

Abstract
More than half of Caenorhabditis elegans pre-mRNAs lose their original 5′ ends in a process termed “trans-splicing” in which the RNA extending from the transcription start site (TSS) to the site of trans-splicing of the primary transcript, termed the “outron,” is replaced with a 22-nt spliced leader. This complicates the mapping of TSSs, leading to a lack of available TSS mapping data for these genes. We used growth at low temperature and nuclear isolation to enrich for transcripts still containing outrons, applying a modified SAGE capture procedure and high-throughput sequencing to characterize 5′ termini in this transcript population. We report from this data both a landscape of 5′-end utilization for C. elegans and a representative collection of TSSs for 7351 trans-spliced genes. TSS distributions for individual genes were often dispersed, with a greater average number of TSSs for trans-spliced genes, suggesting that trans-splicing may remove selective pressure for a single TSS. Upstream of newly defined TSSs, we observed well-known motifs (including TATAA-box and SP1) as well as novel motifs. Several of these motifs showed association with tissue-specific expression and/or conservation among six worm species. Comparing TSS features between trans-spliced and non-trans-spliced genes, we found stronger signals among outron TSSs for preferentially positioning of flanking nucleosomes and for downstream Pol II enrichment. Our data provide an enabling resource for both experimental and theoretical analysis of gene structure and function in C. elegans.
In most organisms, the locations of transcription start sites (TSSs) can be determined by establishing the sequences of mRNA 5′ ends. For a subset of single-cell eukaryotes and animals, however, a processing event known as “trans-splicing” obscures the locations of the TSSs (Sutton and Boothroyd 1986; Krause and Hirsh 1987; Lasda and Blumenthal 2011). Trans-splicing is an efficient process that results in removal of the 5′ end of the pre-mRNA, replacing it with a short 5′ leader that is then retained on the mature mRNA. The removed sequence (between the TSS and the first active 3′ splice site in the newly transcribed mRNA precursor) is called the “outron” (Conrad et al. 1991). Trans-splicing is a spliceosome-catalyzed process that can be thought of as a surrogate use of a mobile 5′ splice site contained on a short (usually ∼100 nt) RNA called an SL RNA. SL RNAs are present in the nucleus in the form of Sm protein-bound small nuclear ribonucleoprotein particles (snRNPs), where the 5′ 22 nt forms the SL exon, with the 3′ portion serving as “intron.”
Caenorhabditis elegans has been a valuable model system for studying a variety of aspects of gene expression (including trans-splicing) (Krause and Hirsh 1987; Lasda and Blumenthal 2011). Approximately 70% of C. elegans genes are subject to trans-splicing (Allen et al. 2011; Lasda and Blumenthal 2011). C. elegans has two types of trans-splicing, each with a distinct splice leader (SL1 and SL2) (Spieth et al. 1993). SL1 is the more common splice leader, with 55% of C. elegans genes spliced to SL1 (Allen et al. 2011). SL1 trans-spliced genes are either singly transcribed genes or the first genes in cotranscribed operons. The SL1 trans-spliced genes are thus those for which the outron 5′ ends correspond to TSS positions. About 15% of all genes are trans-spliced by the spliced leader SL2 or one of its close relatives (Allen et al. 2011). SL2 trans-spliced genes are always downstream in operons, so they generally do not have TSSs present just 5′ of the gene body. Due to the rapidity of trans-splicing and the overwhelming predominance of post-trans-splicing forms among stable cytoplasmic RNAs, the annotation of the worm genome has lacked a global description of TSSs, confounding analysis of cis-regulatory sequences, chromatin configurations around promoters, and identification of specialized features such as bidirectional promoters.
Here we perform a 5′-targeted analysis on nuclear RNA from C. elegans grown under conditions that slow the processing of outrons, enabling the mapping of a large number of TSS for both SL1 trans-spliced and non-trans-spliced genes and a consequent picture of transcriptional initiation repertoire in this system.
