEF1a.FGFR2b.N550K.IRES. neo plasmid DNA using Amaxa nucleofection and selected for 14 days in 1200 µg/ml G418, as previously reported [34]. Stably selected cells were plated at a density of 1 x 105 and 4 x 105 cells/well in six 96-well plates each in BaF3 growth media without IL-3, supplemented with 1 nM FGF10 and 5 µg/ml heparan sulfate. Dovitinib was added to duplicate plates of each cell density at 5, 10, or 15x the inhibitory concentration 50 (IC50) (100, 200, and 300 nM, respectively, for FGFR2b- and S252W-expressing cells, and 2000, 4000, and 6000 nM, respectively, for N550K-expressing cells). Fresh FGF10 and heparan sulfate were added every 2 to 3 days. Colonies that grew out were expanded in media with FGF10 and heparan sulfate, and genomic DNA was extracted using the GenElute Mammalian Genomic DNA Miniprep Kit (Sigma-Aldrich, St Louis, MO). Inserted human FGFR2b was amplified using overlapping primer pairs (5′-ATGCCCAGCCCCACATCCAG-3′ and 5′-GACTGGAAGCCGCCCATTGGTG-3′; 5′-CCCAAGGAGGCGGTCACCG-3′ and 5′-GGCATGGTCTCCCTGCTCAGTG-3′) and sequenced in two directions for mutations in the intracellular domain of FGFR2b (sequencing primers available upon request). Mutations were confirmed in an independent polymerase chain reaction. Amino acid substitutions are listed according to isoform 2 of human FGFR2 (FGFR2b; NP_075259.4).
Site-directed Mutagenesis
Each putative dovitinib-resistant mutation was introduced into full-length FGFR2b by site-directed mutagenesis (SDM), as previously described [35]. Briefly, SDM was performed on 50 ng of pEF1a.FGFR2b.IRES.neo plasmid DNA using the QuikChange II XL Site-Directed Mutagenesis Kit (Agilent Technologies, Santa Clara, CA). SDM primers were designed to introduce the desired dovitinib-resistant mutation as well as a silent mutation to introduce a restriction site for ease in screening (primers available upon request). Plasmid DNA was isolated using the Plasmid DNA Miniprep Kit (Qiagen, Valencia, CA), and diagnostic restriction digests were performed. Plasmid DNA was then isolated from SDM-positive clones using the Qiagen EndoFree Plasmid MaxiPrep Kit (Qiagen). Mutations were confirmed by sequencing of the entire coding region of FGFR2b (primers available upon request).
Generation of BaF3 Cells Stably Expressing Dovitinib-Resistant FGFR2b Mutations
pEF1a.FGFR2b.IRES.neo or the various FGFR2b mutant plasmids were introduced into BaF3 cells using the Amaxa nucleofector kit V, according to the manufacturer's instructions (Amaxa, Walkersville, MD). Cells were selected in growth media containing 1200 µg/ml G418 and 5 ng/ml IL-3 for 14 days and frozen down. Proliferation assays in the presence or absence of drug were performed in BaF3 cells that had not been passaged for more than 5 weeks after this initial freeze.
Generation of JHUEM-2 Cells Stably Expressing WT and Mutant FGFR2b
JHUEM-2 cells were infected with lentiviral particles containing pEF1α.FGFR2b.IRES.neo plasmids encoding WT FGFR2b, FGFR2bY376C, or FGFR2bN550K. A JHUEM-2 line was also infected with an empty pEF1α.IRES.neo vector as a control. Cells were then selected in growth media containing 900 µg/ml G418 for 14 days and frozen down.
IC50 Analysis
BaF3 cells expressing WT or mutant FGFR2b were plated at either 3000 or 10,000 cells per well in 96-well plates in BaF3 media without IL-3, supplemented with 1 nM FGF10 and 5 µg/ml heparan sulfate. Dovitinib and PD173074 were added at half-log dilutions ranging from 10 µM to 3 nM, while ponatinib was added at half-log dilutions ranging from 1 µM to 0.1 nM, respectively. After 72 hours, cell viability was measured using the ViaLight Kit (Lonza, Walkersville, MD). Values were normalized to DMSO vehicle control wells, and IC50 values were generated by nonlinear regression analysis with variable slope using Prism software version 4.0c (GraphPad Software, San Diego, CA). For the ponatinib experiments, 3000 cells per well were seeded and assayed in triplicate on two independent days. As biologic replicate data for dovitinib and PD173074 had been generated with 10,000 cells per well, these assays were repeated a third time with 3000 cells per well with no significant differences observed and the presented IC50 values are the replicates of these three independent experiments.
Parental EC cell lines and stably transfected JHUEM-2 cells were seeded at 3000 cells per well in 96-well plates in their individual growth media. After 24 hours, dovitinib, PD173074, and ponatinib were added at half-log dilutions (1 nM to 10 µM). Following 72 hours of drug treatment, cell viability was assessed using the CyQUANT Cell Proliferation Assay Kit (Life Technologies, Carlsbad, CA). Values were normalized to DMSO vehicle control wells, and IC50 values were calculated as described above. Proliferation assays were performed in triplicate on two independent days and the results were averaged.
Receptor Phosphorylation in Response to Ligand Treatment
BaF3 cells expressing WT or mutant FGFR2 were washed twice in media minus IL-3. Cells were then resuspended in 200 µl of BaF3 media minus IL-3 containing 5 µg/ml heparan sulfate and 16 nM FGF10 for 7.5 minutes. Cells were centrifuged at 1000 rpm for 5 minutes, the supernatant discarded, and the cell pellet resuspended in 200 µl of lysis buffer [1% Triton X-100, 50 mM Tris-HCl (pH 7.4), 150 mM NaCl, 2 mM Na3VO4, and 10 mM NaF]. The protein concentration was determined using a Bio-Rad Quick Start Kit. A total of 150 µg of protein was subjected to sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) on a 4% to 12% bisacrylamide gradient gel, transferred to a nitrocellulose membrane, blocked with odyssey blocking buffer, and incubated with the primary antibody diluted in Odyssey blocking buffer overnight at 4°C. The membranes were washed with TBS-T and incubated with the secondary antibody diluted in Odyssey blocking buffer for 1 hour at room temperature. After another washing step, the membrane was scanned using a Lycor flatbed scanner.
Inhibition of Receptor Phosphorylation in Response to Ligand
BaF3 cells expressing WT or mutant FGFR2 were grown in T75 flasks in 50 ml of BaF3 media. Cells were washed twice with IL-3-free media, resuspended in 35 ml of IL-3-free media, and the cells evenly split into seven T25 flasks. An FGFR inhibitor was added to final concentrations of 1, 10, 30, 100, 300, 1000 nM or DMSO control corresponding to the highest FGFR inhibitor concentration (0.01% vol/vol). Cells were incubated with the inhibitors for 90 minutes at 37°C and pelleted at 1000 rpm, and the pellet was resuspended in BaF3 media minus IL-3 containing 5 µg/ml heparan sulfate and 16 nM FGF10 for 7.5 minutes. After the incubation period, cells were centrifuged at 1000 rpm for 5 minutes, the supernatant discarded, and the cells resuspended in 200 µl of lysis buffer. The cell lysates were processed and subjected to SDS-PAGE as described above.
Structural Modeling
Binding of dovitinib to FGFR2 kinase was modeled by superimposing the structure of the A-loop phosphorylated activated WT FGFR2 kinase domain (PDB ID: 2PVF) [36] onto the structure of the CHK-1 kinase domain in complex with the inhibitor 4-(aminoalkylamino)3-benzimidazole-quinolinones complex structure (PDB ID: 2GDO) [37]. Similarly, binding of ponatinib to FGFR2 kinase was modeled by superimposing the structure of the A-loop phosphorylated activated WT FGFR2 kinase domain (PDB ID: 2PVF) [36] onto the structure of the ABL kinase in complex with ponatinib (PDB ID: 3OY3) [38]. Modeling of the pathogenic mutations was performed and analyzed using O [39]. Atomic superimpositions were performed using program lsqkab [40] in CCP4 Suite [41] and structural representations were prepared using PyMol [42].
