

Abstract
Genomes of eusocial insects code for dramatic examples of phenotypic plasticity and social organization. We compared the genomes of seven ants, the honeybee, and various solitary insects to examine whether eusocial lineages share distinct features of genomic organization. Each ant lineage contains ∼4000 novel genes, but only 64 of these genes are conserved among all seven ants. Many gene families have been expanded in ants, notably those involved in chemical communication (e.g., desaturases and odorant receptors). Alignment of the ant genomes revealed reduced purifying selection compared with Drosophila without significantly reduced synteny. Correspondingly, ant genomes exhibit dramatic divergence of noncoding regulatory elements; however, extant conserved regions are enriched for novel noncoding RNAs and transcription factor–binding sites. Comparison of orthologous gene promoters between eusocial and solitary species revealed significant regulatory evolution in both cis (e.g., Creb) and trans (e.g., fork head) for nearly 2000 genes, many of which exhibit phenotypic plasticity. Our results emphasize that genomic changes can occur remarkably fast in ants, because two recently diverged leaf-cutter ant species exhibit faster accumulation of species-specific genes and greater divergence in regulatory elements compared with other ants or Drosophila. Thus, while the “socio-genomes” of ants and the honeybee are broadly characterized by a pervasive pattern of divergence in gene composition and regulation, they preserve lineage-specific regulatory features linked to eusociality. We propose that changes in gene regulation played a key role in the origins of insect eusociality, whereas changes in gene composition were more relevant for lineage-specific eusocial adaptations.
The insect order Hymenoptera encompasses several lineages, including ants, bees, and aculeate wasps, that independently evolved obligate eusociality. Such eusocial lineages are characterized by reproductive division of labor, cooperative brood care, and overlapping generations (Michener 1969). Ants (Formicidae) represent one of the oldest (∼130 million years) and most successful exclusively eusocial lineages (Cardinal and Danforth 2011). They have colonized every terrestrial habitat except at the highest latitudes, and they have achieved substantial diversity in both individual and colonial traits. The ecological and evolutionary success of the more than 15,000 described extant ant species (http://www.antweb.org) is often attributed to their sociality and ability to engineer environments, e.g., by building elaborate nests, herding aphids for honeydew, or practicing sustainable agriculture (Crozier and Pamilo 1996; Hölldobler and Wilson 2009).
The genomes of seven ant species, representatives of four major lineages that comprise two-thirds of all ant species, have recently been sequenced and characterized independently (for review, see Gadau et al. 2012): Jerdon's jumping ant, Harpegnathos saltator (Ponerinae; n = 1033 extant species) (Bonasio et al. 2010); the globally invasive Argentine ant, Linepithema humile (Dolichoderinae; n = 692) (Smith et al. 2011a), the Florida carpenter ant, Camponotus floridanus (Formicinae; n = 2831) (Bonasio et al. 2010); and four ants within the hyperdiverse subfamily Myrmicinae (n = 6087): the red harvester ant, Pogonomyrmex barbatus (Smith et al. 2011b), the invasive red imported fire ant, Solenopsis invicta (Wurm et al. 2011); and two agricultural leaf-cutter species, Acromyrmex echinatior and Atta cephalotes (Nygaard et al. 2011; Suen et al. 2011). Together with the honeybee, Apis mellifera (The Honeybee Genome Sequencing Consortium 2006), eight eusocial genomes are now available from two evolutionarily independent lineages. While ants and honeybees are both eusocial Hymenoptera, they differ significantly in many aspects. For instance, ants have wingless and often polymorphic worker castes, enjoy long life spans for insects, and are descendants of predatory wasps, whereas honeybees only have winged monomorphic workers with limited life spans and are derived from solitary bees.
Given the remarkable phenotypic diversity among eusocial insects, a key question is to what extent do derived and independent eusocial lineages harbor shared features of genomic organization that enable their eusocial lifestyles (Robinson et al. 2005; Gadau et al. 2012; Ferreira et al. 2013). To address this question, we performed a comprehensive characterization of the genomic basis for eusociality, using eight eusocial insect genomes in addition to 22 available solitary insect genomes. Our results reveal a variety of lineage-specific changes in both gene composition and gene expression regulation that have facilitated the evolution of eusociality.
Results
Ant genomes harbor thousands of taxonomically restricted genes
Comparison of existing gene annotations from the seven ant genomes showed that, while the number of protein-coding genes is only partially explained by genome assembly size (R = 0.32) (Fig. 1A), the number of orthologous genes shared among these ants varies considerably (3.1-fold) (Supplemental Fig. 1). This suggested that either existing annotations lack many valid genes, or ant genomes harbor an abundance of taxonomically restricted genes (TRGs), which have been associated with the evolution of novel functionalities in other systems (for review, see Khalturin et al. 2009). Previous analyses of individual ant genomes predicted up to 8000 species-specific TRGs (e.g., Smith et al. 2011b), in addition to 840 TRGs that are exclusively shared among ants (e.g., Bonasio et al. 2010).
Figure 1.

Overview of protein-coding gene composition and genome size in Hymenoptera. (A) Gene and genome content in seven ant species and honeybee (red), with representative solitary insects (blue) as outgroups. Orthology delineation among protein-coding genes from 12 insects identified orthologs present in all (Universal, n = 12) or almost all (Broad, 10 ≤ n ≤ 11) species, conserved as single-copy genes or with paralogs (with duplications). Differential gene losses leave orthologs shared among fewer species across the phylogeny (Patchy, n < 10). Remaining ant genes exhibit orthology with honeybee ([AMELL] Apis mellifera) and/or jewel wasp ([NVITR] Nasonia vitripennis) (Hymenoptera), among ants (Formicidae), or lack orthology (Undetectable). Total estimated genome sizes vary among Hymenoptera, largely due to repetitive regions (orange bars); however, hymenopterans share a nonrepetitive core of ∼200 Mb (green bars). A maximum-likelihood species tree computed from the concatenated alignment of all universal single-copy orthologs confirms the established ant phylogeny (Moreau et al. 2006). Rates of molecular evolution are comparable to the other hymenopterans, flour beetle ([TCAST] Tribolium castaneum; genome size ∼200 Mb), and body louse ([PHUMA] Pediculus humanus; genome size ∼108 Mb), but are much slower than the dipteran representative ([DMELA] Drosophila melanogaster; genome size 175 Mb). The ant species are (HSALT) Harpegnathos saltator; (LHUMI) Linepithema humile; (CFLOR) Camponotus floridanus; (PBARB) Pogonomyrmex barbatus; (SINVI) Solenopsis invicta; (AECHI) Acromyrmex echinatior; and (ACEPH) Atta cephalotes. (B) Occurrence (blue) and emergence rate (red) of taxonomically restricted genes (TRGs) in different taxonomic clades of Hymenoptera (colors) and Diptera (gray). The youngest clades of both Hymenoptera and Diptera exhibit the highest rates of TRG accumulation. Age is measured as the time between the most distant members of each group and hence does not reflect a clade's absolute age. (C) Rate of change of TRGs versus divergence time, for eight species groupings. Pearson's correlation coefficient is shown. P-value was computed using a two-tailed t-test.
To infer more accurately the origin and abundance of TRGs while minimizing annotation error, we applied two approaches to reannotate ant and honeybee genomes in terms of gene number and model quality. First, by comparing known protein sequences among species, we identified 3313 genes from 2635 orthologous groups that were missing from existing annotations (Supplemental Figs. 2–4; Supplemental Table 1). Second, we developed a broader approach involving 30 published arthropod genomes and conservative filtering steps and identified 5996 additional, previously missing genes (Supplemental Figs. 5–8; Supplemental Tables 2, 3). Thus, our significantly revised genome annotations include 9309 newly annotated genes for the eight eusocial species. These analyses corroborate that the honeybee has an exceptionally low gene number (Fig. 1A): We found only 223 previously missing genes in the honeybee versus 856 on average for ants (Supplemental Fig. 5) and relatively few TRGs compared with other insects. Whether this apparent gene loss is restricted to Apis honeybees or is shared with other corbiculate bees will soon be elucidated by ongoing efforts to sequence multiple bee and bumble bee genomes.
Having identified the missing genes from social insect genome annotations, we delineated TRGs for different clades within Hymenoptera and Diptera. Notably, we found 28,581 TRGs that are restricted to Formicidae (ant TRGs), and 42% of these genes appear to be species-specific, given our current taxon sampling (Fig. 1A,B). Thus, we estimate that each ant genome harbors an average of 4083 TRGs, of which 1715 appear to be species specific. Intriguingly, among the remaining, non-species-specific TRGs (i.e., TRGs present in multiple ant genomes), only 64 are present in all seven ant genomes (Supplemental Text 1). These 64 pan-ant TRGs have a median length of 97 amino acids (25th–75th percentile range of 72–149 amino acids), are well conserved and show strong expression support in two ant species (Fig. 2); however, they generally lack caste-specific expression (n = 3, FDR < 0.05) and do not encode any known protein domains. These results suggest that a broad “social toolkit” of conserved de novo protein-coding genes is not a requirement for eusociality.