Results
Capture of 5′-end RNA reads from nuclei
Animals grown at the low end of the normal temperature range were used to isolate potential pre-trans-splicing intermediates. The choice of lower growth temperatures to support identification of pre-spliced intermediates follows observations that (1) C. elegans strains partially defective in key trans-splicing RNP factors show a cold sensitivity in organismal phenotype (MacMorris et al. 2007), and (2) splicing can be mechanistically sensitive to cold temperatures as a result of numerous base-pairing interactions that must be remodeled during the process (Strauss and Guthrie 1991).
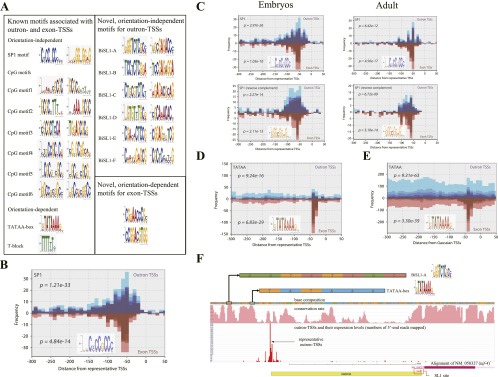
To identify TSSs that are processed before trans-splicing or are free of trans-spliced leaders, we grew populations of animals at 16°C, isolated nuclei from embryos and whole adult tissues, and extracted total nuclear RNA. To minimize contaminating ribosomal RNA (rRNA) molecules, we used hybrid subtraction-based enrichment of RNA with the RiboMinus RNA Kit before sequencing (Ruan et al. 2004; Chen and Duan 2011). We then used a 5′-SAGE method (Fig. 1A) to capture 5′ ends, including DNase treatment and a phosphatase treatment preceding cap removal and linker ligation that strongly enriches for RNA molecules where a 5′ phosphate is only revealed following decapping (protocol described in Hashimoto et al. 2009). Illumina sequencing was then carried out, resulting in approximately 119 million and 76 million 76-bp single-end reads collected from the nuclei of embryo and adult samples, respectively (see Methods). These reads were aligned to the C. elegans genome WS220 (Hillier et al. 2009; Harris et al. 2010) using BWA (Li and Durbin 2009) (see Methods). From this analysis, approximately 55.6 million and 30.4 million reads, respectively, were anchored to unique positions (Table 1), with the approximately 55.6 million reads from embryo samples used as the primary data set for initial TSS identification. As expected from the mass of ribosomal RNA present even after (incomplete) hybrid subtraction, the pool of sequenced reads still included approximately 24.2 million 5′-end rRNA reads. We removed these after their alignment to the genome, leaving a total of 31.4 million reads from the embryonic library.
Figure 1.
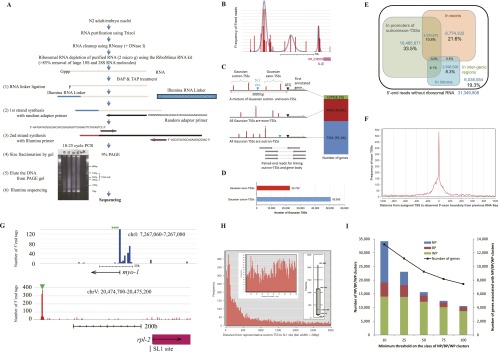
Identification of TSSs from alignments of 5′-end reads. (A) Process for 5′-end library construction for Illumina GA sequencing. See details in Methods. (B) Frequency distribution of reads aligned to genomic locations upstream of NM_058956 (phosphatase). Three peaks were calculated as the best-fitting set of Gaussian distributions according to Bayesian information criteria. (C) Three classes of genes with different patterns of Gaussian TSS distributions in the regions of 3000 bp upstream of the translation initiation sites. (Top figure) Case in which both Gaussian outron/exon-TSSs occur because of the presence of an SL1 site between the two classes of Gaussian TSSs. (Middle and bottom figures) Cases in which all Gaussian TSSs are exon-TSSs and outron-TSSs, respectively. (Vertical bars with asterisk) Representative TSSs with the maximum number of aligned 5′-end tags. (Right bars) The numbers of genes in the three classes. As shown in the bottom, alignments of paired-end reads were useful in linking representative TSSs to their gene bodies over SL1 sites. (D) Frequency of Gaussian exon/outron-TSSs. (E) 5′-End reads excluding rRNA reads were categorized into the four groups of reads that were mapped onto promoters of outron/exon-TSSs, exons, introns, and intergenic regions, respectively. The Venn diagram illustrates the relationship among the four groups. (F) Frequency distribution of distances between 5′-exon