Protein Expression and Purification
The cDNA fragment encoding residues P459 to E769 of human FGFR2c (Accession code: NP_075259) was amplified by polymerase chain reaction and subcloned into pET bacterial expression vector with an NH2 terminal 6x His tag to aid in protein purification. Point mutations (M536I, M538I, I548V, N550H, N550K, N550S, V565I, E566G, L618M, and K660E) were introduced using QuikChange Site-Directed Mutagenesis Kit (Stratagene, La Jolla, CA). The bacterial strain BL21 (DE3) cells were transformed with the expression constructs, and kinase expression was induced with 1 mM isopropyl-L-thio-BDgalactopyranoside overnight at the appropriate temperature. The cells were lysed, and the soluble kinase proteins were purified according to the published protocol [36]. N-terminally His-tagged substrate peptide consisting of residues L762 to T822 of FGFR2 was expressed and purified similar to the kinase domain. The substrate peptide corresponds to the C-terminal tail of FGFR2 and contains five authentic tyrosine phosphorylation sites (Y770, Y780, Y784, Y806, and Y813).
Kinase Assay
WT and mutated FGFR2 kinases were mixed with reaction solutions containing ATP, MgCl2, and the substrate peptide. The final concentrations of the reaction mix are 0.5 mg/ml kinase, 2.17 mg/ml substrate, 10 mM ATP, and 20 mM MgCl2. The reactions were quenched at different time points by adding 100 mM EDTA. The progress of the substrate phosphorylation was followed by native PAGE, and tyrosine phosphorylation content of the substrate peptide was quantified by time-resolved matrix-assisted laser desorption/ionization time-of-flight (MALDI-TOF) mass spectrometry using a Bruker Autoflex mass spectrometer operated in linear mode according to the published protocol by comparing signals from phosphorylated and the cognate nonphosphorylated peptides [43].
In Vitro Kinase Inhibition Assay
WT FGFR2 kinase and the N550H and V565I mutants were incubated for 5 minutes with reaction solutions containing ATP, MgCl2, and increasing concentrations of either dovitinib or ponatinib. The final concentrations of kinase, ATP, and MgCl2 were 90 µM, 5.33 mM, and 10.66 mM, respectively. The molar ratios of kinase/inhibitor in the reaction mix were 1:0, 1:0.2, 1:0.5, 1:1, 1:2, 1:5, or 1:10. The reactions were quenched by adding EDTA to a final concentration of 69.6 mM, and the progress of the kinase autophosphorylation/inhibition was monitored by native PAGE.
Results
BaF3 Screen for Dovitinib-Resistant FGFR2 Mutations
BaF3 cells stably expressing the “b” splice isoform of FGFR2 (FGFR2b, NM_022970) were treated with 100, 200, and 300 nM dovitinib corresponding to concentrations that are 5, 10, and 15 times the IC50 value of dovitinib in this cell line. In the presence of 100 nM dovitinib, 73 of 384 wells grew out, corresponding to a resistant clone frequency of 0.76 per million cells. Nineteen of 384 wells and 9 of 384 wells grew out in the 200 and 300 nM dovitinib-treated groups, respectively, resulting in a resistant clone frequency of 0.20 and 0.09 per million cells. Sequencing of the intracellular domain of FGFR2 from 63 of the dovitinib-resistant BaF3.FGFR2 clones led to the identification of mutations in 26 resistant clones (41%). The mutation frequency increased with dovitinib concentration, with 7 of 35 (20%), 13 of 19 (68%), and 6 of 9 (67%) resistant clones containing an FGFR2 mutation for clones selected at 100, 200, and 300 nM, respectively (Table W1).
Eleven different FGFR2 mutations, affecting nine amino acids, were detected (Figure 1A). Ten mutations map into the kinase domain (M536I, M538I, I548V, N550H/K/S, V565I, E566G, L618M, and E719G), whereas the Y770fsX14 localizes to the C-terminal tail past the kinase domain (Figure 1A). The most commonly mutated codon was N550, with mutations accounting for 73% (19 of 26) of FGFR2 mutations. One resistant clone harbored two mutations, N550H and E719G. Mutation at the gatekeeper residue (V565I) was identified in one clone.
Figure 1.
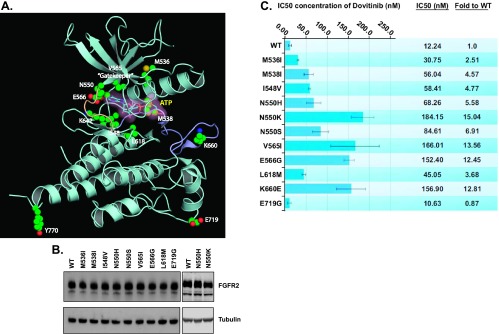
Identified FGFR2 mutations confer dovitinib resistance in stable BaF3.FGFR2 cells. (A) The ribbon diagram of the A-loop phosphorylated activated WT FGFR2 kinase structure (PDB ID: 2PVF) [36] showing the locations of the drug-resistant mutations. The A-loop and kinase hinge are colored light purple and light orange, respectively. The drug-resistant mutations are rendered as ball and stick. The AMP-PCP (the ATP analog) is shown in both ball-and-stick representation and a semitransparent surface. (B) Western blot analysis of stable BaF3 cells expressing WT or mutant FGFR2 using an anti-FGFR2 antibody (BEK-C17) or anti-tubulin antibody as loading control. (C) Stable BaF3-FGFR2 cells were seeded in 96-well plates in media containing 5 µg/ml heparan sulfate and 1 nM FGF10, and dovitinib was added in concentrations ranging from 3 nM to 10 µM. The cells were incubated at 37°C for 72 hours and proliferation was measured using the ViaLight proliferation kit and the IC50 was calculated using GraphPad Prism.
We also performed parallel dovitinib resistance screens using BaF3 cells expressing either of the FGFR2-activating mutations, S252W or N550K. The S252W mutation is the most common FGFR2 mutation seen in endometrial tumors and maps to the extracellular ligandbinding region of FGFR2. Structural and biochemical studies have shown that this mutation results in ligand-dependent receptor activation by introducing additional contacts between FGFR and FGF ligand [44], and as such, we did not expect a different pattern of resistance mutations. N550K is the second most common FGFR2 mutation identified in endometrioid EC [45] and we show herein that this mutation activates the kinase. BaF3 cells expressing S252W mutant FGFR2 show similar dovitinib sensitivity to BaF3 cells expressing WT FGFR2 (Figure W1) and were thus treated in a similar manner with 100, 200, or 300 nM dovitinib. Resistant clones grew out in 51 of 384 wells and the FGFR2 kinase domain was sequenced in 35 resistant clones that grew out at the two highest dovitinib concentrations. FGFR2 mutations were identified in four clones affecting three amino acids: N550T, E566A (two independent clones), and K642N. Although we observed a reduced mutation rate in the resistant BaF3.FGFR2 S252W BaF3 clones, the presence of the S252W mutation did not dramatically alter the spectrum of dovitinib-resistant mutations identified, as two of these codons (N550, E566) were also mutated in the WT FGFR2 BaF3 screen. Moreover, all three mutations were confirmed to confer resistance to dovitinib when expressed in conjunction with the activating S252W mutation in proliferation assays (Figure W1). For the N550K resistance screen, BaF3 cells expressing N550K mutant FGFR2 were treated with 2, 4, or 6 µM dovitinib, corresponding to 5, 10, and 15 times the IC50, because, as noted above, N550K already imparts significant resistance to dovitinib in isolation. No resistant clones were isolated after dovitinib treatment in the N550K resistance screen.
Reintroduction of the Mutations Confirmed the Identified Mutations Induce Dovitinib Resistance
To confirm that the mutations identified in the BaF3 screen were sufficient to confer dovitinib resistance, independent BaF3 cell lines stably expressing FGFR2 harboring the putative drug-resistant mutations identified in the initial BaF3 screen were generated (Figure 1B). As the Y770fsX14 C-terminal deletion mapped away from the ATP-binding site and could not be identified with the C-terminal antibody we used, this mutation was not assessed. The sensitivity of these stable cell lines to dovitinib was then measured by assessing cell viability at increasing dovitinib concentrations (Figure 1C). As the activating N550K mutation was identified in the resistance screen, we also assessed the sensitivity of the other major activating mutation seen in patients, K660E. All mutations with the exception of E719G led to drug resistance as manifested by 2.11 to 15.04-fold increases in IC50 value. The N550K, V565I, K660E, and E566G mutations imparted the greatest magnitude of resistance (Figure 1C). Dovitinib sensitivity of BaF3 cells expressing the E719G mutant FGFR2 was not significantly different than that of those expressing WT FGFR2. The clone where the E719G mutation was identified also harbored an N550H mutation in FGFR2, so presumably the latter N550H mutation conveyed resistance in this clone and the E719G mutation represents a passenger mutation.