Figure 2.
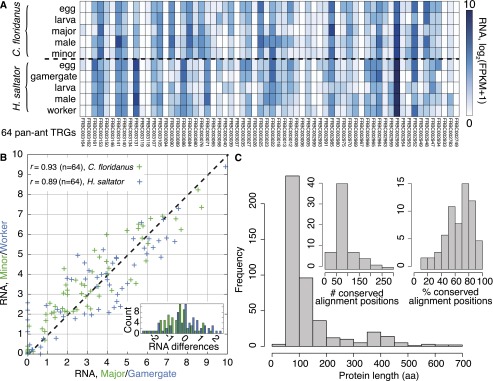
Analysis of 64 pan-ant taxonomically restricted genes (TRGs). (A) RNA expression support for 64 TRGs that are orthologous among all seven ant species but not found in other genomes. RNA expression levels, estimated as log2(FPKM + 1), are shown for various developmental stages and adult castes of C. floridanus and H. saltator. (B) Expression correlation between adult worker castes in C. floridanus (major vs. minor; green) and H. saltator (gamergate vs. worker; blue) for the 64 novel ant TRGs; Pearson's correlation coefficients are shown. (Inset) Histogram of differences in gene expression levels between castes (major − minor in green, gamergate − worker in blue) per gene. (C) Length distribution (in amino acids) of the 64 novel ant TRGs. (Inset) Distribution of the number (left) and percentage (right) of conserved alignment positions (see Supplemental Text 1).
Hymenoptera, especially the two leaf-cutter ants, exhibit a faster emergence rate for taxonomically restricted genes than Diptera
Having multiple genomes for two insect orders allowed us to compare rates of TRG emergence between Hymenoptera (n = 9) and Diptera (n = 15). This comparison revealed that Hymenoptera have both a greater number of TRGs (4658 vs. 3442 average TRGs per species) and a faster TRG emergence rate (31 vs. 14 average TRGs per species per million years) than Diptera (Fig. 1B). Young insect lineages also tend to have a faster TRG emergence rate than older lineages (R = −0.68, P < 0.04) (Fig. 1C). Notably, the two leaf-cutter ant species, which diverged only 8–12 million years (Myr) ago (Schultz and Brady 2008), exhibit the highest number of TRGs (n = 6796) and the fastest TRG emergence rate of any sequenced insect lineage, gaining 340 TRGs per species per million years. In comparison, the Drosophila melanogaster subgroup (n = 6), also having diverged ∼12 Myr ago, gains 115 TRGs per species/Myr. This pattern of rapid but transient expansions of gene content may coincide with dramatic life-history changes associated with early stages of lineage divergence. For example, A. cephalotes is distinguished from A. echinatior by loss of cuticular actinomycete cultures, physically distinct soldier castes, and claustral colony founding (Fernández-Marín et al. 2009; Villesen et al. 2009). In support of this, most leaf-cutter TRGs (68%) are species specific.
We hypothesize that these rapid TRG expansions observed for Hymenoptera may be due to differences in the rate of gene loss rather than gene gain. Natural selection is expected to be less efficient at removing superfluous genes from populations with small effective population sizes. Haplo-diploid sex determination and reproductive division of labor in eusocial Hymenoptera reduce effective population size relative to solitary and diploid Diptera (Crozier and Pamilo 1996; Gadau et al. 2012). The lack of a significant codon usage bias in ant genomes compared with Drosophila further supports the idea of relatively reduced selection efficiency in eusocial Hymenoptera (Supplemental Fig. 9; Supplemental Table 4; Supplemental Text 2).
Extensive gene family evolution in ants targets cytochromes, desaturases, olfactory receptors, and transcription factors
Gene families are sets of paralogs that often display functional similarity. Expansions or contractions in gene families may correspond to adaptive events coupled to life-history transitions (Ranson et al. 2002). To identify gene families in ants that have expanded or contracted due to natural selection, we examined changes in gene family size along branches of the phylogeny for 15 insects and estimated the rates of change, using a null model of gene family evolution that reflects the expected divergence due to neutral mutation and genetic drift (Supplemental Table 5; Supplemental Methods; Hahn et al. 2007). We found hundreds of gene family expansions and contractions along each of the terminal branches (Supplemental Fig. 10A) resulting in significant increases in variation for 281 families (P < 0.01). Along the branch leading to Formicidae, 11 significant expansions and nine significant contractions have occurred. Functional annotation of these 20 families showed that 55% possess DNA-binding capacity, generally characterized by zinc-finger or helix–loop–helix domains (Supplemental Fig. 11); of these 55% of the expanded and 22% of the contracted families may be involved in regulation of transcription. This suggests that changes in the transcription factor (TF) repertoire were important in the initial stages of ant evolution.
In addition, 96 gene families (34% of significant families) show significantly increased variation within Formicidae with several showing repeated expansions and contractions. This includes the P450 cytochrome superfamily, which has been linked to ecdysteroid metabolism and the detoxification of xenobiotics, and odorant receptor and desaturase genes, which are involved in chemical communication, e.g., caste and colony recognition (Nygaard et al. 2011; Smith et al. 2011b; Suen et al. 2011). Repeated changes in these families may reflect adaptations to novel ecological niches (e.g., tropics vs. desert or terrestrial vs. arboreal) and/or changes in social organization (e.g., colony size, mode of reproduction, division of labor). For instance, dietary specialization may conceivably demand novel genes to detoxify or metabolize novel compounds, while existing genes that help process undesirable food items could become unnecessary and therefore lost through genetic drift, e.g., P450 cytochrome pseudogenization in P. barbatus (Smith et al. 2011b) and loss of metabolic pathways in leaf-cutter ants (Nygaard et al. 2011; Suen et al. 2011). Analogously, the efficiency of chemical stimuli varies between environments and communication systems, necessitating tailored changes to groups of desaturases or olfactory receptors.
Desaturase proteins are central to the production of alkenes, a highly variable component of cuticular hydrocarbons reported to transmit complex signals like nest-mate recognition cues in ants (Martin and Drijfhout 2009; van Zweden et al. 2010). Manual annotation and phylogenetic analyses of the Δ9 and Δ11 desaturase gene families revealed that five ancestral subfamilies are present in all holometabolous insects (Supplemental Fig. 10B). Two of these subfamilies experienced multiple episodes of gene expansion at various times during hymenopteran evolution, resulting in 10–23 putatively functional genes in ants, compared with seven genes in D. melanogaster. Large numbers of related but nonfunctional gene fragments scattered throughout ant genomes also suggest that ants frequently altered their desaturase gene repertoire. For example, the invasive species L. humile and S. invicta possess 25 and 15 pseudogenes, respectively, suggesting that drastic changes in habitat and social organization following novel habitat invasion might have an immediate effect on genes implicated in social communication. Overall, the elevated number of desaturase genes and their variability in sequence and expression might reflect increased demand for chemical signal diversity used in ant social communication. Consistent with this, gene families presumably involved in the perception of these signals (e.g., olfactory receptors) (Smith et al. 2011b; Zhou et al. 2012) exhibit similar expansions.
Finally, we reanalyzed immune gene families of social and solitary insects, since the honeybee genome is reported to contain only one-third of the immune genes found in Drosophila (The Honeybee Genome Sequencing Consortium 2006), yet these families did not emerge from our gene family evolution analysis. The immune gene complements of eusocial insects did not differ from solitary insects as dramatically as previously proposed, because only three of 16 immune gene families showed significant changes in size between eusocial and solitary groups (FDR < 0.1) (Supplemental Table 6).
Ant genomes exhibit a strong degree of synteny
Comparative analysis of large-scale genome structure in the Drosophila clade (63 Myr) (Tamura et al. 2004) has revealed broad genome-scale similarity with 66% synteny, less than threefold change in total genome size, and little change in chromosome number (four to six chromosomes) (Drosophila 12 Genomes Consortium 2007). While genome size among the seven ants shows a similar threefold range, ant species cover twice the evolutionary distance, have more variable chromosome numbers (eight to 22 chromosomes) (Gadau et al. 2012), and exhibit extremely high recombination frequencies (e.g., 71 kb/cM for Pogonomyrmex rugosus) (Sirviö et al. 2011). These differences between Formicidae and Drosophila should favor a rapid decline of synteny in ants. To examine this, we first assembled syntenic blocks of homologous sequences using pairwise alignment of single-copy exons among genomes (Supplemental Fig. 12A–C). While only 5.3% of assembled contigs exhibit significant homology among ants, they comprise 3639 syntenic blocks that cover 65% of each genome on average (minimum 57%, H. saltator; maximum 71%, A. cephalotes), largely consistent with estimates from Drosophila (66%) (Drosophila 12 Genomes Consortium 2007). Moreover, average synteny increases to 74% among the four Mymicinae, which have comparable divergence time to the 12 sequenced Drosophila species, and to 86% between the two leaf-cutter ants (Supplemental Fig. 12A). Thus, despite fragmented genome assemblies and deep evolutionary history, ant genomes show moderate to strong genomic synteny, especially in gene-rich, euchromatic regions. Of course, given the variability in repetitive and total genome size in ants (see Fig. 1A), we do suspect that heterochromatic regions may harbor a greater degree of large-scale structural divergence.