boundaries from WS220 (Hillier et al. 2009; Harris et al. 2010) and proximal exon-TSSs identified in our data. (G) Examples of 5′ capture data for genes that have been previous subjects of TSS mapping. (Above) Exon 5′ end for the gene myo-1 with original 5′ region having four candidate 5′ ends (from S1 nuclease mapping with accuracy to within a few bp) (Okkema et al. 1993) marked with green arrows. (Below) Outron 5′ region for rpl-2 with original 5′ end (from 5′ RACE mapping with accuracy to within 1–2 bp) (Sleumer et al. 2012) highlighted by the green arrow. (H) Frequency distribution of the distances between SL1 sites and most abundant representative outron-TSSs supported by 10 or more single-end reads. (Left inset) This magnifies the region ranging from 10 to 100 bp; (right inset) the 90th, 75th, 25th, and 10th percentiles and the median of outron lengths. (I) Numbers of TSS clusters categorized into NP, BP, and WP types in terms of five values of the minimum threshold on the size of TSS clusters. Because multiple TSS clusters may be associated with each gene, the second vertical axis shows the numbers of genes involved. Observe that TSS clusters of WP type are dominant for threshold ≥25.
Table 1.
Statistics of sequenced 5′-end reads from nuclei of embryo and adult samples and their alignment to the worm genome (WS220)
Selecting representative TSSs is not simple because the distribution of TSSs is not necessarily sharp and unique but is often broad and/or multi-modal (Suzuki et al. 2001; Schug et al. 2005; Carninci et al. 2006; Frith et al. 2006; Saxonov et al. 2006; Zhu et al. 2008; Rach et al. 2011), as illustrated in Figure 1B. We take several approaches in working with the multi-modal character of TSS distributions. These include both approaches based on calling one or a few most prominent starts for each gene as representative and approaches based on a base-by-base distribution of TSS frequencies covering an extensive upstream region of every gene.
In calling individual reference starts based on many noisy peaks present in the frequency distribution, one approach is to model start site regions as a mixture of Gaussian distributions; such an approach has been widely used in the recent analysis of TSS distributions (Boyle et al. 2008; Ni et al. 2010; Rach et al. 2011). We calculated a series of best-fitting Gaussian distributions using Bayesian information criteria (see Fig. 1B). We then quantified the expression level of the resulting Gaussian-based TSS models by the number of reads in the corresponding Gaussian window. To eliminate noise in genome-wide analysis, we used 73,277 Gaussian TSS peaks that were supported by 10 or more reads (see their positions in Supplemental Table S1). In addition, we used a noise-filtering method using feature density estimator F-Seq (Boyle et al. 2008) to exclude TSS peaks whose F-Seq values are less than 4 standard deviations above the mean of the background (F-Seq has been widely used in other studies on TSS distributions) (Ni et al. 2010; Rach et al. 2011).
The identified “peaks” from the above analysis ranged from cases showing a small number of starts in a tight cluster to those spread over hundreds of bases of DNA. While the unique-site peaks are readily recognizable as a defined entity, parsing of more disperse TSS regions into individual peaks is by nature dependent on the algorithm and parameters. With >80% of the observed Gaussian peaks showing a dispersion (standard deviation) of <20 bp, the most challenging cases are in the minority (for reference, Supplemental Table S2 shows a list of Gaussian peaks parsed from the data with a maximal standard deviation parameter of 20 bp, and Supplemental Fig. S1 presents a statistical analysis of Supplemental Table S2).