Dovitinib-resistant Mutations at N550 and E566 Disrupt the Molecular Brake to Activate the FGFR2 Kinase
To understand the molecular mechanisms by which these mutations confer resistance to dovitinib, a structural model of FGFR2 kinase bound to dovitinib was created by superimposing the structure of the A-loop phosphorylated activated WT FGFR2 kinase domain (PDB ID: 2PVF) [36] onto the structure of the CHK-1 kinase domain in complex with the inhibitor 4-(aminoalkylamino)-3-benzimidazolequinolinones complex structure (PDB ID: 2GDO) [37]. Structural analysis shows that the V565I gatekeeper mutation could confer drug resistance by sterically hindering access of the drug into the hydrophobic rear corner of the ATP-binding cleft of the kinase (Figure 2, A and B). In contrast, the remaining mutations are unlikely to cause drug resistance by sterically interfering with drug binding. Instead, these mutations appear to impart drug resistance by stabilizing the active conformation of the FGFR2 kinase. Specifically, several of the dovitinib-resistant mutations identified in the original and FGFR2 S252W screen (N550H/K/S/T, E566G/A, K642N) target the triad of residues (N550, E566, and K642) that form an autoinhibitory network of hydrogen bonds, termed the molecular brake, at the kinase hinge region. This molecular brake restricts the motion of the kinase into the active state [36]. Mutation of these residues have been shown to disengage this brake, permitting the kinase to more readily adopt the active conformation (Figure 2, C and D) [36]. These data suggest that the N550H/K/S/T, E566G/A, and K642N dovitinibresistant mutations favor the active conformation of the kinase (Figure 2, E and F), implying that dovitinib does not act on the active kinase conformation.
Figure 2.

The molecular mechanisms by which mutations confer resistance to dovitinib. (A and B) Gatekeeper V565I mutation confers resistance to dovitinib through steric hindrance. Binding of dovitinib [in yellow, partial, taken from the CHK-1 and inhibitor 4-(aminoalkylamino)-3-benzimidazole-quinolinones complex structure, PDB ID: 2GDO] [37] is modeled onto the A-loop phosphorylated activated WT FGFR2 kinase structure (PDB ID: 2PVF) [36]. Hydrogen bonds between the kinase and dovitinib are colored yellow. The molecular surface of dovitinib is shown to emphasize the steric clash with the mutated I565. (C–F) Mutations at N550 and E566 confer dovitinib resistance by activating the kinase through disengagement of the molecular brake. The molecular brake is engaged at the kinase hinge region in the unphosphorylated unactivated WT FGFR2 kinase (C; PDB ID: 2PSQ) [36] and is disengaged by A-loop tyrosine phosphorylation (D; PDB ID: 2PVF) [36] or by mutations at N550 (E; PDB ID: 2PWL) [36] and E566 (F; PDB ID: 2PY3) [36]. (G) Some mutations confer dovitinib resistance by activating the kinase through strengthening the hydrophobic spine of the FGFR2 kinase. The hydrophobic spine is shown as a semitransparent surface. Residues comprising the hydrophobic spine are rendered as sticks. The drug-resistant mutations targeting residues V565, M536, M538, I548, and L618 are colored and labeled green, and the others are labeled white.
Several of the dovitinib-resistant mutations, namely, M536I, M538I, I548V, and L618M, appear to stabilize the active kinase conformation by strengthening the hydrophobic spine of the FGFR2 kinase (Figure 2G). Furthermore, the gatekeeper residue is positioned at the top corner of the hydrophobic spine and its mutation to bulkier hydrophobic residues has been also proposed to activate the kinase through fortifying the hydrophobic spine. Together, these data suggest that dovitinib-resistant mutations act by stabilizing the active kinase conformation either through disengaging the molecular brake or strengthening the hydrophobic spine. The V565I gatekeeper mutation can additionally confer drug resistance through steric hindrance.
Dovitinib-resistant Mutations Elevate the Intrinsic Kinase Activity of FGFR2
To test our structural prediction that the dovitinib-resistant mutations confer resistance by stabilizing the active conformation of the kinase, we decided to study the effect of the drug-resistant mutations on the intrinsic kinase activity of FGFR2 kinase. Recombinant WT and mutated kinase domains harboring the drug-resistant mutations were expressed and purified to homogeneity, and their substrate phosphorylation activities were compared using native PAGE coupled with time-resolved mass spectrometry. Briefly, WT or drug-resistant mutant FGFR2 kinases were incubated with peptide substrate in the presence of ATP and MgCl2. The C-terminal tail of the FGFR2 kinase, which contains five authentic phosphorylation sites, served as the substrate. Phosphorylation reactions were quenched at different time points with the addition of EDTA. The formation of phosphorylated species was monitored by native PAGE and analyzed by mass spectrometry and the percentage of at least one site phosphorylation on the substrate was quantitated using the peak intensity data generated by mass spectrometry (Figures 3A and W2). Compared to WT kinase, the drug-resistant mutant FGFR2 kinases exhibited increased ability to phosphorylate the substrate, demonstrating that the dovitinib-resistant mutations elevate the intrinsic activity of the enzyme.
Figure 3.
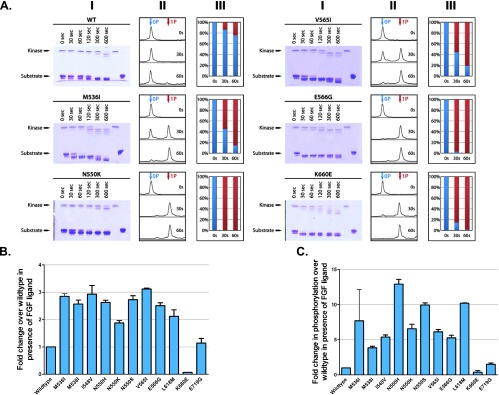
Dovitinib-resistant mutations increase the intrinsic kinase activity of FGFR2. (A) The substrate phosphorylation activities of WT and mutated FGFR2 kinase domain harboring the drug-resistant mutations were compared using native PAGE (panel I) coupled with time-resolved mass spectrometry (MS; panels II and III). For accuracy, only the early time point (30- and 60-second) MS data, which are in the linear phase of the kinase assay, were processed. The percentage of at least one site phosphorylation on the substrate (panel III) was estimated using the peak intensity data generated by mass spectrometry. (B) Stable BaF3.FGFR2 cells were seeded at 10,000 cells/well in 96-well plates in media containing 5 µg/ml heparan sulfate and 1 nM FGF10. The cells were incubated at 37°C for 72 hours and proliferation was measured using the ViaLight proliferation kit. The increase in proliferation compared to FGFR2 WT cells is presented. (C) Stable BaF3 cells expressing WT or mutant FGFR2 were stimulated for 7.5 minutes with media containing 5 µg/ml heparan sulfate and 16 nM FGF10. After the stimulation period, the cells were lysed in lysis buffer containing phosphatase inhibitors, and 150 µg of total protein was separated on an SDS-PAGE, transferred to a nitrocellulose membrane, and probed with an anti-pan-phospho-FGFR, anti-FGFR2 (BEK-C17), and anti-tubulin antibodies. The ratio of phospho to total FGFR2 was calculated by densitometry using Odyssey 3.0 software.
Dovitinib-resistant Mutations Result in Ligand-Dependent Receptor Activation In Vitro
To validate our in vitro data, we next tested the ability of the drug-resistant mutations to elevate the kinase activity of full-length receptor by measuring ligand-independent and ligand-dependent proliferation of the BaF3 cell lines expressing the drug-resistant FGFR2b mutants. None of the dovitinib-resistant mutations was sufficient to significantly drive FGF-independent cell survival and proliferation (Figure W3A). These data may seem conflicting with the in vitro data showing these mutations have constitutive activity; however, comparative data suggest that FGFR2 is not able to drive IL-3-independent BaF3 proliferation in the same way as FGFR1, perhaps reflecting its reduced overall kinase activity. Specifically, BaF3 cells expressing FGFR1c N546K demonstrate significant proliferation compared to WT in the absence of ligand in contrast to the homologous N550K mutation in FGFR2c that does not (Figure W3B). Nevertheless, in the presence of FGF10 (a well-known cognate ligand of FGFR2b), BaF3 cell lines expressing each of the dovitinib-resistant mutants displayed increased proliferation compared to cells expressing WT FGFR2 (Figure 3B), supporting the in vitro findings that the dovitinib-resistant mutations increase the tyrosine kinase activity of full-length FGFR2b.
