

Abstract
Positive-strand RNA viruses encompass more than one-third of known virus genera and include many medically and agriculturally relevant human, animal, and plant pathogens. The nematode Caenorhabditis elegans and its natural pathogen, the positive-strand RNA virus Orsay, have recently emerged as a new animal model to understand the mechanisms and evolution of innate immune responses. In particular, the RNA interference (RNAi) pathway is required for C. elegans resistance to viral infection. Here we report the first genome-wide analyses of gene expression upon viral infection in C. elegans. Using the laboratory strain N2, we identify a novel C. elegans innate immune response specific to viral infection. A subset of these changes is driven by the RNAi response to the virus, which redirects the Argonaute protein RDE-1 from its endogenous small RNA cofactors, leading to loss of repression of endogenous RDE-1 targets. Additionally, we show that a C. elegans wild isolate, JU1580, has a distinct gene expression signature in response to viral infection. This is associated with a reduction in microRNA (miRNA) levels and an up-regulation of their target genes. Intriguingly, alterations in miRNA levels upon JU1580 infection are associated with a transformation of the antiviral transcriptional response into an antibacterial-like response. Together our data support a model whereby antiviral RNAi competes with endogenous small RNA pathways, causing widespread transcriptional changes. This provides an elegant mechanism for C. elegans to orchestrate its antiviral response, which may have significance for the relationship between small RNA pathways and immune regulation in other organisms.
Most cells of a multicellular organism have an extensive armory to fight infection, even without the intervention of specialized cells of the immune system. Many of these innate defense mechanisms respond dynamically to infection, requiring widespread changes in gene expression programs. Therefore, general mechanisms exist to sense infection and transduce this signal into transcriptional and post-transcriptional responses (Hoffmann et al. 2006). Understanding how these responses are triggered is vital to a full understanding of how animals fight infection; it is equally important to learn how they could be quenched because persistent inflammation due to a vigorous response to minor infection can be a cause of pathology in its own right (Park et al. 2012).
Because innate immunity does not necessarily require specialized immune cells, invertebrate model organisms are attractive systems to study the fundamental processes required (Gravato-Nobre and Hodgkin 2005). In this regard, the nematode Caenorhabditis elegans has emerged as a model to understand the innate immune response (Mahajan-Miklos et al. 1999). Important studies analyzing the changes in gene expression upon infection with various pathogens have been performed. Predominantly, this has involved infecting C. elegans with human pathogenic bacteria such as Pseudomonas, which has identified some conserved and some C. elegans–specific pathways (for review, see Irazoqui et al. 2010). Analyses of the transcriptional response to pathogens that appear to infect nematodes in the wild, such as Mycobacterium nematophilum (O'Rourke et al. 2006) and the fungus Drechmeria coniospora (Pujol et al. 2008), have also been performed. Despite identification of several robust gene expression changes, many of which involve the use of conserved signaling pathways, the mechanism of sensing various pathogens is not clear (Partridge et al. 2010). C. elegans does not appear to have obvious homologs of many of the major pattern recognition modules found in other organisms, and there is debate over whether the single Toll-like receptor in C. elegans is of primary importance in pathogen resistance, although it may activate a pathogen-avoidance response and could be important in the response to Salmonella infection (Pujol et al. 2001; Tenor and Aballay 2008).
An ancient innate immune response to viral infection that is conserved in C. elegans is the RNA interference (RNAi) pathway, which in both plants and animals is able to defend against RNA viruses (Hamilton and Baulcombe 1999; Li et al. 2002). Antiviral RNAi in animals depends on members of the Dicer family of RNase III endoribonucleases, which process double-stranded viral RNA to generate small interfering RNAs (siRNAs) against the viral genome, named viRNAs (Galiana-Arnoux et al. 2006). In combination with Argonaute proteins, viRNAs target the viral genome and/or mRNAs for destruction, thus limiting viral replication (Aliyari and Ding 2009).
C. elegans possesses a single Dicer enzyme, DCR-1 (Knight and Bass 2001), that contributes to three small RNA pathways through its ability to form complexes with multiple different interacting proteins (Duchaine et al. 2006). DCR-1 processes stem–loop structures characteristic of precursor microRNAs (pre-miRNAs) to generate mature microRNAs (miRNAs) (Ketting et al. 2001). It is also able to act on both exogenous and endogenous sources of double-stranded RNA, which it processes to form primary small-interfering RNAs (siRNAs). Endogenous siRNAs (endo-siRNAs) arise from several loci across the C. elegans genome and require members of the ERI complex in addition to DCR-1 for generation of primary siRNAs (Kennedy et al. 2004; Pavelec et al. 2009; Conine et al. 2010; Welker et al. 2010). Processing of exogenous dsRNA by DCR-1 requires its binding partner RDE-4 (Parrish and Fire 2001) and generates primary siRNAs that bind to the Argonaute protein RDE-1 (Tabara et al. 1999). In C. elegans as in plants, primary small RNAs from either endogenous or exogenous sources can then be amplified by RNA-dependent RNA polymerases, using the target RNA as a template to generate antisense secondary siRNAs (Sijen et al. 2001). Secondary siRNAs usually have a guanine (G) as their first nucleotide, a length of 22 nucleotides (nt), and have a 5′ triphosphate, which distinguishes them from primary siRNAs (Pak and Fire 2007; Sijen et al. 2007; Gent et al. 2010). Together with a number of different Argonaute proteins, 22G secondary siRNAs are the effectors of RNAi-mediated gene silencing in C. elegans. (Claycomb et al. 2009; Gu et al. 2009).
The RNAi pathway in C. elegans was shown to be involved in restricting accumulation of RNA generated by a transgenic RNA-dependent RNA polymerase (Lu et al. 2005) and defending against infection of C. elegans primary cells with a mammalian virus (Schott et al. 2005; Wilkins et al. 2005). Moreover, recently, the first natural virus to infect C. elegans, a single-stranded RNA virus named Orsay, was identified (Félix et al. 2011). Sensitivity to the Orsay virus is dependent on genetic background: The laboratory reference strain N2 (Bristol) is relatively resistant to Orsay virus (as evidenced by mild pathology and low levels of viral replication), but a wild isolate of C. elegans, JU1580 (Paris), is much more sensitive (Félix et al. 2011). N2 animals generate both primary and secondary antiviral siRNAs, and this response is important for defense against the virus because mutants lacking components of the RNA interference pathway, such as rde-1 mutants lacking the Argonaute protein RDE-1, are sensitive to viral infection. Indeed, defects in the antiviral small RNA response also underpin the sensitivity of the JU1580 wild isolate (Félix et al. 2011). Infection of C. elegans with the Orsay virus therefore offers an opportunity to explore the relationship between innate immune responses and the antiviral RNAi pathway.