We defined outron-TSSs as candidate TSSs by two criteria: (1) They had previously mapped downstream SL1 sites (Allen et al. 2011); and (2) the TSS occurred in a window from 10 bp to 3000 bp upstream of the translation initiation site of the first downstream annotated gene (illustrated in Fig. 1C). The 10-bp lower limit was chosen to avoid putative outrons that would likely be too short to allow trans-splicing (also see below) as well as confounding instances resulting from short upstream near-matches to SL1 sequence; the 3000-bp upper limit was chosen based on general characteristics of C. elegans promoters (Hunt-Newbury et al. 2007). The term “outron-TSS” is derived from the term “outron” (Blumenthal 2005) indicative of the sequence between the natural transcription start site and spliced leader attachment site of a relevant gene (Blumenthal 2005). We defined exon-TSSs as candidate TSSs with an annotated coding region with no SL1 trans-splice sites that were not also intron 3′ splice sites. (A low level of trans-splicing sometimes occurs at intron 3′ splice sites [Allen et al. 2011].) Figure 1D indicates the incidence of all Gaussian outron/exon-TSSs.
Among 13,336 genes having Gaussian TSS peaks in the region of length 3000 bp upstream of their translation initiation sites, 7351 genes (55.1%) had only Gaussian outron-TSSs and were therefore defined as trans-spliced genes, and 4906 genes (36.8%) having only Gaussian exon-TSSs were categorized as non-trans-spliced genes (Fig. 1C). For the other 1079 genes (8.1%) that had both types of Gaussian TSSs in our data, we place the genes in neither category, avoiding for this analysis any decision of whether they were trans-spliced. For genes that could be defined unambiguously as trans-spliced or non-trans-spliced, we assigned gene-unique representative outron/exon-TSSs with the maximum number of aligned reads. This allowed us to pursue further analysis in parallel with (1) all Gaussian peaks as TSSs (“The Gaussian Peak Collection”), and (2) representative gene-unique TSSs (one for each gene).
To assess the specificity of our 5′-SAGE method in detecting TSSs, we examined the balance between ends present within known RNAs and those in regions upstream of annotated genes, assigning the approximately 31.4 million embryonic alignments to one or more of the categories: promoters of outron/exon-TSSs, exons, introns, and intergenic regions. The former three categories are overlapping as illustrated by the Venn diagram in Figure 1E. We compared our results with those by the CAGE method, another widely used method of monitoring 5′ ends of RNA. The ratio of 5′-end tags mapped to promoter regions is 50.2%, while the ratio of 5′-end tags by the CAGE method in the human and mouse genomes was smaller than 30% (Carninci et al. 2006), indicating that the background signal of our 5′-SAGE method is similar to that of CAGE. Although the modest number of 5′ ends present in annotated transcribed regions (21.6% of the total) are under some suspicion as potentially representing capture of fragmented mRNA precursors at positions other than the 5′ end, the fact that the vast majority (78.4%) are in regions with no known abundant transcripts suggests that the latter RNAs are almost certainly TSSs. In particular, we stress that a situation in which 5′ ends were captured at random from fragmented RNA present in the samples would invariably yield a dramatic enrichment for tags within regions annotated in other RNA sequencing projects. That this was not the case argues strongly for the enrichment in this analysis of bona fide capped 5′ ends.