To further corroborate our findings, cell lines expressing drug-resistant FGFR2b mutants were incubated with heparan sulfate and FGF10 for 7.5 minutes, and the receptor phosphorylation was assessed by Western blot analysis using a phospho-FGFR antibody. Densitometric analysis of biologic replicate experiments shows that the drug-resistant FGFR2 mutants exhibited a five-fold to six-fold increase in autophosphorylation compared to the WT FGFR2 (Figure 3C). No increase in BaF3 proliferation or receptor phosphorylation in response to FGF10 was seen in the BaF3 cells transduced with FGFR2 K660E, although this mutated receptor shows strong constitutive activity in the absence of ligand (Figure W3B). This is consistent with mislocalization of this activating mutant to the endoplasmic reticulum (ER)/Golgi, similar to what has been reported previously for the K650E mutation in FGFR3 [46,47]. Taken together with the in vitro kinase assay data, these cell-based data demonstrate that the dovitinib-resistant mutations increase the tyrosine kinase activity of FGFR2.
Mutations Cause Cross-Resistance to PD173074 but Not to Ponatinib
To examine whether the identified dovitinib-resistant FGFR2 mutations can also confer resistance to other FGFR inhibitors, ligand-induced proliferation of BaF3 cells expressing the drug-resistant FGFR2 was measured in the presence of PD173074 and ponatinib. As shown in Figure 4A, the dovitinib-resistant mutations also imparted resistance to PD173074. As with dovitinib, the N550K molecular brake region mutation and the V565I gatekeeper mutation also caused greatest resistance toward PD173074. Interestingly, the N550K mutation provided considerably more resistance than N550H and N550S, perhaps indicating that the conformation of N550K provides resistance through another mechanism, in addition to loss of the molecular brake. In contrast, ponatinib effectively inhibited all the dovitinib-resistant FGFR2 mutants with the exception of the V565I gatekeeper mutant (Figure 4B).
Figure 4.

Dovitinib resistance mutations are similarly resistant to PD173074 but are almost all sensitive to ponatinib. (A) Stable BaF3-FGFR2 cells were seeded in 96-well plates in media containing 5 µg/ml heparan sulfate and 1 nM FGF10, and PD173074 was added in concentrations ranging from 3 nM to 10 µM. The cells were incubated at 37°C for 72 hours and proliferation was measured using the ViaLight proliferation kit and the IC50 was calculated using Prism (GraphPad Software). (B) Cells were treated as in A, but 0.1 nM to 1 µM ponatinib was added to each stable cell line.
To further explore the differential sensitivity of ponatinib to the dovitinib-resistant mutations, BaF3 cell lines expressing WT FGFR2b or N550K and V565I drug-resistant FGFR2b mutants were incubated with dovitinib or ponatinib followed by FGF10 ligand stimulation and the phosphorylation of FGFR2 was examined. Treatment with dovitinib reduced receptor phosphorylation in BaF3.FGFR2 WT cells to ∼50% at a concentration of 52.1 nM (Figure 5, A and B). In contrast, the concentration of dovitinib required to reduce receptor phosphorylation to ∼50% in BaF3.FGFR2 N550K and BaF3.FGFR2 V565I cells was 794 and 954 nM, respectively. Ponatinib inhibited phosphorylation of WT FGFR2b with an IC50 of 30.73 nM that is comparable to that of dovitinib. In stark contrast to dovitinib, ponatinib was highly effective in inhibiting the N550K FGFR2 mutant (IC50 of 5.72 nM), demonstrating the sensitivity of this FGFR2 mutant to ponatinib. Notably, the V565I gatekeeper mutant was still refractory to inhibition by ponatinib (IC50 of 661 nM), emphasizing the potency of this mutation to confer resistance to all three FGFR inhibitors.
Figure 5.

Change in FGFR2 phosphorylation in response to treatment with dovitinib and ponatinib. (A) Stable BaF3.FGFR2 cells were pretreated in IL-3-free BaF3 media for 90 minutes at 37°C. After the 90-minute incubation period, the media were removed and the cells were incubated for 7.5 minutes at 37°C with media containing 5 µg/ml heparan sulfate and 16 nM FGF10. Cells were lysed in lysis buffer containing phosphatase inhibitors and 150 µg of total protein was separated on an SDS-PAGE, transferred to a nitrocellulose membrane, and probed using an anti-pan-phospho-FGFR, anti-FGFR2 (BEK-C17), and anti-tubulin antibodies. (B) Densitometry analysis of the change in phosphorylation due to pretreatment with dovitinib and ponatinib. The ratio of phospho to total FGFR2 was calculated by densitometry using Odyssey 3.0 software and the concentration required to decrease the phosphorylation to 50% compared to WT.
EC Cell Lines Expressing Various FGFR2 Mutations (N550K, S252W, and C383R) Demonstrate Both Sensitivity and Intrinsic Resistance to FGFR Inhibition
Our laboratory and others have reported that the AN3CA and MFE296 cell lines, which carry the N550K FGFR2 mutation, are sensitive to FGFR inhibition with PD174074 [48,49]. To better gauge the relevance of the N550K mutation in resistance to FGFR inhibition in the correct cellular context, we identified four additional cell lines with mutations in FGFR2 (Gordon Mills, personal communication). We hypothesized that a comparison of the mutant FGFR2 cell lines would show that all cell lines would be equally sensitive to ponatinib but that the N550K lines could show relative resistance to PD173074 and dovitinib (compared to cell lines carrying S252W and C383R FGFR2 mutations).
Sensitivity across the panel was in the order ponatinib (most sensitive), then dovitinib, then PD173074 (least sensitive). Ponatinib was more potent than the other FGFR inhibitors in both the FGFR2 mutant and FGFR2 WT cell lines (e.g., Ishikawa), suggesting that its increased potency was not only due to its ability to bind the active FGFR2 but also due to its multi-kinase nature. We should note that the IC50 values we report are higher than those that we and others have reported previously for several cell lines. However, in this report, we directly measured cell proliferation with a nucleic acid-based kit rather than a metabolic-based assay [48,50] or one based on cellular protein content [49]. Within the FGFR2mutant cell lines, three of seven showed marked resistance to PD173074 (IC50 > 4 µM) including EI, EJ, and EN1078D. With the exception of EN1078D treated with dovitinib, these three FGFR2mutant cell lines also showed relative resistance to dovitinib and ponatinib when compared to the average IC50 of the three most sensitive cell lines versus that of the three FGFR2wild-type cell lines (Table 1). As the same cell lines showed similar relative resistance to PD173074, dovitinib, and ponatinib, it suggests that these cell lines have intrinsic resistance to FGFR inhibition that is not overcome by an inhibitor that is capable of binding to the active conformation of the kinase.
Table 1.
Sensitivity of Endometrial Cancer Cell Lines to PD173074, Dovitinib and Ponatinib.