Here, we use experimental infection with the Orsay virus to report the first analysis of the transcriptional response to viral infection in C. elegans. This identifies a largely virus-specific immune response in N2 and rde-1 animals, which involves clusters of coregulated genes that are frequently deleted in C. elegans wild isolates. We also identify a virus-specific infection response that is dependent on the primary Argonaute RDE-1, because viral-response genes are already up-regulated in RDE-1-deficient mutants. Intriguingly the response of the wild isolate JU1580 (Paris) to viral infection is altered relative to either N2 or rde-1 animals, such that a viral-specific response is transformed into a response more similar to bacterial infection of N2 animals. Both of these responses can be explained in terms of a model whereby competition between exogenous and endogenous RNAi pathways regulates gene expression changes in response to viral infection.
Results
The transcriptome response to viral infection in C. elegans
To identify gene expression changes in C. elegans upon Orsay virus infection, we examined microarray data comparing infected to noninfected animals. We selected the laboratory reference strain N2, the rde-1 mutant deficient in the primary Argonaute RDE-1, and the wild isolate JU1580 for this analysis. JU1580 and rde-1 are both ∼100-fold more sensitive to infection with the Orsay virus than N2 (as measured by viral load) (Félix et al. 2011). Both sensitive strains are deficient in the production of viRNAs against the Orsay virus; however, JU1580 appears to be deficient in production of primary viRNAs whilst rde-1 mutants are deficient in the production of secondary viRNAs (Supplemental Fig. 1; Félix et al. 2011; A Ashe, T Belicard, J LePen, P Sarkies, L Frezal, N Lehrbach, M-A Felix, EA Miska, in prep.).
Examination of microarray data showed gene expression changes in each strain that were consistent between biological replicates. For each strain, we identified a set of genes that change upon infection by more than twofold on average across biological replicates, with a Student's t-test, P-value of <0.05 (summarized in Table 1; complete set in Supplemental Table 1). qRT-PCR analysis on biological replicates showed excellent correlation with the microarray data for four of these genes (Supplemental Fig. 2).
Table 1.
The number of genes that are greater than twofold either up-regulated or down-regulated (P < 0.05, Student's t-test) upon viral infection in the indicated strain
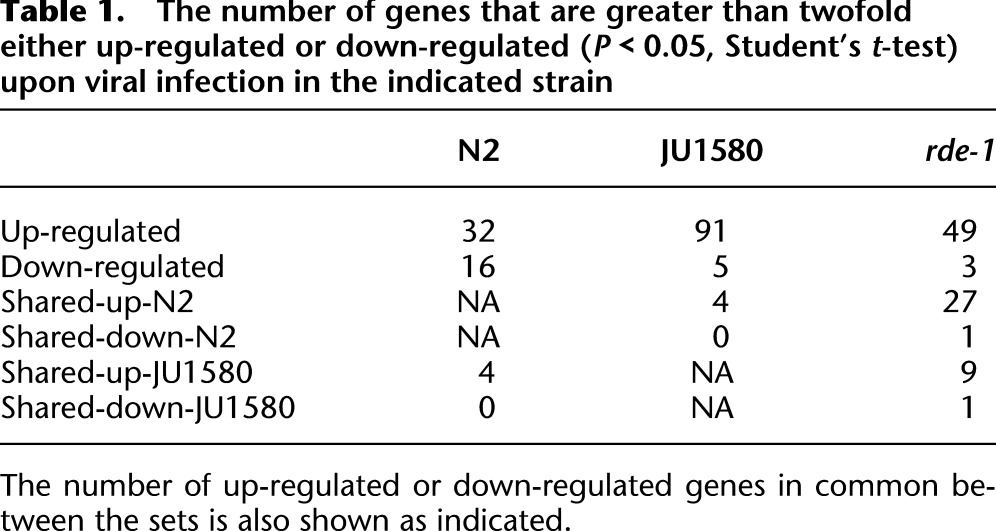
A heatmap comparing the set of genes showing statistically significant alterations in N2, JU1580, or rde-1 upon infection (Fig. 1) showed that perturbations in transcript levels following viral infection were similar between rde-1 mutants and N2, whereas a distinct pattern of changes was seen in JU1580. This difference cannot simply be explained by increased viral load in JU1580, because rde-1 animals are infected to similar levels as JU1580 (Félix et al. 2011).
Figure 1.

Heatmap showing changes in gene expression upon viral infection. The heatmap shows all the genes that change by more than twofold in at least one strain after infection (t-test, P < 0.05). The changes that occur in each infected biological replicate (vertical lanes) relative to the mean value of all biological replicates of that strain without infection are indicated by the color on the red–green scale.
To further investigate the effect of viral infection on gene expression, we used hierarchical cluster analysis across the entire transcriptome (Fig. 2A). This demonstrated that infected JU1580 samples were indeed distinct from both the N2- and rde-1-infected samples. Importantly, however, uninfected N2 and uninfected JU1580 clustered closely together, suggesting that the differences in the response to viral infection are not simply due to differences in the transcriptome of uninfected JU1580 and N2. Finally, the cluster analysis showed an intriguing similarity between the infected N2 sample and the uninfected rde-1 sample. This similarity will be considered further below.
Figure 2.

Similarity between rde-1 mutants and virally infected N2 animals. (A) Hierarchical clustering performed on all the probes, averaged over all biological replicates, for infected and uninfected strains as indicated. (B) The behavior of RDE-1-regulated genes (defined as being 30% or more up-regulated in rde-1 mutants with a P-value of <0.05; two-tailed t-test) in N2 animals upon infection, relative to the behavior of all genes. The density plot uses the mean from all biological replicates. (C) The change in small RNA reads normalized to total library size mapping antisense to either all protein-coding genes or protein-coding genes with RDE-1-dependent small RNAs mapping to them upon infection of N2 animals. Boxes show median and interquartile range, and the whiskers extend to the most extreme data point, which is no more than 1.5 times the interquartile range from the box. Dots represent outliers. (D) The gene expression changes for the same set of genes shown in C upon infection of N2 animals compared with the change in expression between uninfected N2 and uninfected rde-1 animals. (Red) Genes that were up-regulated by >30% (P < 0.05) in rde-1 mutant animals.
Regulation of viral response genes by RDE-1
The cluster analysis indicated that a large set of genes might be responsible for the observed similarity between infected N2 and uninfected rde-1. We therefore searched our data set for genes that were significantly differentially expressed (P < 0.05, Student's t-test) between uninfected rde-1 and uninfected N2 at a relatively low cutoff of 30% difference (Supplemental Table 2). Strikingly, this set of 672 rde-1-regulated genes showed a global increase in expression upon infection of N2 (P < 2 × 10−16, two-tailed signed rank test) (Fig. 2B). The same effect was also seen for larger differences in expression between uninfected N2 and rde-1 (Supplemental Fig. 3). Infected JU1580 animals showed no global up-regulation of RDE-1-regulated genes upon infection (Supplemental Fig. 4). Importantly, RDE-1-regulated genes do not become further up-regulated in the rde-1 mutant animals upon infection (Supplemental Fig. 4a). Thus, rde-1 mutants are epistatic to viral infection for the regulation of this set of genes.