An independent evaluation of 5′-end mapping was possible for known exon-TSSs that are free from trans-splicing. We first evaluated the correspondence of 5′ mapping for several C. elegans genes in which single-gene studies (generally individual primer extension reactions either with a sequencing endpoint or with a PCR/sequencing endpoint, or nuclease protection mapping) had defined an exon 5′ end within a few nucleotides. Although not large in number, such specific examples served as valuable benchmarks. For the set of genes evaluated (unc-54, myo-1, myo-2, sqt-1, col-12), the major 5′ ends defined in earlier studies were within a few base pairs of prevalent 5′ capture sites from this work (Krause and Hirsh 1987; Dibb et al. 1989; Park and Kramer 1990, 1994; Okkema et al. 1993). In addition to this individual gene analysis, we examined 5268 presumed non-trans-spliced first-exon 5′ boundaries that were derived from whole RNA-seq data in WS220 (Hillier et al. 2009; Harris et al. 2010) and that reside ≥100 bp upstream of ATG sites of 1078 genes, analyzing the frequency distribution of distances between the known 5′ ends and their proximal exon-TSSs in our data. This data set would be expected to be highly enriched for exon TSS positions. As would be expected from concordance of the two data sets, we observed a remarkable peak at zero in the distance distribution, confirming the significant concordance between known 5′ ends of mRNAs and our exon-TSSs (Fig. 1F).
Existing outron-TSS mapping studies are much rarer in C. elegans, as noted above, due to the challenges inherent in capturing a transient 5′-end sequence. We know of five genes that have been mapped at high precision through primer extension (rol-6 and col-13) (Park and Kramer 1990, 1994), 5′-anchored PCR and sequencing (rpl-2 and ace-1) (Culetto et al. 1999; Sleumer et al. 2012), or S1 nuclease protection (ubq-1). In our 5′-end repertoires, we see support for the mapped outron-TSSs for rpl-2 (Fig. 1G), col-13, and for two of three 5′ ends mapped for ace-1 outron sequences. Rol-6 expression is very low in the samples used for this work, presumably due to the precise staging of transcription. Thus, little or no signal is seen in this study for any sequence upstream of rol-6. For ubq-1, we observe a different 5′ end from that mapped in the original study by S1 nuclease protection. (The prominent 5′ end inferred from the original study was at −455 nt. We observed a strong end at −294, in a region of the gene that was electrophoresed out of the pictured gel in the S1 mapping study. That study was also carried out with total RNA from heat-shocked embryos, while we used nuclear RNA from embryos grown at the lower end of the C. elegans' viable temperature range.) These comparisons, as well as comparisons with the regional outron mapping of Morton and Blumenthal (2011), serve both to support the utility of the present data and to provide caution in any universal interpretation of data from a limited set of developmental timing and growth conditions. Even if stable mRNA is present in the sample, these assays will only provide a positive outron signal based on recent transcription, specifically addressing the (likely transient) presence of the pre-trans-splicing nuclear precursor under the growth conditions of the assay.
To provide valid outron-TSS candidates, we sought where possible to associate each with the body of the corresponding gene. Exon-TSS assignments have the advantage of potential validation through a continuous set of RNA-seq reads that go into the relevant mRNA. To further evaluate outron-TSS assignments, we performed paired-end sequencing of the embryonic library, generating 42.2 million paired-end reads of 5′-end RNA fragments; 23.1 million pairs mapped to unique positions in the genome (Table 2). We then applied an analysis that checked whether the SL1 site in a gene was sandwiched by the aligned paired-end reads, providing support for the assignment of an outron-TSS to a gene body associated with an SL1 site as shown in Figure 1C. One might be concerned with the difficulty of linking outron-TSSs to SL1 sites because the distance between paired-end reads (∼250 bp being the cDNA insert size in our experiment) could be smaller than the length of typical outrons. Examining the frequency distribution of the distances between the SL1 sites and the most abundant representative outron-TSSs (defined by single-end sequencing) shows that, although many TSSs were located at >250 bp upstream of corresponding SL1 sites, 40.3% of 7351 TSSs were seen within 250 bp of SL1 sites (Fig. 1H), indicating that our paired-end sequencing data will be useful in linking many TSSs to associated gene bodies.
Table 2.