| EC Cell Line | FGFR2 Status | Dovitinib IC50 (nM) | PD173074 IC50 (nM) | Ponatinib IC50 (nM) |
| AN3CA | FGFR2N550K | 449 ± 114 | 761 ± 130 | 58 ± 13 |
| MFE296 | FGFR2N550K | 614 ± 18 | 821 ± 71 | 104 ± 29 |
| EJ | FGFR2N550K | 2374 ± 789 | >10,000 | 949 ± 241 |
| EN1078D | FGFR2N550K | 770 ± 174 | 4475 ± 258 | 614 ± 18 |
| MFE280 | FGFR2S252W | 1503 ± 381 | 821 ± 71 | 230 ± 52 |
| EI | FGFR2S252W | 1268 ± 146 | 5161 ± 149 | 729 ± 21 |
| JHUEM-2 | FGFR2C383R | 558 ± 111 | 183 ± 41 | 145 ± 33 |
| HEC1A | WT | 3572 ± 409 | >10,000 | 1519 ± 260 |
| Ishikawa | WT | 1850 ± 662 | 8749 ± 1251 | 669 ± 222 |
| KLE | WT | 4751 ± 1933 | >10,000 | 1167 ± 167 |
Stable Expression of N550K Mutant FGFR2 in Inhibitor Sensitive JHUEM-2 Cells Confers Resistance to PD173074
As those EC cell lines carrying the N550K mutation had a diverse response to FGFR inhibition (presumably reflecting the acquisition of additional genetic/epigenetic changes), we sought an alternative approach to confirm whether FGFR2N550K is a true resistance mutation. We stably transfected the sensitive JHUEM-2 cell line (FGFR2C383R) with FGFR2N550K. JHUEM-2 cells stably expressing an empty vector control, WT FGFR2, and an extracellular domain activating FGFR2 mutant (Y376C) were also created. We then measured the cell viability of these lines in response to FGFR inhibition with dovitinib, PD173074, and ponatinib. Although expression of FGFR2N550K did not affect the sensitivity of JHUEM-2 cells to dovitinib and ponatinib, it did, however, cause an about five-fold increase in the IC50 to PD173074 (Figure 7A). As demonstrated in Figure 7B, all three WT and mutant FGFR2-transfected cell lines express higher levels of FGFR2 than the empty vector control line. Indeed, the FGFR2N550K expressing cells expressed less FGFR2 than the FGFR2Y376C cell line, and yet only the FGFR2N550K cells showed increased resistance to PD173074. While N550K did not confer resistance to dovitinib and ponatinib when expressed at low levels in JHUEM cells, we were able to confirm that N550H imparts resistance to dovitinib using in vitro kinase assays (Figure 7C). Similar to the N550H/K-expressing BaF3 cells, in vitro kinase assays showed that N550H was more sensitive to ponatinib than dovitinib. Specifically, dovitinib and ponatinib could inhibit the kinase activity of WT FGFR2 when mixed at a kinase/inhibitor molar ratio of 1:2. Ponatinib could inhibit N550H at a similar molar ratio, whereas dovitinib could not provide the same inhibition even at a molar ratio of 1:10. The kinase activity of the V565I mutant was resistant to both dovitinib and ponatinib even when mixed at a molar ratio of 1:10. These results confirm the BaF3 data showing that ponatinib is more effective than dovitinib at inhibiting FGFR2N550K and that the V565I gatekeeper mutant is resistant to both dovitinib and ponatinib. Taken together, this confirms our BaF3 data that FGFR2N550K is indeed a true resistance mutation.
Figure 7.

The N550K mutation confers resistance to PD173074, but not dovitinib or ponatinib, when expressed in FGFR inhibitor-sensitive JHUEM-2 cells. (A) Stably transfected JHUEM-2 cells were seeded in 96-well plates 24 hours before drug addition. Dovitinib, PD173074, and ponatinib were added in increasing concentrations from 1 nM to 10 µM. The cells were incubated at 37°C for 72 hours and proliferation was measured using the CyQUANT Cell Proliferation Assay Kit and the IC50 was calculated. (B) Western blots demonstrating FGFR2 expression levels in stably transfected JHUEM-2 lines. EV, empty vector. (C) Both dovitinib and ponatinib are potent inhibitors of the WT FGFR2 kinase, whereas only ponatinib effectively inhibits the N550H mutant FGFR2. Due to the steric clash, neither dovitinib nor ponatinib is capable of inhibiting the V565I “gatekeeper” mutant. The control lane 1 shows extent of phosphorylation in the absence of inhibitors. In lanes 2 to 7, increasing concentrations of inhibitors were added into the autophosphorylation reactions to inhibit the kinase autophosphorylation. The kinase/inhibitor molar ratios of lanes 2 to 7 are 1:0.2, 1:0.5, 1:1, 1:2, 1:5, and 1:10, respectively. The control lane 0 is the kinase in the absence of ATP/MgCl2.
Discussion
This study provides the first discovery of TKI-resistant mutations in FGFR2, an important drug target in EC. These mutations include M536I, M538I, I548V, N550H/K/S/T, V565I, E566G/A, L618M, and K642N. Given the identification of N550K, we also investigated the clinically relevant activating mutation, K660E, and showed that it was associated with resistance to dovitinib and PD173074. Identification of the V565I mutation in our screen reiterates mutation of the gatekeeper residue as a general mechanism of acquired resistance to TKIs. Importantly, our structural and biochemical data show that these mutations stabilize the active conformation of FGFR2 kinase manifesting in increased intrinsic activity of the drug-resistant FGFR2K mutants. Although several resistance mutations were not functionally tested (N550T, E566A, and K642N), data from the remaining mutations indicate that seven of the identified resistance mutations, namely, N550H/K/S/T, E566G/A, and K642N, drive the enzyme into the active state by disengaging the autoinhibitory molecular brake at the kinase hinge region. The remaining five mutations, namely, M536I, M538I, I548V, V565I, and L618M, stabilize the kinase active conformation by strengthening the hydrophobic spine, a network of hydrophobic packing interactions between the N- and C-lobe of the kinase that characterizes the active conformation of the kinase. It has been suggested that dovitinib may inhibit both the active and inactive forms of VEGFR [4]. However, our findings indicate that dovitinib and PD173074 preferentially bind the inactive form of the FGFR2 kinase. In contrast, ponatinib effectively inhibited all of the FGFR2-activating mutations except the V565I gatekeeper mutation, suggesting that ponatinib is capable of targeting both the inactive and the active conformations of the kinase. Modeling studies suggest that the gatekeeper mutation, in addition to strengthening the hydrophobic spine, may also create a steric conflict for drug binding, explaining the exceptional resistance of this mutation to ponatinib (Figure 6).
Figure 6.

The V565I gatekeeper FGFR2 kinase mutant is also refractory to inhibition by ponatinib due to steric conflicts. Ponatinib taken from the Abl-ponatinib complex structure (PDB ID: 3OY3) [38] was modeled onto the A-loop phosphorylated activated WT FGFR2 kinase structure (PDB ID: 2PVF) [36]. The molecular surface of ponatinib (in yellow) is shown to emphasize the steric clash with the mutated I565.
Amino acids corresponding to all the dovitinib-resistant mutations identified in FGFR2 are conserved among the other three members of the FGFR family. Therefore, it is likely that the corresponding mutations in other FGFR family members could impart dovitinib resistance in tumors that are dependent on these FGFRs. All three constituents of the molecular brake (N550, E566, and K642) and several residues of the hydrophobic spine in FGFR2b are also conserved in VEGFR1, VEGFR2, PDGFRA, PDGFRB, c-KIT, and FLT3. Indeed, mutations at the homologous residue to FGFR2 (N550) in FLT3 (N676) have been linked to resistance to the kinase inhibitor PKC412, including clinical resistance in a patient with acute myeloid leukemia [28,51]. Conversely, N659 in PDGFRA (homologous to N550 in FGFR2) is mutated in a subset of GIST tumors, but this mutation does not result in imatinib resistance in vitro [33].
In addition to being the most commonly mutated codon identified in our resistance screen, N550 is also the second most common amino acid of FGFR2 altered in endometrioid EC [45,52]. As such, cancer patients carrying activating mutations at N550 may be resistant to the anti-FGFR activity of dovitinib, whereas patients harboring FGFR2 mutations outside the kinase domain would be expected to achieve more clinically significant responses. Preliminary findings from a phase I/II pharmacodynamic study indicate that oral administration of dovitinib at 400 mg/day results in plasma Cmax drug levels of 119 to 382 ng/ml (247–792 nM) [15]. With a half-life of approximately 12 hours, dovitinib plasma Cmin levels would be expected to be approximately 25% those of the Cmax levels (in the order of 62–198 nM). On the basis of our in vitro data, effective inhibition of N550K, E566G, K660E, and V565I would not be expected with these plasma concentrations. Thus, we propose that patients presenting with N550 and K660 mutations potentially be treated with higher doses of dovitinib and for their plasma concentrations to be analyzed and correlated with clinical response. It should be noted, however, that dovitinib should still exert anti-angiogenic activity in patients with the N550K mutation owing to its additional inhibition of VEGFR and PDGFR [15] and the established efficacy of bevacizumab in this patient population [53]. That being said, we would predict fewer partial and complete responses in patients carrying kinase domain mutations due to its reduced anti-tumor efficacy. All mutations except V565I (which may possibly arise in the context of ponatinib-selective pressure) are effectively inhibited at achievable plasma levels of ponatinib, and as such, a multi-institutional phase II trial of ponatinib in FGFR2 mutation-positive EC patients is currently in development.