These data suggested that repression of these genes by RDE-1 in wild-type animals might be relieved upon infection. RDE-1 lies downstream from DCR-1 function both in the response to exogenous dsRNA (Parrish and Fire 2001) and in the antiviral RNAi response (Supplemental Fig. 1; A Ashe, T Belicard, J LePen, P Sarkies, L Frezal, N Lehrbach, M-A Felix, EA Miska, in prep.). Thus, one possibility is that DCR-1 products generated upon viral infection replication displace endogenous small RNAs from RDE-1, leading to gene expression changes. We therefore sought to identify whether any RDE-1-dependent small RNAs would be depleted in N2 animals upon viral infection.
We considered two possible sources of RDE-1-dependent small RNAs. Although most miRNAs are associated with the Argonaute proteins ALG-1 and ALG-2, certain miRNAs have been shown to bind to RDE-1 (Corrêa et al. 2010). However, in agreement with this study, we did not find a clear difference in overall read counts of these miRNAs in rde-1 mutants, nor in the expression of predicted target genes of these miRNAs upon infection of N2 (data not shown). This suggests that alterations in miRNA expression are not a major source of RDE-1-dependent changes in gene expression upon viral infection.
In C. elegans, another potential source of regulatory RNAs is endogenous short interfering RNAs (endo-siRNAs). To investigate whether changes in this population of small RNAs might occur in rde-1 mutants, we analyzed small RNA sequencing data from uninfected N2 and rde-1 animals. We identified genes for which antisense small RNA reads were more than fourfold depleted in rde-1 compared with N2 (Supplemental Table 3) with a negative binomial test P-value of <0.05. The majority of these sequences were 22 nt long and mapped antisense to target loci, suggesting that they are downstream from RDE-1 function (for an example locus, see Supplemental Fig. 5). These RDE-1-dependent small RNAs showed reduced numbers of reads in infected N2 animals compared with uninfected N2 animals (P < 1 × 10−10, two-tailed signed rank test) (Fig. 2C; Supplemental Fig. 3). This suggests that the changes in small RNAs that occur in rde-1 mutants are mirrored in N2 upon infection, just as are the changes in gene expression.
Again in agreement with previous work (Corrêa et al. 2010), we did not observe a direct correlation between expression level of genes in rde-1 mutants and reduced antisense small RNA reads. However, 5% of the genes showing robustly reduced antisense small RNA reads in rde-1 mutants also showed >30% increased expression in rde-1 mutants relative to N2. These genes also all showed increased expression in N2 after infection (P = 0.01, two-tailed signed rank test; red dots in Fig. 2D). Interestingly, the most strongly up-regulated gene within this set in both uninfected rde-1 mutants and infected N2 animals, Y47H10A.5, was previously identified as a robust target of RDE-1-dependent small RNAs (Corrêa et al. 2010). Consistent with the trend seen for all rde-1-dependent small RNA loci, normalized antisense small RNAs mapping to this gene were reduced on average by around twofold in N2 upon infection (Supplemental Fig. 5). Taken together, this suggests that some gene expression changes in N2 on infection are caused by reduction in small RNA reads due to alterations in RDE-1 activity.
Up-regulation of miRNA targets in JU1580 animals upon infection
Although global up-regulation of RDE-1-dependent genes was not seen in JU1580 animals upon infection, JU1580 animals did show up-regulation of DCR-1-dependent genes upon infection, defined as those up-regulated by more than twofold in dcr-1 mutants (Welker et al. 2007). This was not the case for either N2 or rde-1 mutants upon infection (Fig. 3A). As discussed above, changes in gene expression in JU1580 upon infection were not linked to RDE-1-regulated genes. Equally, RDE-4-dependent genes were not globally up-regulated in JU1580, and neither were presumed DCR-1-dependent endo-siRNA targets that are dependent on the DCR-1 helicase domain (Supplemental Fig. 4; Welker et al. 2010). We therefore tested whether this up-regulation might reflect up-regulation of miRNA target genes. We generated a list of all potential miRNA targets using PicTar (http://pictar.mdc-berlin.de/) (Krek et al. 2005; Lall et al. 2006). Mean expression of these genes was increased by 19% on average in JU1580 upon infection (P < 2 × 10−16, two-tailed signed rank test), whereas there was no increase in expression in N2 or rde-1-infected animals (P = 0.6 and 0.5, respectively) (Fig. 3B). Thus, increased expression of DCR-dependent genes in JU1580 is likely to be explained by increased expression of miRNA targets.
Figure 3.

miRNA targets are up-regulated in JU1580 animals upon infection. (A) The behavior of genes up-regulated in dcr-1 mutants in N2, rde-1, and JU1580 upon infection (Welker et al. 2007). (B) Expression changes of all predicted miRNA targets (PicTar) in the three strains upon infection. (C) miR-80 predicted targets (PicTar) in JU1580 upon infection. All panels use the mean of all biological replicates.
We hypothesized that up-regulation of miRNA target gene levels might be caused by reduced levels of mature miRNAs. We therefore examined deep sequencing data to look for changes in miRNA levels upon viral infection in JU1580. We used miRDeep2 (Friedländer et al. 2012) using miRNA sequences downloaded from miRBase (Kozomara and Griffiths-Jones 2011) to quantify the levels of miRNAs in infected versus uninfected animals. This identified 41 miRNAs that were more than twofold altered in infected JU1580 compared with noninfected (Fig. 4). Of these miRNAs, 79% were down-regulated, a statistically significant bias (P = 0.012, χ2 test). Down-regulated miRNAs included the abundant miRNAs miR-71 and miR-80, which we confirmed via qPCR (Supplemental Fig. 6). Consistently, predicted targets of miR-80 showed up-regulation in JU1580 animals upon infection (P < 1 × 10−8, signed rank test) (Fig. 3C). Moreover, down-regulated miRNAs after infection in JU1580 were not generally down-regulated in either N2 or rde-1 animals after infection (Fig. 4), consistent with the lack of changes in DCR-1-regulated genes in N2 or rde-1 mutants (Fig. 3A). Taken together, these data support the idea that the up-regulation of DCR-1-regulated genes in JU1580 upon infection reflects alterations in miRNA levels.
Figure 4.