Using a subset of 287 syntenic blocks showing strongest synteny in ants (Supplemental Fig. 13), we evaluated the extent of gene inversions and rearrangements in ants and other insects, using the A. echinatior genome as a common reference (the assembly of this species has the greatest N50 contig size) (Gadau et al. 2012). These highly syntenic blocks average 300 kb, include 10–15 genes per block, and harbor 8749 genes, including 5202 (91%) single-copy genes found in all seven ant genomes (Supplemental Fig. 1). As expected, both inversions and rearrangements increase with evolutionary distance from A. echinatior, although inversions appear to be more common overall (Supplemental Fig. 14). Notably, gene order in the hox cluster is identical among ants and is consistent with the ordering in Drosophila (Supplemental Fig. 15). Interestingly, all ant species display a lower percentage of gene rearrangements (<4%) compared with D. melanogaster or A. mellifera (∼7%) and much lower compared with the parasitoid wasp Nasonia vitripennis (11%) (Supplemental Fig. 14B). In contrast, D. melanogaster shows 2.5-fold more gene inversions compared with A. mellifera, ants, and N. vitripennis, as expected phylogenetically. Thus, some lineages of Hymenoptera may have accumulated specific kinds of structural divergence, including rearrangements, at a faster rate than Drosophila and independent of eusociality.
Ant conserved regions harbor an abundance of regulatory elements and are enriched near neuronal genes
Leveraging the high structural homology among ant genomes, we generated global multiple sequence alignments of all 3639 syntenic blocks (Supplemental Fig. 12D) and identified more than 1.7 million conserved elements (CEs), including 424 CEs that span at least 1 kb (Supplemental Fig. 16). After conservatively comparing CEs against all annotated exonic sequences, including those from TRGs, and masking likely untranslated regions (UTRs), nearly half (49%) of the CEs appear to be intergenic (Supplemental Fig. 16D). By counting nucleotides delimited by CEs, we estimated that, on average, 18.6% of each ant genome undergoes purifying selection; similar analysis restricted to Myrmicinae (using 74% of the four genome sequences, compared with 65% for seven ants) yielded approximately the same estimate of 20.7%. Purifying selection is greatest for exons (59%), microRNAs (miRNAs) (92%), and tRNAs (28%), i.e., explicitly functional DNA sequences (Supplemental Fig. 16D,E). These estimates may be overestimated, because syntenic blocks are generally depleted for repetitive DNA, a major source of evolutionary variation. Thus, ants appear to exhibit 1.8-fold to 2.8-fold less purifying selection than D. melanogaster (37%–53%) (Siepel et al. 2005; Sella et al. 2009) and threefold more than Homo sapiens (5.5%) (Lindblad-Toh et al. 2011), consistent with the hypothesis that eusocial insects have reduced selection efficiency (see above).
When grouping genes by the location of proximal CEs, we recovered several significant functional categories. For example, 2883 genes harboring promoter CEs are enriched for 35 categories pertaining to system, organ, and anatomical structure development, signal transduction, and cell differentiation (FDR < 0.05). Also, the 2721 genes associated with the top 5% of CEs (ultraconserved elements) (Supplemental Fig. 16F) are enriched for 113 categories that are not represented among all CEs (FDR < 0.05) (Supplemental Table 7), 24 (21%) of which identify nervous system regulation as a key process associated with strongest conservation in ants. This is consistent with the significant differences in brain structure seen in many ants between workers and queens, worker subcastes, and age-dependent worker task groups (Gronenberg et al. 1996).
To examine whether CEs exhibit conservation beyond primary sequence, we predicted their secondary structures and identified 3318 significant structural CEs (Supplemental Methods). Most of these structures are short (91% <15 nt), likely forming hairpins, and the majority are located near protein-coding genes (61% ≤5 kb, 37% ≤1 kb) (Supplemental Table 8). While structural CEs are enriched in likely 3′ UTRs (P < 10−15), similar to vertebrate genomes (Parker et al. 2011), 60% are intergenic, suggestive of functional small noncoding RNAs (see below). Genes near structural CEs are enriched for functional categories related to development (e.g., imaginal disc–derived wing morphogenesis, specification of segmental identity and head) and cellular dynamics (cell motility, cell migration) (Supplemental Table 9). These results indicate that DNA sequences conserved among ants identify genes and regulatory processes known to be involved in the transition to and elaboration of eusociality.
Given the abundance of conserved regulatory elements in ant genomes, we examined three mechanisms previously implicated in the regulation of social traits or phenotypic plasticity: direct modification of DNA by methylation (Kucharski et al. 2008; Bonasio et al. 2012; Smith et al. 2012), transcriptional and translational regulation by small RNAs (Pauli et al. 2011), and transcriptional regulation by transcription factors (TFs) (Rebeiz et al. 2011).
Ant genomes exhibit distinct signatures of DNA methylation
DNA methylation has been implicated in regulating gene function in social insects (Glastad et al. 2011). For example, relative depletion of CpG dinucleotides (CpG O/E, a sequence-based signature of DNA methylation) correlates negatively with DNA methylation and distinguishes classes of genes that are differentially expressed between honeybee queens and workers (Elango et al. 2009). We found that all seven ants are distinct from the honeybee in exhibiting unimodal CpG O/E distributions and significantly less CpG depletion over exons genome-wide (i.e., higher mean CpG O/E) (Fig. 3A). Interestingly however, H. saltator (a basal ant in our analysis) exhibits moderately greater exonic CpG depletion than the six other ants (Fig. 3A). In contrast, CpG O/E patterns over introns and promoters are broadly similar across Hymenoptera with little CpG depletion. Moreover, ant CEs do not differ substantially from genomic background in terms of CpG depletion (Fig. 3A). These observations are consistent with a coarser, domain-scale analysis of GC bias (Supplemental Figs. 17–21; Supplemental Table 10; Supplemental Text 3), in which eusocial insects, especially ants, show a negative relationship between genome-wide GC content and exonic bias toward GC-rich domains (R = −0.93, P < 0.0007) (Supplemental Fig. 21). These analyses confirm that the honeybee is an outlier among hymenopterans in terms of sequence-based patterns of DNA methylation.
Figure 3.

DNA methylation profiles in ant genomes. (A) Normalized CpG content (CpG O/E) of different genomic elements, including exons, introns, and promoter regions (1.5 kb upstream of coding sequence start sites) for protein-coding genes, nongenic conserved elements, and genome-wide background (1-kb fragments). Exons show the strongest evidence of CpG depletion in ants, indicating that they are the most highly methylated regions of the genome in all taxa (confirmed by Bisulfite-seq; below). Introns also show slight depletion of CpGs in ants, suggesting some intron methylation. (B) Scatterplot of log10(mCG/CG) methylation levels estimated by Bisulfite-seq versus CpG O/E for coding sequences in S. invicta reveals a bimodal distribution of gene body methylation. (C) Average methylation levels (mCG/CG) for protein-coding genes in S. invicta males, grouped according to the number of taxa in Hymenoptera with orthologs for each gene; indicating that conserved genes tend to be highly methylated. Error bars indicate 95% confidence intervals for the mean.
To confirm statistical patterns of DNA methylation experimentally, we generated a complete bisulfite-sequence map for S. invicta. We found that high levels of DNA methylation (mCG/CG) correspond well with CpG depletion in exons (Spearman's R = −0.53) (Fig. 3B; Supplemental Tables 11,12), indicating that some genes in ants are distinguished by DNA methylation, similar to the honeybee (see also Bonasio et al. 2012; Smith et al. 2012). Functional analysis of genes putatively methylated in all seven ant genomes (low CpG O/E) revealed enrichment for housekeeping functions, including transcription, translation, and cellular metabolic function (FDR < 0.05) (Supplemental Tables 13–15), as reported for the honeybee (Elango et al. 2009). Next, we computed the average methylation level of genes, grouped by the number of hymenopteran species with orthologs. Interestingly, levels of DNA methylation (mCG/CG and CpG O/E) increase with evolutionary conservation of protein-coding genes, especially when genes have orthologs in more distant Hymenoptera (Fig. 3C). Thus, highly conserved genes are preferentially targeted by DNA methylation. We also note that CpG O/E is a poor predictor of methylation for genes with paralogs (i.e., multicopy genes), regardless of orthology (Supplemental Figs. 22–24; Supplemental Tables 11, 12). Thus, while ants and the honeybee exhibit significantly distinct statistical patterns predictive of DNA methylation, all insects that possess DNA methyltransferases likely methylate exons of highly conserved genes, indicating a common role for DNA methylation independent of eusociality.