The prevalence of drug-resistant mutations affecting N550 was somewhat surprising, considering the fact that we, and others, have previously shown that EC cell lines with FGFR2b N550K mutation are sensitive to PD173074 [48,49]. This inconsistency is an important one as our BaF3 resistance screen results may not be readily translated into the clinic in EC patients treated with dovitinib. To better gauge the relevance of the N550K resistance mutation in EC cell lines, we ran all three FGFR inhibitors in this study across a number of N550K mutant, non-N550K mutant, and FGFR2 WT cell lines. While we confirmed the JHUEM-2 cell line as an additional sensitive cell line, the EJ, EI, and, to a lesser extent, EN1078D cell lines showed resistance to the panel of kinase inhibitors. Of these EJ and EN1078D carried an N550K mutation and EI carried an S252W mutation. Previous data indicate that the MFE319 cell line carrying an S252W mutation is also resistant to FGFR inhibition [54]. Therefore, only four of eight (50%) of the EC cell lines with known activating mutations show sensitivity to FGFR inhibition, contrasting with our original findings where two of two cell lines showed sensitivity. As sensitivity/resistance does not correlate with any specific mutation, sensitivity to FGFR inhibition appears to be more complex than simply which FGFR2 mutation is present. Perhaps intuitively, sensitivity is greater for those FGFR inhibitors, like dovitinib and ponatinib, with multiple kinase targets. Importantly, our cell line findings highlight the importance of intrinsic resistance to FGFR inhibition in EC, such that additional assessment of biomarkers of sensitivity and resistance may be required before the clinical success of FGFR inhibition is observed in patients.
To better approach the question as to whether FGFR2N550K is a true resistance mutation, we stably transfected the sensitive non-N550K mutant (FGFR2C383R) EC cell line JHUEM-2 with an FGFR2N550K expressing construct and compared its response to FGFR inhibition with that of other JHUEM-2 lines similarly transfected with relevant controls. Strikingly, the presence of FGFR2N550K in JHUEM-2 cells conferred an about five-fold increase in IC50 to PD173074 in this cell line, confirming our BaF3 screen results that FGFR2N550K is indeed a true resistance mutation. Although there was no increase in resistance to dovitinib in the FGFR2N550K-transfected JHUEM-2 cells, we hypothesize that this is due to a combination of low levels of FGFR2N550K combined with receptor heterodimerization and the fact that the N550K allele provides relatively less resistance to dovitinib. Specifically, in the BaF3 assay, N550K provides less resistance to dovitinib than PD173074 (∼15x WT IC50 vs ∼250x WT IC50), but we must be cognizant of the fact that BaF3 cells express no endogenous FGFR2 and so we are measuring the drug resistance associated with homodimerization of the FGFR2N550K allele. In contrast, the JHUEM-2 cell line expresses a high level of endogenous FGFR2C383R and there are relatively low expression levels of FGFR2N550K compared to endogenous FGFR2C383R (see Figure 7). In this case, we presume that the relative small proportion of receptor dimers carrying FGFR2N550K on a background of high FGFR2C383R expression is sufficient to provide resistance to PD173074 but not to dovitinib. To provide additional data that FGFR2N550K provides resistance to dovitinib but not ponatinib, additional in vitro kinase assays were performed. These showed that while both inhibitors showed poor activity against the gatekeeper mutation, ponatinib showed greater activity than dovitinib against the N550H mutation. Taken together, this confirms our BaF3 data that FGFR2N550K is indeed a true resistance mutation; however, we must await the clinical data to see to what extent this is reflected in patients.
In conclusion, we have identified FGFR2 mutations, including the common N550K mutation capable of conferring resistance to dovitinib in BaF3 assays. These drug-resistant mutations increase RTK activity by disengaging the molecular brake or by stabilizing the hydrophobic spine of FGFR2. Introduction of the drug-resistant FGFR2N550K allele into the FGFR2C383R JHUEM cell line resulted in a six-fold increase in resistance to PD173074. With the evaluation of four additional FGFR2 mutant EC cell lines and the finding that only two of four EC cell lines with N550K mutations and one of three cell lines with S252W mutations are sensitive to dovitinib and ponatinib, additional markers of drug sensitivity and/or resistance are required. We look forward to the ongoing dovitinib trial in EC patients to see if patients with heterozygous or homozygous activating kinase domain mutations respond as well as those with mutations in other domains. If previous experience with other kinase inhibitors can be assumed to hold true for anti-FGFR agents, future drug design should focus on inhibiting the active conformation of the FGFRs, as well as the development of second-generation inhibitors targeting the gatekeeper form of FGFR2.
Supplementary Material
Abbreviations
- EGFR
epidermal growth factor receptor
- FGFR
fibroblast growth factor receptor
- RTK
receptor tyrosine kinase
- SDM
site-directed mutagenesis
Footnotes
This work was supported by a Sylvia Chase Postdoctoral Fellowship (S.A.B.) and an American Cancer Society Postdoctoral Fellowship (PF-07-215-01-TBE to S.A.B.), National Institutes of Health (NIH) grant R01 DE13686 (to M.M.), NIH grant P30 CA016087 and 100 Women in Hedge Funds Foundation (to T.A.N.), a QUT Vice Chancellors Fellowship (to P.M.P.), and a National Health and Medical Research Council CDF2 (to P.M.P.).
This article refers to supplementary materials, which are designated by Table W1 and Figures W1 to W3 and are available online at www.neoplasia.com.
References
- 1.Turner N, Grose R. Fibroblast growth factor signalling: from development to cancer. Nat Rev Cancer. 2010;10(2):116–129. doi: 10.1038/nrc2780. [DOI] [PubMed] [Google Scholar]
- 2.Wesche J, Haglund K, Haglund EM. Fibroblast growth factors and their receptors in cancer. Biochem J. 2011;437(2):199–213. doi: 10.1042/BJ20101603. [DOI] [PubMed] [Google Scholar]
- 3.Brooks AN, Kilgour E, Smith PD. Molecular pathways: fibroblast growth factor signaling: a new therapeutic opportunity in cancer. Clin Cancer Res. 2012;18(7):1855–1862. doi: 10.1158/1078-0432.CCR-11-0699. [DOI] [PubMed] [Google Scholar]
- 4.Renhowe PA, Pecchi S, Shafer CM, Machajewski TD, Jazan EM, Taylor C, Antonios-McCrea W, McBride CM, Frazier K, Wiesmann M, et al. Design, structure-activity relationships and in vivo characterization of 4-amino-3-benzimidazol-2-ylhydroquinolin-2-ones: a novel class of receptor tyrosine kinase inhibitors. J Med Chem. 2009;52(2):278–292. doi: 10.1021/jm800790t. [DOI] [PubMed] [Google Scholar]
- 5.Huynh H, Chow PK, Tai WM, Choo SP, Chung AY, Ong HS, Soo KC, Ong R, Linnartz R, Shi MM. Dovitinib demonstrates antitumor and anti-metastatic activities in xenograft models of hepatocellular carcinoma. J Hepatol. 2012;56(3):595–601. doi: 10.1016/j.jhep.2011.09.017. [DOI] [PubMed] [Google Scholar]