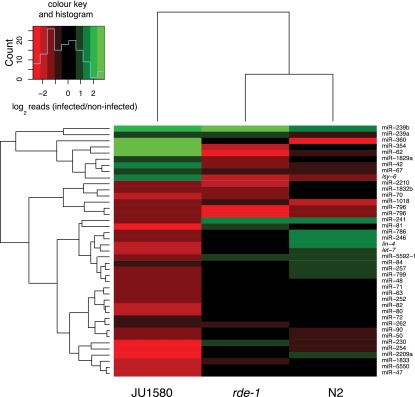
Heatmap showing changes in small RNA reads for individual microRNAs after infection. The heatmap shows all microRNAs for which at least a twofold change in normalized reads was seen upon infection in JU1580 animals.
Reduced miRNA levels transform the antiviral gene expression response into an antibacterial response in JU1580
Previously, DCR-1 and RDE-1 have been suggested to regulate innate immunity genes (Welker et al. 2007); thus, we decided to investigate how known sets of C. elegans innate immunity genes alter in expression in response to infection with Orsay virus. We compared the list of genes that were twofold up-regulated in response to viral infection in N2, rde-1, or JU1580 to genes previously annotated as being up-regulated in response to various other pathogens. In N2 and rde-1, only a small proportion of genes up-regulated in response to the Orsay virus were up-regulated in response to at least one other pathogen (2/48 [4%] and 4/97 [4%], respectively). In contrast, a much higher proportion of genes up-regulated in response to viral infection of JU1580 (147/655 [22%]) were also up-regulated in response to at least one other pathogen. This difference was statistically significant (P < 1 × 10−10, Fisher's exact test). These data suggest that the response to viral infection in N2 and rde-1 is largely specific, whereas JU1580 appears to show up-regulation of other pathogen-response genes after Orsay virus infection.
To investigate this in more detail, we looked at global gene expression changes upon viral infection for the individual sets of previously annotated pathogen-response genes. We first considered changes in genes up-regulated in N2 animals in response to bacterial infection with either Pseudomonas aeruginosa (Troemel et al. 2006) or M. nematophilum (O'Rourke et al. 2006). P. aeruginosa infection-response genes showed markedly increased expression upon viral infection of JU1580 (P < 2 × 10−16, signed rank test). In contrast, increased expression was not observed in virus-infected N2 or rde-1 animals, although in rde-1 animals a small subset of genes was up-regulated (Fig. 5A; see also Supplemental Fig. 7). Similarly, increased expression of M. nematophilum–induced genes was seen in JU1580 upon infection by the Orsay virus (P < 2 × 10−16, signed rank test), but this was not generally so for N2 or rde-1 (Fig. 5B).
Figure 5.

Behavior of previously annotated pathogen response genes upon viral infection of C. elegans. (A–D) Changes upon viral infection in various sets of pathogen-regulated genes, as indicated above the panels. Density plots show the mean changes across all biological replicates. (E,F) The gene expression changes upon viral infection for microRNA targets divided into pathogen response genes and genes not previously annotated as pathogen response genes, respectively. Boxes show median and interquartile range, and the whiskers extend to the most extreme data point, which is no more than 1.5 times the interquartile range from the box. Dots represent outliers.
We also considered genes up-regulated in response to a pore-forming toxin Cry5B (Kao et al. 2011). Interestingly, these genes showed a similar pattern to the genes up-regulated upon bacterial infection, with a marked increase in expression in JU1580 after infection by the Orsay virus (P < 2 × 10−16, signed rank test), not seen in either N2 or rde-1 (Fig. 5C). Finally, we examined genes up-regulated in response to infection with fungus (Pujol et al. 2008). In contrast to bacterial and pore-forming toxin-induced genes, we did not observe a global up-regulation of fungal response genes in JU1580 after infection by the Orsay virus; however, a subset of genes was up-regulated upon infection in JU1580. Again, no up-regulation was observed for fungal response genes in either N2 or rde-1 (Fig. 5D).
Recently certain microRNAs have been suggested to repress antibacterial response genes (Kudlow et al. 2012). We therefore wondered whether the up-regulation of bacterial response genes seen upon viral infection of JU1580 might be due to the reduced levels of miRNAs shown in the previous section. Accordingly, miRNA targets that were also pathogen-response genes were up-regulated upon viral infection in JU1580 (Fig. 5E) (P < 1 × 10−10, signed rank test) but not in N2 or rde-1. Importantly, however, pathogen-response genes were not the only miRNA targets up-regulated because the general increase in expression of miRNA targets seen in JU1580 upon viral infection was also seen for miRNA targets that were not up-regulated in response to infection with any other pathogen (P < 1 × 10−8, signed rank test) (Fig. 5F). Thus, the antibacterial response seen in JU1580 upon viral infection is likely to be a consequence of widespread changes in miRNA target gene expression upon infection.
The virus-specific innate immune response in C. elegans
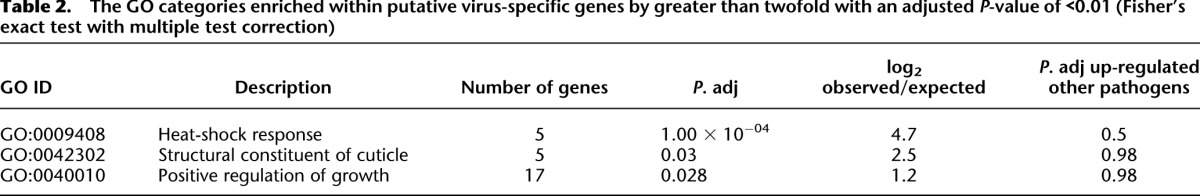
Given that the response of JU1580 to viruses might be abnormal, we decided to focus on genes that were up-regulated in either N2 or rde-1 or both, but not affected by infection with any of the other pathogens, as potential virus-specific response genes. We performed Gene Ontology analysis of this set of genes, which showed statistically significant enrichment (P < 0.1, Fisher's exact test with Benjamani and Hochberg multiple test correction) for positive regulation of growth rate, structural constituent of cuticle, and the heat shock response (Table 2). Supporting the idea that these viral-response genes might represent a novel innate immune response, none of these enriched categories were enriched in the set of previously annotated pathogen response genes (Table 2), nor were enriched categories for previously annotated pathogen-response genes enriched within N2 or rde-1 viral-response genes.
Table 2.