Conserved miRNAs and small noncoding RNAs exhibit caste differential expression
Recent investigations have uncovered novel regulatory roles for various classes of noncoding RNAs (e.g., Loewer et al. 2011). We first evaluated known miRNA genes (Bonasio et al. 2010) and found that 63 are highly conserved among ants (average ∼80% conservation) (Supplemental Fig. 16D). We then reannotated the seven ant genomes for miRNAs and uncovered 24 novel loci, 18 of which are specific to ants (Supplemental Table 16). Using RNA-seq gene expression data, we confirmed that a total of 115 miRNAs are expressed in C. floridanus, including 20 of the novel miRNAs. Several miRNAs show stage- and caste-specific expression (Supplemental Fig. 25), and many, including 12 of the novel ant-specific miRNAs, are predicted to share orthologous gene targets among ant species, typically located in 3′ UTRs (data not shown).
We also used several small and poly(A)+ RNA-seq data sets (Bonasio et al. 2010) to identify more than 70,000 CEs that overlap transcribed sequences in C. floridanus and H. saltator. Most transcribed CEs (∼64%) are intergenic, including 23,000 CEs located more than 2 kb upstream of protein-coding genes (Supplemental Table 17); these CEs comprise a class of predominantly small, conserved noncoding RNAs. Many conserved noncoding RNAs show moderate differences in expression level between adult worker castes, notably a group of 2290 RNAs that overlap CpG islands and show the highest median expression difference between C. floridanus worker castes (Supplemental Fig. 26A,C). Interestingly, CpG islands are also the only nonexonic regions significantly enriched for CEs (P < 0.01) (Supplemental Fig. 16D, top), and CpG island RNA expression levels correlate positively with expression levels of the nearest downstream protein-coding genes (0.1 ≤ R ≤ 0.5), with stronger correlations generally found for CpG islands closer to genes (Supplemental Fig. 26B). Functional analysis of these downstream genes revealed a striking enrichment for regulatory processes targeting neuron differentiation and neurogenesis, steroid hormone signaling, cell differentiation, and gene expression (FDR < 0.1) (Supplemental Table 18). These results support the notion that ant CpG islands, which are broadly hypomethylated in ants (Bonasio et al. 2012; this study), may serve a regulatory role, perhaps by harboring enhancer binding sequences or by being preferentially targeted by regulators of chromatin structure (Ramirez-Carrozzi et al. 2009), where small RNAs may be directly or indirectly involved (Kim et al. 2010; Pauli et al. 2011).
Genome-wide evolution of TF-binding sites is more divergent within ants than between eusocial and solitary insects
The genomic organization of sequence-specific TF-binding sites (TFBSs) represents a profound source for transcriptional regulatory variation potentially used during the evolution of insect sociality (Gadau et al. 2012). We evaluated the extent of TF-mediated regulatory evolution by analyzing the genome-wide occurrence and distribution of 59 TFBSs (Supplemental Fig. 27), corresponding to developmentally expressed TFs that exhibit broad conservation in their genomic copy number (i.e., they do not belong to evolutionarily variable gene families) (Supplemental Table 19) and nonadaptive coding sequence evolution (93% of branch tests for positive selection in TF loci were not significant) among insects (Supplemental Fig. 28; Supplemental Table 20). Indeed, 26% of all CEs and 48% of ultraconserved CEs harbor at least one TFBS (Supplemental Fig. 29A), confirming that conserved regions are broadly enriched for regulatory elements in ants. In fact, most of these CEs harbor multiple TFBSs (average 4.3 TFBSs/CE) (Supplemental Fig. 29B), suggesting that preservation of TF coregulation is also important.
We proceeded to examine whether ant genes that harbor conserved TFBSs in their promoters (0–2 kb upstream of ORFs) exhibit evolutionary changes in TF regulation among insects, including the eight eusocial species and 20 solitary species. For most TFs, the average number of promoter binding sites remains similar across insects, although a few TFs do show overall gains in Hymenoptera (e.g., Antennapedia, Giant) (Fig. 4A). To evaluate the extent of divergence of TF regulation, we compared genome-wide TFBS profiles by computing Euclidean distances between species using the number of TFBSs per gene per TF (Fig. 4A, right; Supplemental Fig. 30). Eusocial species (especially ants) exhibit striking divergence in promoter TFBSs that is greater than their divergence from solitary species. Moreover, the two leaf-cutter ants differ more from each other than the two most divergent flies, and the honeybee shows greater divergence from ants than from flies. This suggests that while many TFBSs remain conserved in ants, the overall architecture of TF-mediated gene regulation is highly variable across insects, especially between convergently evolved eusocial lineages in Hymenoptera.
Figure 4.

Evolution of transcription factor binding sites (TFBS) in insects. (A) Heatmap showing number of promoter TFBSs per gene for 59 TFs in 28 insect species (n = 4189 genes associated with 2-kb promoter CEs in ants). Species (rows), ordered by phylogenetic grouping, are denoted as solitary (blue) or eusocial (red). TFs were clustered hierarchically using average linkage by computing Euclidean distance between TFBS profiles over all queried genes. (Right) Boxplots show distributions of Euclidean distance values for pairs of species, computed using genome-wide TFBS abundance profiles over genes and TFs (see Supplemental Fig. 30). Each boxplot reflects a group of paired comparisons. P-values estimated by two-tailed Mann–Whitney U-test. (**) P < 10−5; (***) P < 10−10. (B) Genes and TFs exhibiting significant TFBS evolution between solitary and eusocial groups. Three thousand two hundred and thirty-one of 4189 genes had sufficient data for significance testing. (*)TF with significant promoter TFBS evolution (two-tail Mann–Whitney U-test; FDR < 0.25). Top two rows indicate numbers of genes showing significant gain or loss of binding sites for the specified TF. Bottom row indicates proportion of significant genes showing more TFBSs in eusocial compared with solitary insects. More than 93% of tested genes are single-copy in the ant genomes. Bottom panels show the mean and standard error of the standard deviation in RNA expression levels (y-axis) for 96 genes with greatest significance in multiple TFs (top 5%), grouped by TF. Expression levels estimated by log2(FPKM + 1). (FPKM) Fragments per kilobase per million reads. (*) Significantly increased caste variation in RNA expression (compared with all ant orthologs, Background, P < 0.05). (C) TFBS abundance profiles for significant genes, shown for three TFs. Species order (x-axis) as in A. (D) TFBS abundance profiles for two neuronal genes with significance in multiple TFs. Cell colors are row-normalized. Periods (.) Missing data. P-values were computed by a Mann–Whitney U-test. (E) mRNA expression level estimates for the genes in D, shown for different worker castes in H. saltator (reproductive/nonreproductive) and C. floridanus (major/minor). Error bars indicate standard error over three biological replicates. (**) FDR < 0.01; (*) FDR < 0.25.
Ant genomes exhibit similar patterns of cis-regulatory evolution associated with evolutionary increased gene expression plasticity between worker castes
To evaluate whether gains or losses of TFBSs are specifically maintained in eusocial insects but not solitary insects, we examined whether individual gene promoters exhibit changes in TFBS abundance between eusocial (n = 8) and solitary (n = 20) species. Indeed, we identified nearly 2000 significant genes (FDR < 0.25) (Supplemental Fig. 29C; Supplemental Tables 21, 22), which represent potential drivers of the genome-wide divergence pattern (see above). This analysis implicates 30 TFs, 16 of which are associated with more than 100 significant genes each (Fig. 4B). Most of the significant TFs show either predominant gains (n = 7) or losses (n = 8) of TFBSs in the eusocial genomes (e.g., SHN and EMS), although a few TFs show more complex patterns of gains and losses (e.g., CREB) (Fig. 4C). We also identified 292 genes that exhibit significant changes in TFBS abundance for multiple TFs, i.e., apparent targets of concentrated cis-regulatory rewiring (Fig. 4D). These 292 genes are enriched for 41 functional categories involved in hormone regulation and transcription factor activity (FDR < 0.05) (Supplemental Table 23) and include nervous wreck, which regulates synaptic growth and neurotransmission (Coyle et al. 2004), and choline O-acetyltransferase, a key enzyme for synthesizing the neurotransmitter acetylcholine (Fig. 4D). This suggests that the insect neuroendocrine system has been targeted for regulatory changes during eusocial evolution. Thus, specific TF regulatory proteins conserved among insects exhibit significant divergence in their targets of regulation between eusocial and solitary lineages.