- 6.Sivanand S, Pena-Llopis S, Zhao H, Kucejova B, Spence P, Pavia-Jimenez A, Yamasaki T, McBride DJ, Gillen J, Wolff NC, et al. A validated tumor-graft model reveals activity of dovitinib against renal cell carcinoma. Sci Transl Med. 2012;4(137):137ra75. doi: 10.1126/scitranslmed.3003643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Dey JH, Bianchi F, Voshol J, Bonenfant D, Oakeley EJ, Hynes NE. Targeting fibroblast growth factor receptors blocks PI3K/AKT signaling, induces apoptosis, and impairs mammary tumor outgrowth and metastasis. Cancer Res. 2010;70(10):4151–4162. doi: 10.1158/0008-5472.CAN-09-4479. [DOI] [PubMed] [Google Scholar]
- 8.Taeger J, Moser C, Hellerbrand C, Mycielska ME, Glockzin G, Schlitt HJ, Geissler EK, Stoeltzing O, Lang SA. Targeting FGFR/PDGFR/VEGFR impairs tumor growth, angiogenesis, and metastasis by effects on tumor cells, endothelial cells, and pericytes in pancreatic cancer. Mol Cancer Ther. 2011;10(11):2157–2167. doi: 10.1158/1535-7163.MCT-11-0312. [DOI] [PubMed] [Google Scholar]
- 9.Trudel S, Li ZH, Wei E, Wiesmann M, Chang H, Chen C, Reece D, Heise C, Stewart AK. CHIR-258, a novel, multitargeted tyrosine kinase inhibitor for the potential treatment of t(4;14) multiple myeloma. Blood. 2005;105(7):2941–2948. doi: 10.1182/blood-2004-10-3913. [DOI] [PubMed] [Google Scholar]
- 10.Lopes de Menezes DE, Peng J, Garrett EN, Louie SG, Lee SH, Wiesmann M, Tang Y, Shephard L, Goldbeck C, Oei Y, et al. CHIR-258: a potent inhibitor of FLT3 kinase in experimental tumor xenograft models of human acute myelogenous leukemia. Clin Cancer Res. 2005;11(14):5281–5291. doi: 10.1158/1078-0432.CCR-05-0358. [DOI] [PubMed] [Google Scholar]
- 11.Xin X, Abrams TJ, Hollenbach PW, Rendahl KG, Tang Y, Oei YA, Embry MG, Swinarski DE, Garrett EN, Pryer NK, et al. CHIR-258 is efficacious in a newly developed fibroblast growth factor receptor 3-expressing orthotopic multiple myeloma model in mice. Clin Cancer Res. 2006;12(16):4908–4915. doi: 10.1158/1078-0432.CCR-06-0957. [DOI] [PubMed] [Google Scholar]
- 12.Chase A, Grand FH, Cross NC. Activity of TKI258 against primary cells and cell lines with FGFR1 fusion genes associated with the 8p11 myeloproliferative syndrome. Blood. 2007;110(10):3729–3734. doi: 10.1182/blood-2007-02-074286. [DOI] [PubMed] [Google Scholar]
- 13.Lamont FR, Tomlinson DC, Cooper PA, Shnyder SD, Chester JD, Knowles MA. Small molecule FGF receptor inhibitors block FGFR-dependent urothelial carcinoma growth in vitro and in vivo. Br J Cancer. 2011;104(1):75–82. doi: 10.1038/sj.bjc.6606016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Deng N, Goh LK, Wang H, Das K, Tao J, Tan IB, Zhang S, Lee M, Wu J, Lim KH, et al. A comprehensive survey of genomic alterations in gastric cancer reveals systematic patterns of molecular exclusivity and co-occurrence among distinct therapeutic targets. Gut. 2012;61(5):673–684. doi: 10.1136/gutjnl-2011-301839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sarker D, Molife R, Evans TR, Hardie M, Marriott C, Butzberger-Zimmerli P, Morrison R, Fox JA, Heise C, Louie S, et al. A phase I pharmacokinetic and pharmacodynamic study of TKI258, an oral, multitargeted receptor tyrosine kinase inhibitor in patients with advanced solid tumors. Clin Cancer Res. 2008;14(7):2075–2081. doi: 10.1158/1078-0432.CCR-07-1466. [DOI] [PubMed] [Google Scholar]
- 16.Branford S, Rudzki Z, Walsh S, Grigg A, Arthur C, Taylor K, Herrmann R, Lynch KP, Hughes TP. High frequency of point mutations clustered within the adenosine triphosphate-binding region of BCR/ABL in patients with chronic myeloid leukemia or Ph-positive acute lymphoblastic leukemia who develop imatinib (STI571) resistance. Blood. 2002;99(9):3472–3475. doi: 10.1182/blood.v99.9.3472. [DOI] [PubMed] [Google Scholar]
- 17.Mauro MJ. Defining and managing imatinib resistance. Hematology Am Soc Hematol Educ Program. 2006:219–225. doi: 10.1182/asheducation-2006.1.219. [DOI] [PubMed] [Google Scholar]
- 18.Engelman JA, Janne PA. Mechanisms of acquired resistance to epidermal growth factor receptor tyrosine kinase inhibitors in non-small cell lung cancer. Clin Cancer Res. 2008;14(10):2895–2899. doi: 10.1158/1078-0432.CCR-07-2248. [DOI] [PubMed] [Google Scholar]
- 19.Yun CH, Mengwasser KE, Toms AV, Woo MS, Greulich H, Wong KK, Meyerson M, Eck MJ. The T790M mutation in EGFR kinase causes drug resistance by increasing the affinity for ATP. Proc Natl Acad Sci USA. 2008;105(6):2070–2075. doi: 10.1073/pnas.0709662105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kosaka T, Yatabe Y, Endoh H, Yoshida K, Hida T, Tsuboi M, Tada H, Kuwano H, Mitsudomi T. Analysis of epidermal growth factor receptor gene mutation in patients with non-small cell lung cancer and acquired resistance to gefitinib. Clin Cancer Res. 2006;12(19):5764–5769. doi: 10.1158/1078-0432.CCR-06-0714. [DOI] [PubMed] [Google Scholar]
- 21.Gibbons DL, Pricl S, Kantarjian H, Cortes J, Quintas-Cardama A. The rise and fall of gatekeeper mutations? The BCR-ABL1 T315I paradigm. Cancer. 2012;118(2):293–299. doi: 10.1002/cncr.26225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Azam M, Seeliger MA, Gray NS, Kuriyan J, Daley GQ. Activation of tyrosine kinases by mutation of the gatekeeper threonine. Nat Struct Mol Biol. 2008;15(10):1109–1118. doi: 10.1038/nsmb.1486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Rosti G, Castagnetti F, Gugliotta G, Palandri F, Martinelli G, Baccarani M. Dasatinib and nilotinib in imatinib-resistant Philadelphia-positive chronic myelogenous leukemia: a ‘head-to-head comparison’. Leuk Lymphoma. 2010;51(4):583–591. doi: 10.3109/10428191003637282. [DOI] [PubMed] [Google Scholar]
- 24.Blencke S, Zech B, Engkvist O, Greff Z, Orfi L, Horvath Z, Keri G, Ullrich A, Daub H. Characterization of a conserved structural determinant controlling protein kinase sensitivity to selective inhibitors. Chem Biol. 2004;11(5):691–701. doi: 10.1016/j.chembiol.2004.02.029. [DOI] [PubMed] [Google Scholar]
- 25.von Bubnoff N, Barwisch S, Speicher MR, Peschel C, Duyster J. A cell-based screening strategy that predicts mutations in oncogenic tyrosine kinases: implications for clinical resistance in targeted cancer treatment. Cell Cycle. 2005;4(3):400–406. doi: 10.4161/cc.4.3.1560. [DOI] [PubMed] [Google Scholar]
- 26.Warmuth M, Kim S, Gu XJ, Xia G, Adrian F. Ba/F3 cells and their use in kinase drug discovery. Curr Opin Oncol. 2007;19(1):55–60. doi: 10.1097/CCO.0b013e328011a25f. [DOI] [PubMed] [Google Scholar]
- 27.von Bubnoff N, Veach DR, van der Kuip H, Aulitzky WE, Sanger J, Seipel P, Bornmann WG, Peschel C, Clarkson B, Duyster J. A cell-based screen for resistance of Bcr-Abl-positive leukemia identifies the mutation pattern for PD166326, an alternative Abl kinase inhibitor. Blood. 2005;105(4):1652–1659. doi: 10.1182/blood-2004-06-2445. [DOI] [PubMed] [Google Scholar]
- 28.von Bubnoff N, Engh RA, Aberg E, Sanger J, Peschel C, Duyster J. FMS-like tyrosine kinase 3-internal tandem duplication tyrosine kinase inhibitors display a nonoverlapping profile of resistance mutations in vitro. Cancer Res. 2009;69(7):3032–3041. doi: 10.1158/0008-5472.CAN-08-2923. [DOI] [PubMed] [Google Scholar]
- 29.von Bubnoff N, Gorantla SP, Engh RA, Oliveira TM, Thone S, Aberg E, Peschel C, Duyster J. The low frequency of clinical resistance to PDGFR inhibitors in myeloid neoplasms with abnormalities of PDGFRA might be related to the limited repertoire of possible PDGFRA kinase domain mutations in vitro. Oncogene. 2011;30(8):933–943. doi: 10.1038/onc.2010.476. [DOI] [PubMed] [Google Scholar]