The GO categories enriched within putative virus-specific genes by greater than twofold with an adjusted P-value of <0.01 (Fisher's exact test with multiple test correction)
Within this set of genes, as is apparent from the heatmap in Figure 1, there is a subset of genes that change consistently in N2 and rde-1 mutants but do not change upon viral infection in JU1580. We wondered whether this might reflect a viral-response signaling pathway that is deficient in JU1580. We therefore selected genes from the list of up-regulated genes in rde-1 and N2 that were unaltered (by <25%) in JU1580 upon infection (Supplemental Table 4). Intriguingly, we found that many of these genes appeared to be clustered together along the genome (Fig. 6A), such that genes up-regulated in rde-1 and N2 but not JU1580 were 10 times more likely to be within 20 kb of one other than genes up-regulated in JU1580 and either rde-1 or N2 or both upon viral infection (P = 0.01, Fisher's exact test) (Fig. 6B). Indeed, there were no genes that were up-regulated in JU1580 and rde-1 or N2 or both that appeared to be clustered together. This is consistent with the idea that a signaling pathway, deficient in JU1580, acts coordinately on certain genes positioned closely together. Interestingly, manual examination of these clusters showed that all of them carry deletions previously annotated by comparative genome hybridization and PCR-based mapping in other wild isolates (example in Fig. 6C and Table 3; Maydan et al. 2010). We carried out PCR analysis of the clusters in JU1580 and failed to obtain amplification for two out of the four clusters in at least two distinct regions within the cluster, suggesting deletions or substantial sequence divergence (Fig. 6B; Supplemental Fig. 8; summarized in Table 3). Intriguingly, both of these regions were also found to be deleted in the Hawaiian wild isolate CB4856, despite the fact that N2 and JU1580 are more closely related than either is to CB4856 (Andersen et al. 2012). Taken together, these observations point to a specific transcriptome response to viral infection that occurs in N2 and rde-1 animals and targets clusters of genes that have been lost from certain wild isolates of C. elegans.
Figure 6.

Clusters of genes are up-regulated in response to pathogen infection in N2 and rde-1, but not in JU1580. (A) The distance between any greater than twofold up-regulated gene and its nearest neighbor that is also greater than twofold up-regulated in N2 and rde-1 as a histogram. (B) The histogram of distances for genes that are greater than twofold up-regulated in JU1580 as well as N2 and/or rde-1. (C) A diagram of the sdz-6 locus as an example of a cluster up-regulated upon viral infection. (Boxes) Gene bodies; (gray box) the region deleted in the Hawaiian strain CB4856. Information was taken from WormBase (http://www.wormbase.org). (D) The expression changes in the sdz-6 cluster from the array data in N2, rde-1, and JU1580. Bars represent the mean of all biological replicates. (E) PCR amplification of genomic DNA from JU1580 or N2 for three regions within the cluster or the nearby gene srh-208.
Table 3.
Clusters of viral response genes in C. elegans

Discussion
The data presented are in support of a model whereby at least three pathways regulating C. elegans gene expression may become activated in response to viral infection. In the first pathway, RDE-1 becomes occupied by antiviral small RNAs, resulting in the loss of RDE-1-dependent endogenous small RNAs and up-regulation of RDE-1-dependent genes. We observed this pathway upon viral infection of N2, but not upon infection of either rde-1 or JU1580 animals. In rde-1 animals, the lack of this response is due to the fact that the changes have already taken place before infection occurs. In JU1580, the absence of appropriate DCR products (Supplemental Fig. 1; Félix et al. 2011) means that RDE-1 is not fully engaged upon viral entry.
The second pathway is one that occurs in JU1580 but not in either N2 or rde-1 mutants upon infection. This involves up-regulation of DCR-regulated genes, more particularly involving genes targeted by miRNAs. We saw no alteration in DCR-1 mRNA levels by microarray in JU1580 upon infection. However, we have shown that JU1580 shows an abnormal profile of small RNAs mapping to the viral genome compared with N2 and rde-1. In particular, abundant small RNAs are generated from the sense strand of the viral genome (Félix et al. 2011). These appear to come predominantly from regions of strong secondary structure (A Ashe, T Belicard, J LePen, P Sarkies, L Frezal, N Lehrbach, M-A Felix, EA Miska, in prep.). One possible explanation, therefore, for the down-regulation of miRNAs that we observed in JU1580 upon viral infection is that DCR-1 is titrated away from miRNA precursors to engage nonproductively with the sense strand of the viral genome.
It is interesting that the JU1580-specific pathway of gene up-regulation seems to affect several genes implicated in responses to other pathogens, in particular, infection with bacteria. It is tempting to speculate that this is an adaptive response that aids in fighting pathogenic assault in JU1580. However, this response is not seen in either N2 or rde-1 animals. We therefore favor the interpretation that this response only occurs in the context of defective DCR-1 activity and does not represent a mechanism to regulate genes in response to viral infection in normal animals. The fact that bacterial response genes are affected would therefore be an indirect result of the general negative regulation of bacterial response genes by miRNAs in the C. elegans intestine (Kudlow et al. 2012). Two further points can be made in support of this idea. First, although different bacteria have been shown to instigate largely different responses in C. elegans (O'Rourke et al. 2006; Troemel et al. 2006), viral infection of JU1580 appears to affect both P. aeruginosa and M. nematophilum response genes equally. Second, the up-regulation of miRNA targets in JU1580 upon infection affects miRNA targets generally rather than specifically affecting those involved in innate immunity. If the antibacterial response seen is, indeed, aberrant, an intriguing possibility is that the titration of DCR-1 away from miRNAs to the viral genome represents an evasion strategy by the virus, causing the cell to respond to it as if it were a bacterial pathogen.
What might be the cause of the aberrant small RNA response to viral infection in JU1580? We have found that JU1580 carries a deletion in the C-terminal RIG-I domain of the gene drh-1, a homolog of the mammalian gene RIG-I. DRH-1 interacts with DCR-1 (Duchaine et al. 2006) and has been implicated in the small RNA response to artificial viral infection of C. elegans (Lu et al. 2009), and indeed we have found that this deletion is responsible for the defects in primary antiviral siRNA production in JU1580 animals (A Ashe, T Belicard, J LePen, P Sarkies, N Lehrbach, M-A Felix, and E Miska, in prep.). Consistent with this, when we tested the expression changes of five antibacterial response genes induced on viral infection of JU1580 but not N2 or rde-1 in N2 animals carrying a deletion in drh-1 (drh-1 mutants), 4/5 genes showed greater than twofold up-regulation upon infection of drh-1 mutant animals (Supplemental Fig. 7). This suggests that the abnormalities in DCR-dependent gene expression in JU1580 animals upon infection are largely similar in drh-1 mutants, thus implying that the defect in this gene may be responsible for the alterations in both viral-derived and endogenous small RNAs in JU1580 upon infection.