Statistics of sequenced paired-end reads for TSSs from embryo nuclei and their alignment to the worm genome
In Figure 1H, we find that lengths of outrons, the region between the TSS and the trans-splice site, vary greatly, from a 90th percentile of 2099 to a 10th percentile of 59, with a median length of 369. Whereas the long outrons are similar in length to long introns in worms, the putative outrons defined by these data were in some cases much shorter than introns can be, consistent with the idea that the constraint on the lower limit of intron length results from a minimum distance between the two splice sites needed for spliceosome assembly. While the possibility of short outrons is intriguing, we note that the type of data acquired in this work (a static picture of 5′-end structures) does not provide information about precursor–product relationships. Apparent short outrons, based on the distance from the predominant TSS to the trans-splice site, may lead to misinterpretation. The true outrons may derive from initiation events upstream. Indeed, we observed upstream TSSs in 118 (76%) among 154 genes for which the predominant TSS was within 20 bp upstream of the SL1 site (Supplemental Table S2). In the remaining 36 genes, because such outrons may be processed efficiently, these upstream TSSs might either not appear in our data or appear as rarely used sites. Conversely, the outron sites that appear in nuclear RNA would have been enriched for slower 5′ processing, potentially enriching the signal for any short outrons that are spliced inefficiently.
An intriguing diversity among initiation elements comes in the degree to which initiation is specified at a single position in the DNA sequence versus a range of positions. Classifiers for peak breadth have been applied extensively in such analyses, including previous studies on human, mouse, and fruit fly genomes: In each case, once a set of reference TSSs was determined, shapes of TSS distributions in promoters of individual reference TSSs would be assigned (Carninci et al. 2006; Frith et al. 2006; Ni et al. 2010; Rach et al. 2011). We classified C. elegans TSS distributions into three types: NP (Narrow with Peak), BP (Broad with Peak), and WP (Weak Peak) according to the definitions proposed by Ni et al. (2010). The precise category fractions varied somewhat depending on the minimum threshold on the size of the TSS cluster (e.g., 10, 25, 50, 75, and 100). For start sites with modest-to-high expression (thresholds of more than 25 reads observed) started, WP-type shapes were dominant (Fig. 1I). This observation is consistent with WP dominance in other studies of human, mouse, and fruit fly genomes (Carninci et al. 2006; Frith et al. 2006; Ni et al. 2010; Rach et al. 2011). TSS distribution of WP type frequently ranged over a long region (typically of length >500 bp) and had multimodal peaks; for some analyses, we thus decompose each WP TSS distribution into Gaussian peaks to examine characteristics as noted below.
Nucleosome and Pol II positioning around TSSs and SL1 sites
Some types of modified histones and RNA Pol II are known to accumulate at promoter regions (Whittle et al. 2008; Baugh et al. 2009), with information related to this accumulation having been used to approximate the locations of transcription initiation sites. To check whether the TSS collection defined in this study shares these characteristics with other species, we examined the publicly available data of nucleosome positioning estimated from MNase digestion of whole chromatin (Valouev et al. 2008) or of chromatin pre-selected for its association with the histone modification mark H3K4me2/3 (Gu and Fire 2010), also examining Pol II positioning (Gerstein et al. 2010) surrounding representative TSSs. Using either representative TSSs (Fig. 2A,B) or the full Gaussian peak collection (Supplemental Fig. S2A,B), we observed arrays of positioned and phased nucleosomes, with an apparent nucleosome-depleted region immediately upstream of TSSs. The upstream nucleosome depletion is a conserved feature that has also been reported in mammals and other eukaryotes (Mavrich et al. 2008; Kaplan et al. 2009; Sasaki et al. 2009; Weiner et al. 2010; Valouev et al. 2011). The characteristic features of nucleosome positioning relative to representative TSSs were also reconfirmed by comparison with H3K4me2/3 data (Supplemental Fig. S2D). The period of the nucleosome positioning around representative TSSs was estimated as ∼160 bp, as evaluated using an autocorrelation measure (Fig. 2D). The ∼160-bp spacing around representative TSSs was smaller than the ∼175-bp spacing in the entire genome (Valouev et al. 2008). Nucleosome positioning scores were more pronounced for representative TSSs of higher expression (Fig. 2A,B). These observations indicate that transcription correlates with more consistent packing of nucleosomes. This observation is consistent with barrier nucleosome models (Mavrich et al. 2008) in which Pol II stalled at the TSSs potentially serves as a barrier that can facilitate packing of nucleosomes downstream from TSSs. Indeed, Figure 2E exhibits Pol II enrichment at ∼170 bp and ∼100 bp downstream from representative outron/exon-TSSs, respectively. Interestingly, the distance between TSS and maximum Pol II enrichment differed between outron-TSS sites (∼170 bp) and exon-TSSs (∼100 bp), respectively. Although the mechanistic basis for this difference was not clear, the existence of a difference suggested the possibility that the biological system would have the opportunity, if needed, to distinguish functionally between the two types of 5′ end.