- 30.Tiedt R, Degenkolbe E, Furet P, Appleton BA, Wagner S, Schoepfer J, Buck E, Ruddy DA, Monahan JE, Jones MD, et al. A drug resistance screen using a selective MET inhibitor reveals a spectrum of mutations that partially overlap with activating mutations found in cancer patients. Cancer Res. 2011;71(15):5255–5264. doi: 10.1158/0008-5472.CAN-10-4433. [DOI] [PubMed] [Google Scholar]
- 31.Avizienyte E, Ward RA, Garner AP. Comparison of the EGFR resistance mutation profiles generated by EGFR-targeted tyrosine kinase inhibitors and the impact of drug combinations. Biochem J. 2008;415(2):197–206. doi: 10.1042/BJ20080728. [DOI] [PubMed] [Google Scholar]
- 32.Deshpande A, Reddy MM, Schade GO, Ray A, Chowdary TK, Griffin JD, Sattler M. Kinase domain mutations confer resistance to novel inhibitors targeting JAK2V617F in myeloproliferative neoplasms. Leukemia. 2012;26(4):708–715. doi: 10.1038/leu.2011.255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Corless CL, Schroeder A, Griffith D, Town A, McGreevey L, Harrell P, Shiraga S, Bainbridge T, Morich J, Heinrich MC. PDGFRA mutations in gastrointestinal stromal tumors: frequency, spectrum and in vitro sensitivity to imatinib. J Clin Oncol. 2005;23(23):5357–5364. doi: 10.1200/JCO.2005.14.068. [DOI] [PubMed] [Google Scholar]
- 34.Byron SA, Gartside MG, Wellens CL, Goodfellow PJ, Birrer MJ, Campbell IG, Pollock PM. FGFR2 mutations are rare across histologic subtypes of ovarian cancer. Gynecol Oncol. 2010;117(1):125–129. doi: 10.1016/j.ygyno.2009.12.002. [DOI] [PubMed] [Google Scholar]
- 35.Gartside MG, Chen H, Ibrahimi OA, Byron SA, Curtis AV, Wellens CL, Bengston A, Yudt LM, Eliseenkova AV, Ma J, et al. Loss-of-function fibroblast growth factor receptor-2 mutations in melanoma. Mol Cancer Res. 2009;7(1):41–54. doi: 10.1158/1541-7786.MCR-08-0021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Chen H, Ma J, Li W, Eliseenkova AV, Xu C, Neubert TA, Miller WT, Mohammadi M. A molecular brake in the kinase hinge region regulates the activity of receptor tyrosine kinases. Mol Cell. 2007;27(5):717–730. doi: 10.1016/j.molcel.2007.06.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ni ZJ, Barsanti P, Brammeier N, Diebes A, Poon DJ, Ng S, Pecchi S, Pfister K, Renhowe PA, Ramurthy S, et al. 4-(Aminoalkylamino)-3-benzimidazolequinolinones as potent CHK-1 inhibitors. Bioorg Med Chem Lett. 2006;16(12):3121–3124. doi: 10.1016/j.bmcl.2006.03.059. [DOI] [PubMed] [Google Scholar]
- 38.Zhou T, Commodore L, Huang WS, Wang Y, Thomas M, Keats J, Xu Q, Rivera VM, Shakespeare WC, Clackson T, et al. Structural mechanism of the Pan-BCR-ABL inhibitor ponatinib (AP24534): lessons for overcoming kinase inhibitor resistance. Chem Biol Drug Des. 2011;77(1):1–11. doi: 10.1111/j.1747-0285.2010.01054.x. [DOI] [PubMed] [Google Scholar]
- 39.Jones TA, Zou JY, Cowan SW, Kjeldgaard M. Improved methods for building protein models in electron density maps and the location of errors in these models. Acta Crystallogr A. 1991;47(pt 2):110–119. doi: 10.1107/s0108767390010224. [DOI] [PubMed] [Google Scholar]
- 40.Kabsch W. A solution for the best rotation to relate two sets of vectors. Acta Crystallogr Sect A. 1976;32:922–923. [Google Scholar]
- 41.Collaborative Computational Project, Number 4, author. The CCP4 suite: programs for protein crystallography. Acta Crystallogr D Biol Crystallogr. 1994;50(pt 5):760–763. doi: 10.1107/S0907444994003112. [DOI] [PubMed] [Google Scholar]
- 42.DeLano WL. The PyMOL User's Manual. San Carlos, CA: DeLano Scientific; 2002. [Google Scholar]
- 43.Chen H, Xu CF, Ma J, Eliseenkova AV, Li W, Pollock PM, Pitteloud N, Miller WT, Neubert TA, Mohammadi M. A crystallographic snapshot of tyrosine trans-phosphorylation in action. Proc Natl Acad Sci USA. 2008;105(50):19660–19665. doi: 10.1073/pnas.0807752105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ibrahimi OA, Zhang F, Eliseenkova AV, Itoh N, Linhardt RJ, Mohammadi M. Biochemical analysis of pathogenic ligand-dependent FGFR2 mutations suggests distinct pathophysiological mechanisms for craniofacial and limb abnormalities. Hum Mol Genet. 2004;13(19):2313–2324. doi: 10.1093/hmg/ddh235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Byron SA, Gartside M, Powell MA, Wellens CL, Gao F, Mutch DG, Goodfellow PJ, Pollock PM. FGFR2 point mutations in 466 endometrioid endometrial tumors: relationship with MSI, KRAS, PIK3CA, CTNNB1 mutations and clinicopathological features. PLoS One. 2012;7(2):e30801. doi: 10.1371/journal.pone.0030801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lievens PM, Liboi E. The thanatophoric dysplasia type II mutation hampers complete maturation of fibroblast growth factor receptor 3 (FGFR3), which activates signal transducer and activator of transcription 1 (STAT1) from the endoplasmic reticulum. J Biol Chem. 2003;278(19):17344–17349. doi: 10.1074/jbc.M212710200. [DOI] [PubMed] [Google Scholar]
- 47.Lievens PM, Mutinelli C, Baynes D, Liboi E. The kinase activity of fibroblast growth factor receptor 3 with activation loop mutations affects receptor trafficking and signaling. J Biol Chem. 2004;279(41):43254–43260. doi: 10.1074/jbc.M405247200. [DOI] [PubMed] [Google Scholar]
- 48.Dutt A, Salvesen HB, Chen TH, Ramos AH, Onofrio RC, Hatton C, Nicoletti R, Winckler W, Grewal R, Hanna M, et al. Drug-sensitive FGFR2 mutations in endometrial carcinoma. Proc Natl Acad Sci USA. 2008;105(25):8713–8717. doi: 10.1073/pnas.0803379105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Byron SA, Gartside MG, Wellens CL, Mallon MA, Keenan JB, Powell MA, Goodfellow PJ, Pollock PM. Inhibition of activated fibroblast growth factor receptor 2 in endometrial cancer cells induces cell death despite PTEN abrogation. Cancer Res. 2008;68(17):6902–6907. doi: 10.1158/0008-5472.CAN-08-0770. [DOI] [PubMed] [Google Scholar]
- 50.Gozgit JM, Wong MJ, Moran L, Wardwell S, Mohemmad QK, Narasimhan NI, Shakespeare WC, Wang F, Clackson T, Rivera VM. Ponatinib (AP24534), a multitargeted pan-FGFR inhibitor with activity in multiple FGFR-amplified or mutated cancer models. Mol Cancer Ther. 2012;11(3):690–699. doi: 10.1158/1535-7163.MCT-11-0450. [DOI] [PubMed] [Google Scholar]
- 51.Heidel F, Solem FK, Breitenbuecher F, Lipka DB, Kasper S, Thiede MH, Brandts C, Serve H, Roesel J, Giles F, et al. Clinical resistance to the kinase inhibitor PKC412 in acute myeloid leukemia by mutation of Asn-676 in the FLT3 tyrosine kinase domain. Blood. 2006;107(1):293–300. doi: 10.1182/blood-2005-06-2469. [DOI] [PubMed] [Google Scholar]
- 52.Pollock PM, Gartside MG, Dejeza LC, Powell MA, Mallon MA, Davies H, Mohammadi M, Futreal PA, Stratton MR, Trent JM, et al. Frequent activating FGFR2 mutations in endometrial carcinomas parallel germline mutations associated with craniosynostosis and skeletal dysplasia syndromes. Oncogene. 2007;26:7158–716. doi: 10.1038/sj.onc.1210529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Aghajanian C, Sill MW, Darcy KM, Greer B, McMeekin DS, Rose PG, Rotmensch J, Barnes MN, Hanjani P, Leslie KK. Phase II trial of bevacizumab in recurrent or persistent endometrial cancer: a Gynecologic Oncology Group study. J Clin Oncol. 2011;29(16):2259–2265. doi: 10.1200/JCO.2010.32.6397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Harding TC, Palencia S, Long L, Finer JT, Keer HN, Baker KP, Kavanaugh WM. Preclinical efficacy of FP-1039 (FGFR1:Fc) in endometrial carcinoma models with activating mutations in FGFR2; American Association for Cancer Research (AACR) 101st Annual Meeting 2010; Washington, DC: American Association for Cancer Research; 2010. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.