The deletion in drh-1 in JU1580 might also relate to the third pathway of gene expression changes upon viral infection, involving the up-regulation of closely clustered genes that appear to be evolving rapidly. Because this pathway is common to N2 and rde-1 but not seen in JU1580, this may reflect a signaling pathway that is not active in JU1580. In mammals, RIG-I acts to induce the interferon response upon viral infection (Yoneyama et al. 2004). Thus, in addition to its role in antiviral small RNA biogenesis, drh-1 may be involved in regulation of genes in response to viral infection. It is therefore possible that drh-1 induces the expression of the antiviral response genes that are up-regulated in N2 and rde-1 but not JU1580. In support of this idea, drh-1 mutants do not up-regulate the antiviral response gene sdz-6, which is deleted in JU1580, in response to infection (Supplemental Fig. 9). This suggests that the gene-regulation function of RIG-I might be widely conserved through evolution despite the absence of interferon in C. elegans. Further work using the C. elegans viral infection paradigm will therefore be a useful model to understand the evolution of transcriptional responses to viral infection in a wider context.
Taken altogether, these three pathways suggest a model in which the response to viral infection is controlled by the activity of DCR-1 and its accessory factors (Fig. 7). Competition between DCR-1 substrates has already been documented as an important regulatory mechanism for the starvation response in C. elegans (Hellwig and Bass 2008), and competition between endogenous and exogenous small RNAs is known to affect the efficiency of the RNAi response (Kennedy et al. 2004). Here, we suggest that this kind of competition can also apply to regulation of viral response genes acting through engagement of RDE-1 with its substrate.
Figure 7.

Model for competition between small RNA pathways in response to viral infection of C. elegans. (Left) Competition between endogenous RNAi pathways and exogenous RNAi in wild-type animals. (Right) Competition between antiviral RNAi and the microRNA pathway in JU1580 animals.
The simplicity of the mode of regulation we propose, in which the “sensor” and the “processor” of viral infection in C. elegans are the same molecules (Fig. 7), implies that it may be an evolutionarily ancient way for cells to detect viral infection. Conceptually, this is similar to a model proposed recently to apply to bacterial infection of C. elegans, whereby some antibacterial genes are induced by the effects of bacterial toxins on normal cellular pathways as opposed to direct recognition of the toxins themselves (Dunbar et al. 2012; McEwan et al. 2012). Interestingly, inflammatory responses involving up-regulation of innate immunity genes have been reported to occur in response to overexpression of endogenous double-stranded RNA in retinal pigment epithelium (Tarallo et al. 2012), implying that the regulation of innate immunity genes through competition between small RNA pathways might be conserved in higher organisms.
Methods
C. elegans culture and strains
The strains used were as follows: N2, JU1580, rde-1 (ne219), and drh-1 (ok3495). Animals were cultured at 20°C on HB101 and NGM according to standard methods.
Viral infection of C. elegans
C. elegans was infected with the Orsay virus as previously described (Félix et al. 2011). Total RNA for microRNA hybridization was extracted using TRIzol (Invitrogen) from mixed-stage animals after 4 d of infection, or from uninfected control samples. At least three biological replicates were performed for each strain.
Microarray hybridization and data collection
Microarray hybridization was carried out by the Microarray Service at the European Molecular Biology Laboratory, Heidelberg, Germany.
Microarray data processing
Microarray data were processed using Bioconductor. RMA analysis was performed on the samples to give log2 probe intensity values for each of the samples. Custom scripts in R were used to calculate the Student's t-test and mean expression changes for each probe. The biomaRt package was used to identify genes corresponding to each probe from Ensembl.
For the analysis of gene expression changes of various different sets of genes upon viral infection, the mean change in infection for each strain was calculated by subtracting the mean log2 probe intensity level after infection from the mean log2 probe intensity level before infection. Statistical significance of changes was performed by using the mean probe intensity before and after infection for each gene as paired samples, allowing the nonparametric Wilcoxon signed rank test to be used.
Gene Ontology analysis was performed using a custom script in R employing the biomaRt package to obtain Gene Ontology information for each gene within a particular set of interest or all the genes represented on the array as a control. The number of genes within each category was obtained for both test and control sets, and the significance of this was calculated by using a Fisher's exact test and a Benjamani–Hoffmann multiple test correction. Enrichment was defined as greater than twofold, with a P-value of <0.1 after multiple test correction, requiring at least four genes to appear within the category.
qRT-PCR analysis of gene expression
Total RNA was extracted from infected and noninfected samples using TRIzol, and 1 μg of this RNA was used to make cDNA using random hexamer primers (Invitrogen) and the SuperScript III reverse transcriptase (Invitrogen). qRT-PCR was carried out on this cDNA using SYBR-Green. GAPDH was used as a reference. Primers used for qRT-PCR validation are listed in Supplemental Table 5.
qRT-PCR analysis of microRNAs
qRT-PCR analysis of the expression of microRNAs was performed as described previously (Clark et al. 2010). Primers are listed in Supplemental Table 5.
PCR analysis of genomic DNA
Genomic DNA was extracted from 20 adult worms by freeze-cracking once, followed by proteinase K digestion for 20 min at 60°C. PCR used standard Taq amplification for 40 cycles with annealing at 57°C. Primers are listed in Supplemental Table 5. PCR products were run on a 1% agarose gel at 90 V and visualized with ethidium bromide staining.
Small RNA sequencing analysis
Small RNA libraries were constructed, sequenced, and processed to remove adaptors and select small RNAs between 18 and 30 nt in length as described previously (Das et al. 2008). Short read sequencing data for JU1580 with and without infection is under accession number GSE21736 (publicly accessible). To quantify microRNAs, miRDeep2's Quantifier module (Friedländer et al. 2012) was used according to the manual with C. elegans microRNAs downloaded from miRBase (Kozomara and Griffiths-Jones 2011) as the reference. We then used DESeq (Anders and Huber 2010) to produce normalized counts for the 190 identified miRNAs in reads from JU1580 animals. To select miRNAs with high expression, we looked at miRNAs with greater than 100 reads in uninfected samples, identifying 114 miRNAs. To identify small RNAs antisense to protein-coding genes, the small RNAs were mapped to the genome using Bowtie selecting for those that mapped uniquely and with no mismatches as previously described (Bagijn et al. 2012). DESeq was then used to identify genes that showed statistically significantly different numbers of reads between N2 and rde-1 requiring a greater than fourfold difference and a Negative Binomial test P-value of <0.01.
Data access
Newly generated data for this study have been submitted to the NCBI Gene Expression Omnibus (GEO) (http://www.ncbi.nih.gov/geo/) under accession number GSE41058. Short read sequencing and microarray data are under subseries accessions GSE41056 and GSE41057, respectively.
Acknowledgments
We thank Mélanie Tanguy for insights into viral biology, Sylviane Moss for laboratory management, and Charles Bradshaw and Milan Malinsky for bioinformatics advice. This work was supported by a Cancer Research UK Program Grant to E.A.M. P.S. was supported by a Gonville and Caius College Research Fellowship. A.A. was supported by a Herchel-Smith Postdoctoral Research Fellowship. J.L.P. was supported by a Wellcome Trust PhD Scholarship.