We next assessed whether any genes exhibit regulatory changes specifically in ants, and we found 141 genes with significant TFBS gains in ants but zero predicted binding sites for the majority (≥80%) of other species (Supplemental Fig. 31), including the honeybee (Supplemental Fig. 32). Intriguingly, evolution of binding sites for CREB, a TF regulator of long-term memory (CREBB, Ishimoto et al. 2009) and secretory activity (CREBA, Abrams and Andrew 2005) in insects, affects the most genes in this analysis (n = 31). This suggests that ants may have preferentially altered the binding distribution of CREB as a possible means to achieve gene expression plasticity between specialized castes (see below). Indeed, evolutionary gains/losses of CREB-binding sites perfectly discriminate eusocial from solitary species (and ants from the honeybee) in a Principle Components Analysis (Supplemental Fig. 33). Moreover, one of CREB's cofactors, the transcriptional coactivator and histone acetyltransferase CBP, was recently reported to play a role in maintaining caste-specific gene expression patterns in C. floridanus (Simola et al. 2013). These results suggest that while many genes show significant cis-regulatory changes specific to ants, the majority (>90%) of genes with significant eusocial-associated regulatory evolution tend to exhibit similar changes in both ants and the honeybee, broadly suggestive of the importance of cis-regulatory changes in the evolutionary origins of or convergence on eusociality.
Since TF-binding events regulate gene expression levels, we proceeded to examine whether changes in TFBS abundance between ant species may be indicative of evolutionary increases in gene expression plasticity between castes within a species. Interestingly, genes with the most significant changes in TFBS abundance between eusocial and solitary insects show elevated levels of plasticity in a socially sophisticated ant, C. floridanus, compared to H. saltator, whose colonies are smaller and exhibit less reproductive division of labor (average 0.39 vs. 0.27, P < 10−8) (Fig. 4B, bottom). Furthermore, different TFs show significantly different levels of plasticity in C. floridanus (P < 0.05) but not H. saltator (P < 0.13) (Fig. 4B, bottom), as well as correlations in plasticity between species when grouped by TF (R = 0.65, P = 0.002) or for individual genes (R = 0.30, P = 0.003) (Supplemental Fig. 34). Gene targets of nine TFs show greater plasticity compared with all ant orthologs (P < 0.05) (Fig. 4B, asterisks), notably for Empty spiracles (EMS), which regulates brain morphogenesis and antennae development in Drosophila (Cohen and Jürgens 1990), and CREB (see above), which shows the second largest effect (albeit not significant in this analysis). These results suggest that caste-associated gene expression plasticity is a continuously evolving trait in eusocial insects that is partly determined by TFBS abundance (see also Supplemental Fig. 33).
Known eusocial pathways exhibit cis and trans regulatory evolution for several TFs
Finally, we analyzed patterns of regulatory evolution in the salivary gland and wing development regulatory networks, which are known to exhibit phenotypic plasticity between workers and queens and between different worker castes and task groups (Abouheif and Wray 2002; Li and White 2003). We were struck by the over-representation of TFs associated with eusocial regulatory evolution (see above) among key regulators of these networks (Fig. 5A,B; Supplemental Text 4). In particular, fork head (FKH), an essential regulator of insect salivary glands and labial silk gland development in Bombyx mori (Mach et al. 1995), has undergone considerable loss of TFBSs in its own promoter in the eusocial genomes (Fig. 5C)—an example of trans-regulatory evolution that may confer pleiotropic effects. Furthermore, the regulatory network for wing development in ants harbors three TFs—abdominal A (abdA), snail (sna), and engrailed (en)—for which we found significant changes in TFBS abundance; engrailed was previously shown to be down-regulated in wing discs of workers compared with queens during larval development in two ant species (Abouheif and Wray 2002). These observations support the hypothesis that regulatory rewiring both in promoters of loci encoding TF regulators (trans-regulatory evolution) and in promoters of target loci (cis-regulatory evolution) may serve as an evolutionary driving force enabling morphological and behavioral plasticity in eusocial insects.
Figure 5.

Transcription-factor-mediated signaling pathways controlling salivary gland development. (A) Sex combs reduced (SCR) in combination with extradenticle (EXD) and homothorax (HTH) direct the specification of cells to the salivary gland fate in PS2 of the Drosophila embryo; these TFs are essential for the downstream regulation of genes required for gland cell differentiation and morphogenesis. Boundaries of salivary gland development are restricted along the anterior/posterior axis by abdominal B (Abd-B) and teashirt (TSH), along the dorsal axis by decapentaplegic (DPP) signaling, and along underlying mesoderm by twist (TWI) and snail (SNA). Epidermal growth factor (EGF) signaling determines the decision to differentiate into duct or secretory cells. (B) Regulation of programmed cell death of embryonic salivary gland by 20-hydroxyecdysone (20E). Cell death is inhibited by fork head (FKH) expression in embryonic salivary gland cells. A pulse of 20E at the late larval stage triggers broad (BR-C)–mediated FKH inhibition. A second pulse of 20E at the prepupal stage leads to BR-C and ecdysone-induced protein 74EF (EIP74EF) directed transcription of apoptotic genes, including wrinkled (HID). TFs associated with TFBS evolution in more than 100 target genes (cis-regulatory evolution) (see Fig. 4) (red). The promoter of the fork head locus (yellow), which encodes a TF involved in the regulation of secretory cells, shows significant changes in the gain or loss of promoter TFBSs in ants (trans-regulatory evolution), as detailed in C. (Right) mRNA expression level estimates for fork head, shown for different worker castes in H. saltator (reproductive/non-reproductive) and C. floridanus (major/minor). Error bars indicate standard error over three biological replicates.
Discussion
Given the remarkable phenotypic diversity among eusocial insects, a key question is to what extent do derived and independent eusocial lineages harbor shared features of genomic organization that enable a eusocial lifestyle. Our analysis of eight “socio-genomes” (Robinson et al. 2005), together with several solitary insect genomes, suggests that despite a pattern of broad sequence divergence expected by deep evolutionary history, key changes in gene regulation (both in cis and in trans) may have convergently evolved in the early stages of eusocial evolution, whereas changes in gene composition may have been more important for lineage-specific social and ecological adaptations. Ant intergenic sequences are enriched for regulatory elements, including miRNAs, noncoding RNAs, and TF-binding sites that are conserved in both DNA sequence and homologous genomic position. Nearly 2000 genes share similar cis-regulatory changes in eusocial compared with solitary insects, and most of these changes are similar in both eusocial lineages of ants and honeybees. These genes are enriched for neuronal and hormonal functions and implicate a few specific TFs in eusocial evolution, notably EMS and CREB. Changes in TFBS abundance are linked to evolutionary increases in phenotypic plasticity from ants with a simpler social organization to those with more complex social organization. The locus encoding one key TF, fork head, also shows significant regulatory evolution in ants, which has potentially facilitated pleiotropic effects in trans for regulatory networks relevant to eusociality, e.g., exocrine gland and wing development. Finally, six gene families with putative transcription regulatory activity experienced significant gains in family size during early ant evolution.
Our analysis of evolutionary changes in gene composition in ants contextualizes these results. We found an abundance of taxonomically restricted genes (TRGs) in ants as well as a higher rate in the emergence of TRGs in ants compared with flies, suggesting functional ties to eusocial adaptations. In other systems, TRGs can comprise 10%–33% of a species' protein-coding gene complement and have been linked both to morphological adaptations (Khalturin et al. 2009; Tautz and Domazet-Lošo 2011) and to eusocial traits, including caste differentiation (Kamakura 2011) and complex behavioral repertoires (Johnson and Tsutsui 2011). Importantly, while TRGs likely play important roles involved in the elaboration of social adaptations in individual lineages, TRGs that are critical for early eusocial evolution or the maintenance of eusociality should be conserved in multiple ant genomes. We found 64 ant-specific TRGs that are conserved in all seven ant genomes; however, these genes show limited differential expression between adult worker castes in two different species, at least using pooled tissue data. These novel genes may be relevant for the evolution of eusociality in ants, but their specific functional significance remains unclear.
In conclusion, evolutionary changes in gene regulation seem to dominate our view of the shared genomic features associated with the origins of eusociality. However, the broad spectrum of changes observed in eusocial insect genomes suggests that the origin, maintenance, and elaboration of insect eusociality was not necessarily restricted to a small set of genes or regulatory elements. Instead, the organization of eusocial insect genomes appears to harbor sufficient degrees of freedom to allow convergence of higher-order complex traits, such as eusociality, from unique, lineage-specific evolutionary trajectories that involve distinct genes and modes of regulation. Such genomic complexity may be especially engendered by ants, where extreme reproductive divisions of labor resulting from a eusocial lifestyle may effectively reduce the strength of natural selection, thereby facilitating rapid sequence divergence among lineages.