Figure 2.
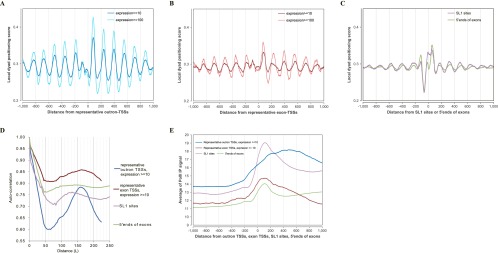
(A,B) The distributions of nucleosome positioning (local dyad positioning score) around representative outron-TSSs (A) and exon-TSSs (B) of expression levels ≥10 and ≥100 (see the distribution around Gaussian TSSs in Supplemental Fig. S2A,B). From these comparisons, note that the periodic pattern of nucleosome positioning was of substantially greater amplitude for TSSs of greater expression. (C) Nucleosome positioning (local dyad positioning score) around SL1 sites and 5′ ends of exons. We used all available SL1 sites without considering their expression levels. (D) Autocorrelation analysis: Let s(x) denote the local dyad positioning score. The autocorrelation defined by 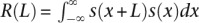 was calculated around representative outron/exon-TSSs, SL1 sites, and 5′ ends of exons, where L is displayed in the x-axis. Sharp peaks at ∼160 bp for outron-TSSs and broad peaks surrounding ∼160 bp for exon-TSSs highlight that arrays of positioned and phased nucleosomes are more prominent around outron-TSSs than around exon-TSSs. Broad peaks at ∼140 bp are also seen for SL1 sites and 5′ ends of exons. (E) Pol II occupancy around representative outron/exon-TSSs of expression level ≥10, SL1 sites, and 5′ ends of exons.
was calculated around representative outron/exon-TSSs, SL1 sites, and 5′ ends of exons, where L is displayed in the x-axis. Sharp peaks at ∼160 bp for outron-TSSs and broad peaks surrounding ∼160 bp for exon-TSSs highlight that arrays of positioned and phased nucleosomes are more prominent around outron-TSSs than around exon-TSSs. Broad peaks at ∼140 bp are also seen for SL1 sites and 5′ ends of exons. (E) Pol II occupancy around representative outron/exon-TSSs of expression level ≥10, SL1 sites, and 5′ ends of exons.
It has been shown that nucleosomes have a tendency to associate with DNA sequences exhibiting high G/C nucleotide and dinucleotide content, while being excluded from DNA sequences exhibiting high A/T nucleotide and dinucleotide content (Bernstein et al. 2004; Field et al. 2008; Hughes and Rando 2009; Segal and Widom 2009; Tillo et al. 2010). We therefore examined the correlation of this underlying nucleotide signal to nucleosome positioning around newly identified representative TSSs (Fig. 3A,B) and around the full collection of Gaussian peaks (Supplemental Fig. S2E,F); in both analyses, we observed an unexpected phenomenon, a significant peak of C/G rate and a remarkable valley of A/T rate around ∼100 bp upstream of outron- and exon-TSSs, corresponding to a site where observed nucleosomes were depleted.
Figure 3.