Footnotes
[Supplemental material is available for this article.]
Article published online before print. Article, supplemental material, and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.153296.112.
Freely available online through the Genome Research Open Access option.
References
- Aliyari R, Ding SW 2009. RNA-based viral immunity initiated by the Dicer family of host immune receptors. Immunol Rev 227: 176–188 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anders S, Huber W 2010. Differential expression analysis for sequence count data. Genome Biol 11: R106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andersen EC, Gerke JP, Shapiro JA, Crissman JR, Ghosh R, Bloom JS, Félix M-A, Kruglyak L 2012. Chromosome-scale selective sweeps shape Caenorhabditis elegans genomic diversity. Nat Genet 44: 285–290 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bagijn MP, Goldstein LD, Sapetschnig A, Weick E-M, Bouasker S, Lehrbach NJ, Simard MJ, Miska EA 2012. Function, targets, and evolution of Caenorhabditis elegans piRNAs. Science 337: 574–578 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark AM, Goldstein LD, Tevlin M, Tavaré S, Shaham S, Miska EA 2010. The microRNA miR-124 controls gene expression in the sensory nervous system of Caenorhabditis elegans. Nucleic Acids Res 38: 3780–3793 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Claycomb JM, Batista PJ, Pang KM, Gu W, Vasale JJ, van Wolfswinkel JC, Chaves DA, Shirayama M, Mitani S, Ketting RF, et al. 2009. The Argonaute CSR-1 and its 22G-RNA cofactors are required for holocentric chromosome segregation. Cell 139: 123–134 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conine CC, Batista PJ, Gu W, Claycomb JM, Chaves DA, Shirayama M, Mello CC 2010. Argonautes ALG-3 and ALG-4 are required for spermatogenesis-specific 26G-RNAs and thermotolerant sperm in Caenorhabditis elegans. Proc Natl Acad Sci 107: 3588–3593 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corrêa RL, Steiner FA, Berezikov E, Ketting RF 2010. MicroRNA-directed siRNA biogenesis in Caenorhabditis elegans. PLoS Genet 6: e1000903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Das PP, Bagijn MP, Goldstein LD, Woolford JR, Lehrbach NJ, Sapetschnig A, Buhecha HR, Gilchrist MJ, Howe KL, Stark R, et al. 2008. Piwi and piRNAs act upstream of an endogenous siRNA pathway to suppress Tc3 transposon mobility in the Caenorhabditis elegans germline. Mol Cell 31: 79–90 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duchaine TF, Wohlschlegel JA, Kennedy S, Bei Y, Conte D, Pang K, Brownell DR, Harding S, Mitani S, Ruvkun G, et al. 2006. Functional proteomics reveals the biochemical niche of C. elegans DCR-1 in multiple small-RNA-mediated pathways. Cell 124: 343–354 [DOI] [PubMed] [Google Scholar]
- Dunbar TL, Yan Z, Balla KM, Smelkinson MG, Troemel ER 2012. C. elegans detects pathogen-induced translational inhibition to activate immune signaling. Cell Host Microbe 11: 375–386 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Félix M-A, Ashe A, Piffaretti J, Wu G, Nuez I, Bélicard T, Jiang Y, Zhao G, Franz CJ, Goldstein LD, et al. 2011. Natural and experimental infection of Caenorhabditis nematodes by novel viruses related to nodaviruses. PLoS Biol 9: e1000586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedländer MR, Mackowiak SD, Li N, Chen W, Rajewsky N 2012. miRDeep2 accurately identifies known and hundreds of novel microRNA genes in seven animal clades. Nucleic Acids Res 40: 37–52 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galiana-Arnoux D, Dostert C, Schneemann A, Hoffmann JA, Imler J-L 2006. Essential function in vivo for Dicer-2 in host defense against RNA viruses in Drosophila. Nat Immunol 7: 590–597 [DOI] [PubMed] [Google Scholar]
- Gent JI, Lamm AT, Pavelec DM, Maniar JM, Parameswaran P, Tao L, Kennedy S, Fire AZ 2010. Distinct phases of siRNA synthesis in an endogenous RNAi pathway in C. elegans soma. Mol Cell 37: 679–689 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gravato-Nobre MJ, Hodgkin J 2005. Caenorhabditis elegans as a model for innate immunity to pathogens. Cell Microbiol 7: 741–751 [DOI] [PubMed] [Google Scholar]
- Gu W, Shirayama M, Conte D Jr, Vasale J, Batista PJ, Claycomb JM, Moresco JJ, Youngman EM, Keys J, Stoltz MJ, et al. 2009. Distinct Argonaute-mediated 22G-RNA pathways direct genome surveillance in the C. elegans germline. Mol Cell 36: 231–244 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamilton AJ, Baulcombe DC 1999. A species of small antisense RNA in posttranscriptional gene silencing in plants. Science 286: 950–952 [DOI] [PubMed] [Google Scholar]
- Hellwig S, Bass BL 2008. A starvation-induced noncoding RNA modulates expression of Dicer-regulated genes. Proc Natl Acad Sci 105: 12897–12902 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffmann A, Natoli G, Ghosh G 2006. Transcriptional regulation via the NF-κB signaling module. Oncogene 25: 6706–6716 [DOI] [PubMed] [Google Scholar]
- Irazoqui JE, Urbach JM, Ausubel FM 2010. Evolution of host innate defence: Insights from Caenorhabditis elegans and primitive invertebrates. Nat Rev Immunol 10: 47–58 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kao C-Y, Los FCO, Huffman DL, Wachi S, Kloft N, Husmann M, Karabrahimi V, Schwartz J-L, Bellier A, Ha C, et al. 2011. Global functional analyses of cellular responses to pore-forming toxins. PLoS Pathog 7: e1001314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kennedy S, Wang D, Ruvkun G 2004. A conserved siRNA-degrading RNase negatively regulates RNA interference in C. elegans. Nature 427: 645–649 [DOI] [PubMed] [Google Scholar]
- Ketting RF, Fischer SE, Bernstein E, Sijen T, Hannon GJ, Plasterk RH 2001. Dicer functions in RNA interference and in synthesis of small RNA involved in developmental timing in C. elegans. Genes Dev 15: 2654–2659 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knight SW, Bass BL 2001. A role for the RNase III enzyme DCR-1 in RNA interference and germ line development in Caenorhabditis elegans. Science 293: 2269–2271 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kozomara A, Griffiths-Jones S 2011. miRBase: Integrating microRNA annotation and deep-sequencing data. Nucleic Acids Res 39: D152–D157 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krek A, Grün D, Poy MN, Wolf R, Rosenberg L, Epstein EJ, MacMenamin P, da Piedade I, Gunsalus KC, Stoffel M, et al. 2005. Combinatorial microRNA target predictions. Nat Genet 37: 495–500 [DOI] [PubMed] [Google Scholar]