Methods
In addition to the Methods described below, the Supplemental Material includes information for the following: quality assessment of annotated genes (see also Supplemental Figs. 3, 4; Supplemental Table 1), identification of paralogous genes (see also Supplemental Figs. 7, 8), codon usage bias (see also Supplemental Fig. 9; Supplemental Text 2; Supplemental Table 4), gene family evolution (see also Supplemental Fig. 10), multiple genome alignment (see also Supplemental Fig. 12), synteny analysis (see also Supplemental Figs. 13, 14), conserved structural RNA analysis (see also Supplemental Table 8), GC compositional analysis (see also Supplemental Figs. 17–21; Supplemental Text 3), and testing positive selection of single-copy orthologs (see also Supplemental Fig. 28).
Assessing homology of known genes among insect species (AntOrthoDB)
The OrthoDB orthology delineation procedure (Waterhouse et al. 2011) was used to delineate orthologous genes at each radiation along the insect species phylogeny, which includes seven sequenced ant and five outgroup insect species (Supplemental Table 1). OrthoDB has been updated to include these results, along with protein descriptors, Gene Ontology, and InterPro attributes (http://cegg.unige.ch/orthodbants) (Supplemental Fig. 2). See Quality Assessment of Annotated Gene Sets in AntOrthoDB in the Supplemental Material for additional information.
Identification of taxonomically restricted genes
Taxonomically restricted genes (TRGs) were identified as protein-coding genes that lack sequence similarity to annotated proteins outside of a focal taxonomic group (e.g., Formicidae) (Fig. 1B), using the official gene sets for 30 arthropod species. Ortholog identification was based on all versus all BLASTP searches among all annotated proteins (E < 1 × 10−3). For Formicidae TRGs, BLAST hits within ant genomes were ignored. Among TRGs, subsets of genes that only occur within an individual species were identified and denoted as lineage-specific genes (LSGs), given our taxon sampling. Among LSGs, genes were conservatively filtered if the gene (1) matched in other proteomes when low complexity filtering was deactivated, (2) matched proteins in SwissProt taxonomic divisions (except invertebrates) as potential contaminations, or (3) appear not to be lineage specific if a similar sequence with matching gene model was found in the genome. To control for false LSGs, putative homologs of genes that were predicted in only one of the ant species were screened against the genomes of the other eight Hymenoptera using a custom-built pipeline. True missing genes had to (1) yield a significant BLAST hit in another genome using its predicted peptide sequence as query for TBLASTN (E ≤ 1 × 10−5, low-complexity filtering activated), and (2) yield a seemingly functional gene model based on the alignment of the protein query against the genomic sequence. GeneWise v2.4.1 was used to align the protein query against the scaffold in a strand- and position-specific manner. Strandedness and position (±50 kb) were derived from the TBLASTN hit. Only GeneWise models with a score >35, coverage of the query sequence >75%, and zero indels (ORF-disrupting frameshift mutations) were accepted as valid gene models. Applying this procedure yielded a total of 2936 (previously lineage-specific) genes, along with 6369 newly identified genes that produced valid gene models in multiple Hymenoptera species (Supplemental Fig. 5). This yields a total of 12,054 LSGs within Formicidae (42.2% of all 28,581 Formicidae TRGs).
Insect phylogeny
The phylogeny shown in Figure 1 was estimated by maximum likelihood from the concatenated alignment of conserved protein sequences of 2756 single-copy orthologs across 12 insect species, comprising 792,477 well-aligned amino acids. Sequence alignments were performed with MAFFT (Katoh et al. 2002), conserved cores were selected with Gblocks (Castresana 2000), and the phylogeny was built with PhyML (Guindon et al. 2010). Using 4346 single-copy orthologs defined across the seven ant species, A. mellifera and N. vitripennis, protein lengths were compared to examine the agreement of ant genes with those of their bee and wasp orthologs (Supplemental Fig. 6; Supplemental Table 2).
Conserved elements
Conserved elements were identified from whole-genome sequence alignments using phastCons (Siepel et al. 2005). Conserved and nonconserved HMMs were estimated with parameters (-target -coverage 0.25 -expected -length 12 -estimate-trees), given a phylogenetic tree (described above) for initialization. Resulting conserved elements (CEs) were filtered to remove any regions whose consensus sequence consisted of gaps only. A subset of ultra-conserved elements (UCEs) were identified as those CEs having length ≥5 nt and LOD/length scores in the top fifth percentile of all CEs, where LOD denotes the log-odds ratio of the posterior probability of conservation to that of nonconservation across the nucleotides delimited by the CE. Noncoding conserved RNAs are defined as transcribed ant CEs and do not overlap annotated ant exonic sequences. Specifically, for each CE, associated DNA sequences from each of seven ant genomes as well as their consensus sequences were locally aligned to all annotated exon sequences from each of the genomes using nucleotide BLAST (E-value of 10). A CE was considered to be exonic if any one associated DNA sequence showed significant alignment to any one exon from any ant genome. CEs were annotated as transcribed if at least 25% of the element overlaps an annotated transcript whose expression level is greater than the fifth percentile of genome-wide expression levels in at least two independent biological samples, for each species (see RNA Expression Analysis, below).
DNA methylation
Normalized CpG dinucleotide content (CpG O/E) was calculated using the equation:
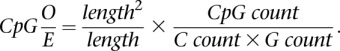 |
Bisulfite-seq data were obtained using genomic DNA from six pooled whole-body haploid males of S. invicta from a single colony (NCBI GEO accessions GSE39959). Bisulfite conversion and sequencing were performed by the Beijing Genomics Institute (Shenzhen, China). Bisulfite treatment was performed using the EZ DNA Methylation-Gold Kit (Zymo Research Corporation). Sequencing was performed using the Illumina HiSeq2000. Reads passing quality control were mapped using Bowtie and Bismark (Langmead et al. 2009; Krueger and Andrews 2011). Aligned reads were processed using SAMtools to remove PCR duplicated reads (rmdup) and parsed using custom Perl scripts to obtain methylated and unmethylated read counts on a per-nucleotide basis. Fractional methylation was calculated for each CpG site with three or more reads as mCG/CG, where mCG is the number of reads with methylated cytosine at a CpG site (according to bisulfite conversion), and CG is the total number of reads with either unmethylated or methylated (converted and unconverted) cytosine at the same CpG site. Fractional methylation values were averaged across each annotated element with data for three or more sites (otherwise the element was discarded). Functional enrichment was performed using Gene Ontology annotation of single-copy D. melanogaster orthologs of H. saltator genes belonging to single-copy seven-ant orthologs, as analyzed by the DAVID (Huang et al. 2009a; Huang et al. 2009b) functional annotation tool.
MicroRNA identification
Hymenoptera small RNA sequences were downloaded from the NCBI Sequence Read Archive (SRA) database (Accession numbers: SRX018737, SRX023147–SRX023156) and searched against the ant genome assemblies using BLASTN. Alignments having ≤2-nt mismatches were retained and analyzed using MIREAP with a minimum folding energy of −18 kcal/mol (http://sourceforge.net/projects/mireap/). An miRNA was considered ant specific when the precursor hairpin and subsequent mature miRNA were present in ≥4/7 ant genomes. This conservative strategy ignores other likely ant-specific miRNAs absent in the current assemblies. Novel miRNA coordinates are listed in Supplemental Table 16.
Target analysis was performed after aligning novel, ant-specific miRNAs to the annotated ant genes. As a proxy for 3′ UTRs, 750 bp of sequence downstream from each stop codon was extracted. miRNA:mRNA target analysis was performed using miRanda (Enright et al. 2003) with parameters: -sc 140 -en 20. Target predictions were considered conserved if at least four ant orthologous ant genes were targeted by the respective miRNA using OrthoDB.
Identification and analysis of transcription factor–binding sites
Identification of TFBS in insects
Position weight matrices (PWMs) for 56 transcription factors (TFs) were taken from DMMPMM (Bigfoot) and iDMMPMM (Kulakovskiy et al. 2009). PWMs for CREB (CREBA and CREBB) and the promoter elements CPE and DPE were obtained from Transfac (http://www.gene-regulation.com). Sequence motifs showing significant similarity to each PWM were predicted using pwm_scan (Levy and Hannenhalli 2002). For each of 28 insect species (as described in text), each PWM was scanned across the 2-kb promoters of protein-coding genes whose orthology among species was established by OrthoDB. To obtain stringent TFBS predictions, an empirical score distribution was estimated for each PWM for each species' genome as the set of all nominally significant (P < 0.05) motif scores identified within the target set of promoter sequences [pwm_scan -s 1 -p ln(0.05)]. Candidate binding sites were then selected for each genome as those scoring in the top 0.02% among this set of nominally significant sites [pwm_scan -s 2 -p ln(1/5000)], where 1/5000 is the recommended P-value roughly estimating expected frequency (Levy and Hannenhalli 2002). (See also Supplemental Figs. 27, 29.)
Between species comparison of number of TFBSs
Each gene of interest was tested for showing a difference in the number of proximal promoter-binding sites for each TF in eight eusocial species compared with 20 solitary species using a two-tailed Mann–Whitney (rank-sum) U-test. To control for potential bias due to variation in nucleotide frequency, GC bias was estimated from the promoter sequences of each genome and used to scale the number of binding sites for each motif x: GC(x)/avg(GC). Only genes having TFBS estimates for at least three eusocial and five solitary (or vice versa) species were used for evaluation. Significant genes were identified using a Benjamini–Hochberg false discovery rate (FDR) of 25%.
Genes with TFBS changes for multiple TFs
Each gene showing a significant change in TFBS abundance between eusocial and solitary genomes was also tested for significant change for multiple TFs by summing the number of 2-kb promoter TFBSs for all TFs and testing for significant difference in total TFBS abundance between eusocial and solitary genomes using a t-test. Genes with |T| values in the top 15% overall were retained.
RNA expression analysis
Raw RNA-seq expression data for C. floridanus and H. saltator were downloaded from NCBI GEO using accession number GSE22680 (Bonasio et al. 2010) or GSE37523 (Simola et al. 2013). Raw sequence reads were mapped using Bowtie + TopHat (Langmead et al. 2009) allowing one mismatch and up to 50 alignments per read (-v 1 -k 50 -best) and default parameter values otherwise. Expression levels for previously annotated gene models were quantified with these maps using Cufflinks (Trapnell et al. 2010), correcting for fragment bias (-frag -bias -correct) and uncertain alignment location (-multi-read -correct) and default parameter values otherwise. Expression levels are reported as log2(FPKM+ 1) unless otherwise stated.
Data access
Sequencing data for DNA methylation in S. invicta have been deposited in the NCBI Gene Expression Omnibus (GEO; http://www.ncbi.nlm.nih.gov/geo/) under accession number GSE39959. Additional supplemental files are freely available for download from the Hymenoptera Genome Database (http://hymenopteragenome.org/ant_genomes/?q=consortium_datasets) (Munoz-Torres et al. 2011).
Acknowledgments
This work was supported by grants from NSF to J.G. and C.R.S. (IOS-0920732) and a Howard Hughes Medical Institute Collaborative Innovation Award #2009005 to D.R., S.L.B., and J.L. D.F.S. was supported in part by an NRSA post-doctoral fellowship from the University of Pennsylvania Department of Cell and Developmental Biology. L.V. was supported in part by Academy of Finland grant #130290. B.H., K.G., and M.G. were supported by the U.S. National Science Foundation (grant numbers DEB-1011349, DEB-0640690, and IOS-0821130) and the Georgia Tech-Elizabeth Smithgall Watts endowment. Y.W. was supported in part by the BBSRC (grant BB/K004204/1). S.N. and J.J.B. were supported by the Danish National Research Foundation. R.M.W. was supported by SNSF 125350 and 143936 to E.M.Z. J.R., E.P., L.K., and Y.W. were supported by grants from the Swiss NSF and En ERC advanced grant. We thank Mira Han for help with running and interpreting the gene family evolution analyses.
Footnotes
[Supplemental material is available for this article.]
Article published online before print. Article, supplemental material, and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.155408.113.
References
- Abouheif E, Wray GA 2002. Evolution of the gene network underlying the wing polyphenism in ants. Science 297: 249–252 [DOI] [PubMed] [Google Scholar]
- Abrams EW, Andrew DJ 2005. CrebA regulates secretory activity in the Drosophila salivary gland and epidermis. Development 132: 2743–2758 [DOI] [PubMed] [Google Scholar]
- Bonasio R, Zhang G, Ye C, Mutti NS, Fang X, Qin N, Donahue G, Yang P, Li Q, Li C, et al. 2010. Genomic comparison of the ants Camponotus floridanus and Harpegnathos saltator. Science 329: 1068–1071 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonasio R, Li Q, Lian J, Mutti NS, Jin L, Zhao H, Zhang P, Wen P, Xiang H, Ding Y, et al. 2012. Genome-wide and caste-specific DNA methylomes of the ants Camponotus floridanus and Harpegnathos saltator. Curr Biol 22: 1755–1764 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cardinal S, Danforth BN 2011. The antiquity and evolutionary history of social behavior in bees. PLoS ONE 66: e21086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castresana J 2000. Selection of conserved blocks from multiple alignments for their use in phylogenetic analysis. Mol Biol Evol 17: 540–552 [DOI] [PubMed] [Google Scholar]
- Cohen SM, Jürgens G 1990. Mediation of Drosophila head development by gap-like segmentation genes. Nature 346: 482–485 [DOI] [PubMed] [Google Scholar]
- Coyle IP, Koh YH, Lee WCM, Slind J, Fergestad T, Littleton JT, Ganetzky B 2004. Nervous wreck, an SH3 adaptor protein that interacts with Wsp, regulates synaptic growth in Drosophila. Neuron 41: 521–534 [DOI] [PubMed] [Google Scholar]
- Crozier RH, Pamilo P 1996. Evolution of social insect colonies: Sex allocation and kin selection. Oxford University Press, New York [Google Scholar]
- Drosophila 12 Genomes Consortium 2007. Evolution of genes and genomes on the Drosophila phylogeny. Nature 450: 203–218 [DOI] [PubMed] [Google Scholar]
- Elango B, Hunt BG, Goodisman MAD, Yi SV 2009. DNA methylation is widespread and associated with differential gene expression in castes of the honeybee, Apis mellifera. Proc Natl Acad Sci 106: 11206–11211 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Enright AJ, John B, Gaul U, Tuschl T, Sander C, Marks DS 2003. MicroRNA targets in Drosophila. Genome Biol 5: R1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernández-Marín H, Zimmerman JK, Nash DR, Boomsma JJ, Wcislo WT 2009. Reduced biological control and enhanced chemical pest management in the evolution of fungus farming in ants. Proc Biol Sci 276: 2263–2269 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferreira PG, Patalano S, Chauhan R, Ffrench-Constant R, Gabaldon T, Guigo R, Sumner S 2013. Transcriptome analyses of primitively eusocial wasps reveal novel insights into the evolution of sociality and the origin of alternative phenotypes. Genome Biol 14: R20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gadau J, Helmkampf M, Nygaard S, Roux J, Simola DF, Smith CR, Suen G, Wurm Y, Smith CD 2012. The genomic impact of 100 million years of social evolution in seven ant species. Trends Genet 28: 14–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glastad KM, Hunt BG, Yi SV, Goodisman MAD 2011. DNA methylation in insects: On the brink of the epigenomic era. Insect Mol Biol 20: 553–565 [DOI] [PubMed] [Google Scholar]
- Gronenberg W, Heeren S, Hölldobler B 1996. Age-dependent and task-related morphological changes in the brain and the mushroom bodies of the ant Camponotus floridanus. J Exp Biol 199: 2011–2019 [DOI] [PubMed] [Google Scholar]
- Guindon S, Dufayard JF, Lefort V, Anisimova M, Hordijk W, Gascuel O 2010. New algorithms and methods to estimate maximum-likelihood phylogenies: Assessing the performance of PhyML 3.0. Syst Biol 59: 307–321 [DOI] [PubMed] [Google Scholar]
- Hahn MW, Han MV, Han SG 2007. Gene family evolution across 12 Drosophila genomes. PLoS Genet 3: e197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hölldobler B, Wilson EO 2009. The superorganism. W.W. Norton, New York [Google Scholar]
- The Honeybee Genome Sequencing Consortium 2006. Insights into social insects from the genome of the honeybee Apis mellifera. Nature 443: 931–949 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang DW, Sherman BT, Lempicki RA 2009a. Bioinformatics enrichment tools: Paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res 37: 1–13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang DW, Sherman BT, Lempicki RA 2009b. Systematic and integrative analysis of large gene lists using DAVID Bioinformatics Resources. Nature Protoc 4: 44–57 [DOI] [PubMed] [Google Scholar]
- Ishimoto H, Sakai T, Kitamoto T 2009. Ecdysone signaling regulates the formation of long-term courtship memory in adult Drosophila melanogaster. Proc Natl Acad Sci 106: 6381–6386 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson BR, Tsutsui ND 2011. Taxonomically restricted genes are associated with the evolution of sociality in the honey bee. BMC Genomics 12: 164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamakura M 2011. Royalactin induces queen differentiation in honeybees. Nature 473: 478–483 [DOI] [PubMed] [Google Scholar]