- Kudlow BA, Zhang L, Han M 2012. Systematic analysis of tissue-restricted miRISCs reveals a broad role for microRNAs in suppressing basal activity of the C. elegans pathogen response. Mol Cell 46: 530–541 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lall S, Grün D, Krek A, Chen K, Wang Y-L, Dewey CN, Sood P, Colombo T, Bray N, MacMenamin P, et al. 2006. A genome-wide map of conserved microRNA targets in C. elegans. Curr Biol 16: 460–471 [DOI] [PubMed] [Google Scholar]
- Li H, Li WX, Ding SW 2002. Induction and suppression of RNA silencing by an animal virus. Science 296: 1319–1321 [DOI] [PubMed] [Google Scholar]
- Lu R, Maduro M, Li F, Li HW, Broitman-Maduro G, Li WX, Ding SW 2005. Animal virus replication and RNAi-mediated antiviral silencing in Caenorhabditis elegans. Nature 436: 1040–1043 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu R, Yigit E, Li WX, Ding SW 2009. An RIG-I-Like RNA helicase mediates antiviral RNAi downstream of viral siRNA biogenesis in Caenorhabditis elegans. PLoS Pathog 5: e1000286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahajan-Miklos S, Tan MW, Rahme LG, Ausubel FM 1999. Molecular mechanisms of bacterial virulence elucidated using a Pseudomonas aeruginosa–Caenorhabditis elegans pathogenesis model. Cell 96: 47–56 [DOI] [PubMed] [Google Scholar]
- Maydan JS, Lorch A, Edgley ML, Flibotte S, Moerman DG 2010. Copy number variation in the genomes of twelve natural isolates of Caenorhabditis elegans. BMC Genomics 11: 62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McEwan DL, Kirienko NV, Ausubel FM 2012. Host translational inhibition by Pseudomonas aeruginosa exotoxin A triggers an immune response in Caenorhabditis elegans. Cell Host Microbe 11: 364–374 [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Rourke D, Baban D, Demidova M, Mott R, Hodgkin J 2006. Genomic clusters, putative pathogen recognition molecules, and antimicrobial genes are induced by infection of C. elegans with M. nematophilum. Genome Res 16: 1005–1016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pak J, Fire A 2007. Distinct populations of primary and secondary effectors during RNAi in C. elegans. Science 315: 241–244 [DOI] [PubMed] [Google Scholar]
- Park H, Bourla AB, Kastner DL, Colbert RA, Siegel RM 2012. Lighting the fires within: The cell biology of autoinflammatory diseases. Nat Rev Immunol 12: 570–580 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parrish S, Fire A 2001. Distinct roles for RDE-1 and RDE-4 during RNA interference in Caenorhabditis elegans. RNA 7: 1397–1402 [PMC free article] [PubMed] [Google Scholar]
- Partridge FA, Gravato-Nobre MJ, Hodgkin J 2010. Signal transduction pathways that function in both development and innate immunity. Dev Dyn 239: 1330–1336 [DOI] [PubMed] [Google Scholar]
- Pavelec DM, Lachowiec J, Duchaine TF, Smith HE, Kennedy S 2009. Requirement for the ERI/DICER complex in endogenous RNA interference and sperm development in Caenorhabditis elegans. Genetics 183: 1283–1295 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pujol N, Link EM, Liu LX, Kurz CL, Alloing G, Tan MW, Ray KP, Solari R, Johnson CD, Ewbank JJ 2001. A reverse genetic analysis of components of the Toll signaling pathway in Caenorhabditis elegans. Curr Biol 11: 809–821 [DOI] [PubMed] [Google Scholar]
- Pujol N, Zugasti O, Wong D, Couillault C, Kurz CL, Schulenburg H, Ewbank JJ 2008. Anti-fungal innate immunity in C. elegans is enhanced by evolutionary diversification of antimicrobial peptides. PLoS Pathog 4: e1000105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schott DH, Cureton DK, Whelan SP, Hunter CP 2005. An antiviral role for the RNA interference machinery in Caenorhabditis elegans. Proc Natl Acad Sci 102: 18420–18424 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sijen T, Fleenor J, Simmer F, Thijssen KL, Parrish S, Timmons L, Plasterk RH, Fire A 2001. On the role of RNA amplification in dsRNA-triggered gene silencing. Cell 107: 465–476 [DOI] [PubMed] [Google Scholar]
- Sijen T, Steiner FA, Thijssen KL, Plasterk RHA 2007. Secondary siRNAs result from unprimed RNA synthesis and form a distinct class. Science 315: 244–247 [DOI] [PubMed] [Google Scholar]
- Tabara H, Sarkissian M, Kelly WG, Fleenor J, Grishok A, Timmons L, Fire A, Mello CC 1999. The rde-1 gene, RNA interference, and transposon silencing in C. elegans. Cell 99: 123–132 [DOI] [PubMed] [Google Scholar]
- Tarallo V, Hirano Y, Gelfand BD, Dridi S, Kerur N, Kim Y, Cho WG, Kaneko H, Fowler BJ, Bogdanovich S, et al. 2012. DICER1 loss and Alu RNA induce age-related macular degeneration via the NLRP3 inflammasome and MyD88. Cell 149: 847–859 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tenor JL, Aballay A 2008. A conserved Toll-like receptor is required for Caenorhabditis elegans innate immunity. EMBO Rep 9: 103–109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Troemel ER, Chu SW, Reinke V, Lee SS, Ausubel FM, Kim DH 2006. p38 MAPK regulates expression of immune response genes and contributes to longevity in C. elegans. PLoS Genet 2: e183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Welker NC, Habig JW, Bass BL 2007. Genes misregulated in C. elegans deficient in Dicer, RDE-4, or RDE-1 are enriched for innate immunity genes. RNA 13: 1090–1102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Welker NC, Pavelec DM, Nix DA, Duchaine TF, Kennedy S, Bass BL 2010. Dicer's helicase domain is required for accumulation of some, but not all, C. elegans endogenous siRNAs. RNA 16: 893–903 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilkins C, Dishongh R, Moore SC, Whitt MA, Chow M, Machaca K 2005. RNA interference is an antiviral defence mechanism in Caenorhabditis elegans. Nature 436: 1044–1047 [DOI] [PubMed] [Google Scholar]
- Yoneyama M, Kikuchi M, Natsukawa T, Shinobu N, Imaizumi T, Miyagishi M, Taira K, Akira S, Fujita T 2004. The RNA helicase RIG-I has an essential function in double-stranded RNA-induced innate antiviral responses. Nat Immunol 5: 730–737 [DOI] [PubMed] [Google Scholar]