- Katoh K, Misawa K, Kuma K, Miyata T 2002. MAFFT: A novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic Acids Res 30: 3059–3066 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khalturin K, Hemmrich G, Fraune S, Augustin R, Bosch TC 2009. More than just orphans: Are taxonomically-restricted genes important in evolution? Trends Genet 25: 404–413 [DOI] [PubMed] [Google Scholar]
- Kim TK, Hemberg M, Gray JM, Costa AM, Bear DM, Wu J, Harmin DA, Laptewicz M, Barbara-Haley K, Kuersten S, et al. 2010. Widespread transcription at neuronal activity-regulated enhancers. Nature 465: 182–187 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krueger F, Andrews SR 2011. Bismark: A flexible aligner and methylation caller for Bisulfite-Seq applications. Bioinformatics 27: 1571–1572 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kucharski R, Maleszka J, Foret S, Maleszka R 2008. Nutritional control of reproductive status in honeybees via DNA methylation. Science 319: 1827–1830 [DOI] [PubMed] [Google Scholar]
- Kulakovskiy IV, Favorov AF, Makeev VJ 2009. Motif discovery and motif finding from genome-mapped DNase footprint data. Bioinformatics 25: 2318–2325 [DOI] [PubMed] [Google Scholar]
- Langmead B, Trapnell C, Pop M, Salzberg SL 2009. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol 10: R25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levy S, Hannenhalli S 2002. Identification of transcription factor binding sites in the human genome. Mamm Genome 13: 510–514 [DOI] [PubMed] [Google Scholar]
- Li T-R, White KP 2003. Tissue-specific gene expression and ecdysone-regulated genomic networks in Drosophila. Dev Cell 5: 59–72 [DOI] [PubMed] [Google Scholar]
- Lindblad-Toh K, Garber M, Zuk O, Lin MF, Parker BJ, Washietl S, Kheradpour P, Ernst J, Jordan G, Mauceli E, et al. 2011. A high-resolution map of human evolutionary constraint using 29 mammals. Nature 478: 476–482 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loewer S, Cabili MN, Guttman M, Loh Y-H, Thomas K, Park IH, Garber M, Curran M, Onder T, Agarwal S, et al. 2011. Large intergenic non-coding RNA-RoR modulates reprogramming of human induced pluripotent stem cells. Nat Genet 42: 1113–1117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mach V, Takiya S, Ohno K, Handa H, Imai T, Suzuki Y 1995. Silk Gland Factor-1 involved in the regulation of Bombyx Sericin-1 gene contains Fork Head motif. J Biol Chem 270: 9340–9346 [DOI] [PubMed] [Google Scholar]
- Martin S, Drijfhout F 2009. A review of ant cuticular hydrocarbons. J Chem Ecol 35: 1151–1161 [DOI] [PubMed] [Google Scholar]
- Michener CD 1969. Comparative social behavior of bees. Annu Rev Entomol 14: 299–342 [Google Scholar]
- Moreau CS, Bell CD, Vila R, Archibald SB, Pierce NE 2006. Phylogeny of the ants: Diversification in the age of angiosperms. Science 312: 101–104 [DOI] [PubMed] [Google Scholar]
- Munoz-Torres MC, Reese JT, Childers CP, Bennett AK, Sundaram JP, Childs KL, Anzola JM, Milshina N, Elsik CG 2011. Hymenoptera genome database: Integrated community resources for insect species of the order Hymenoptera. Nucleic Acids Res 39: D658–D662 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nygaard S, Zhang G, Schiott M, Li C, Wurm Y, Hu H, Zhou J, Ji L, Qiu F, Rasmussen M, et al. 2011. The genome of the leaf-cutting ant Acromyrmex echinatior suggests key adaptations to advanced social life and fungus farming. Genome Res 21: 1339–1348 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parker BJ, Moltke I, Roth A, Washietl S, Wen J, Kellis M, Breaker R, Pedersen JS 2011. New families of human regulatory RNA structures identified by comparative analysis of vertebrate genomes. Genome Res 21: 1929–1943 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pauli A, Rinn JL, Schier AF 2011. Non-coding RNAs as regulators of embryogenesis. Nat Rev Genet 12: 136–149 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramirez-Carrozzi VR, Braas D, Bhatt DM, Cheng CS, Hong C, Doty KR, Black JC, Hoffmann A, Carey M, Smale ST 2009. A unifying model for the selective regulation of inducible transcription by CpG islands and nucleosome remodeling. Cell 138: 114–128 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ranson H, Claudianos C, Ortelli F, Abrall C, Hemingway J, Sharakhova MV, Unger MF, Collins FH, Feyereisen R 2002. Evolution of supergene families associated with insecticide resistance. Science 298: 179–181 [DOI] [PubMed] [Google Scholar]
- Rebeiz M, Jikomes N, Kassner VA, Carroll SB 2011. Evolutionary origin of novel gene expression pattern through co-option of the latent activities of existing regulatory sequences. Proc Natl Acad Sci 108: 10036–10043 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson GE, Grozinger CM, Whitfield CW 2005. Sociogenomics: Social life in molecular terms. Nat Rev Genet 6: 257–270 [DOI] [PubMed] [Google Scholar]
- Schultz TR, Brady SG 2008. Major evolutionary transitions in ant agriculture. Proc Natl Acad Sci 105: 5435–5440 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sella G, Petrov DA, Przeworski M, Andolfatto P 2009. Pervasive natural selection in the Drosophila genome? PLoS Genet 56: e1000495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siepel A, Bejerano G, Pedersen JS, Hinrichs AS, Hou M, Rosenbloom K, Clawson H, Spieth J, Hillier LW, Richards S, et al. 2005. Evolutionarily conserved elements in vertebrate, insect, worm, and yeast genomes. Genome Res 15: 1034–1050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simola DF, Ye C, Mutti NS, Dolezal K, Bonasio R, Liebig J, Reinberg D, Berger S 2013. A chromatin link to caste identity in the carpenter ant Camponotus floridanus. Genome Res 23: 486–496 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sirviö A, Pamilo P, Johnson RA, Page RE Jr, Gadau J 2011. Origin and evolution of the dependent lineages in the genetic caste determination system of Pogonomyrmex spp. Evolution 65: 869–884 [DOI] [PubMed] [Google Scholar]
- Smith CD, Zimin A, Holt C, Abouheif E, Benton R, Cash E, Croset V, Currie CR, Elhaik E, Elsik CG, et al. 2011a. Draft genome of the globally widespread and invasive Argentine ant (Linepithema humile). Proc Natl Acad Sci 108: 5673–5678 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith CR, Smith CD, Robertson HM, Helmkampf M, Zimin A, Yandell M, Holt C, Hu H, Abouheif E, Benton R, et al. 2011b. Draft genome of the red harvester ant Pogonomyrmex barbatus. Proc Natl Acad Sci 108: 5667–5672 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith CR, Mutti NS, Jasper WC, Naidu A, Smith CD, Gadau J 2012. Patterns of DNA methylation in development, division of labor and hybridization in an ant with genetic caste determination. PLoS ONE 78: e42433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suen G, Teiling C, Li L, Holt C, Abouheif E, Bornberg-Bauer E, Bouffard P, Caldera EJ, Cash E, Cavanaugh A, et al. 2011. The genome sequence of the leaf-cutter ant Atta cephalotes reveals insights into its obligate symbiotic lifestyle. PLoS Genet 7: e1002007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamura K, Subramanian S, Kumar S 2004. Temporal patterns of fruit fly Drosophila evolution revealed by mutation clocks. Mol Biol Evol 21: 36–44 [DOI] [PubMed] [Google Scholar]
- Tautz D, Domazet-Lošo T 2011. The evolutionary origin of orphan genes. Nat Rev Genet 12: 692–702 [DOI] [PubMed] [Google Scholar]
- Trapnell C, Williams BA, Pertea G, Mortazavi A, Kwan G, van Baren MJ, Salzberg SL, Wold BJ, Pachter L 2010. Transcript assembly and quantification by RNA-seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat Biotechnol 28: 511–515 [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Zweden JS, Brask JB, Christensen JH, Boomsma JJ, Linksvayer TA, d'Ettorre P 2010. Blending of heritable recognition cues among ant nestmates creates distinct colony gestalt odours but prevents within-colony nepotism. J Evol Biol 23: 1498–1508 [DOI] [PubMed] [Google Scholar]
- Villesen P, Murakami T, Schultz TR, Boomsma JJ 2009. Identifying the transition between single and multiple mating of queens in fungus-growing ants. Proc Biol Sci 269: 1541–1548 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waterhouse RM, Zdobnov EM, Tegenfeldt F, Li J, Kriventseva EV 2011. OrthoDB: The hierarchical catalog of eukaryotic orthologs in 2011. Nucleic Acids Res 39: D283–D288 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wurm Y, Wang J, Riba-Grognuz O, Corona M, Nygaard S, Hunt BG, Ingram KK, Falquet L, Nipitwattanaphon M, Gotzek D, et al. 2011. The genome of the fire ant Solenopsis invicta. Proc Natl Acad Sci 108: 5679–5684 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou X, Slone JD, Rokas A, Berger SL, Liebig J, Ray A, Reinberg D, Zwiebel L 2012. Phylogenetic and transcriptomic analysis of chemosensory receptors in a pair of divergent ant species reveals sex-specific signatures of odor coding. PLoS Genet 8: e1002930. [DOI] [PMC free article] [PubMed] [Google Scholar]