(A,B) A/T and C/G dinucleotide incidence rates around representative outron-TSSs (A) and exon-TSSs (B) of expression levels ≥10 and ≥100 (see the distribution for Gaussian TSSs in Supplemental Fig. S2E,F). AT richness immediately upstream of outron-TSSs is due to the requirement for SL1 splicing motif “TTTTCAG.” In both types of TSSs, a significant valley in the A/T rate and a significant peak in the C/G rate were observed ∼100 bp upstream of TSSs. (C) A/T and C/G dinucleotide incidence rates around SL1 sites and 5′ ends of exons. In all graphs, a running average over a 20-bp window is shown for smoothing lines.
Nucleosome positioning became more prominent around outron-TSSs than around exon-TSSs (Supplemental Fig. S2A,B), suggesting that a higher Pol II signal and a more pronounced array of positioned nucleosomes were associated with trans-splicing activity. We therefore examined nucleosome positioning around SL1 sites and found that nucleosome cores were enriched ∼40 bp downstream from SL1 sites (Fig. 2C). One possibility was that this enrichment might reflect a combination of the distance distribution between TSSs and SL1 sites and the phased array of nucleosomes downstream from TSSs; however, this hypothesis was not supported by our statistical analysis (Supplemental Fig. S2G,H).
As an alternative and more direct link, we considered the possibility that genomic signals for splicing machinery and nucleosome constraint might be associated. Such an association has been observed at cis-spliced sites (Kolasinska-Zwierz et al. 2009), hence an expectation that a disambiguation of TSSs and SL1 spliced sites might show such an effect. Consistent with such an expectation, nucleosome positioning around 5′ ends of exons was quite close to that around SL1 sites (Fig. 2C). Furthermore, we also observed RNA Pol II enrichment at ∼100 bp downstream from SL1 sites and 5′ ends of exons (Fig. 2E), and a strong correlation of A/T and C/G dinucleotide rates to the nucleosome enrichment downstream from SL1 sites and 5′ ends of exons (Fig. 3C). These observations suggest an available confluence in biological regulation in which shared genomic signals and underlying dependencies on the underlying sequence can be used to coordinate nucleosome positioning, splicing, and polymerase initiation.
Evolutionarily conserved sequence motifs around TSSs
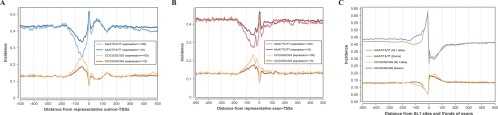
The large number of newly identified TSSs allowed us to search regions surrounding the TSSs for sequence motifs that might serve as cis-regulatory elements, in particular, transcription factor binding sites. Genomic regions ranging from −300 bp to +50 bp of representative TSSs (and in a separate analysis around the full collection of Gaussian TSS peaks) were selected as putative promoters. Both analyses were of use, although we note with the full Gaussian-TSS collection that multiple closely spaced TSSs in this collection would allow some single cis-elements to be multiply counted, providing some impetus to mine the representative TSS collection preferentially. Candidate promoter regions were analyzed using MEME (Bailey and Elkan 1994), a widely used program that identifies over-represented sequence motifs within a set of input sequences (see Methods). We used MEME to search for statistically significant motifs of ≤10 bp (E-value < 10−20), noting that these computationally predicted motifs may not necessarily be functional despite their high statistical significance.
The evolutionary conservation of a motif is one of the strongest lines of evidence for its functional importance. We therefore assessed the conservation of each motif occurrence with respect to the average conservation rate among the genomes of six related nematode species for all positions in the occurrence (see Methods). Seventeen evolutionarily conserved motifs were detected in this analysis; of these, nine were previously known, and the remaining eight were novel (Fig. 4A). SP1 is a family of transcription factors involved in transcriptional control in several different systems. Figure 4B illustrates the frequency distribution of SP1 motif (Huber et al. 1998; Xi et al. 2007) occurrences around representative TSSs. Highly conserved SP1 motif occurrences were significantly enriched in promoter regions of representative outron-TSSs (P = 1.21 × 10−33) and exon-TSSs (P = 4.18 × 10−14) according to the Wilcoxon rank-sum test (see Methods). Since all motifs were of length ≤10, the number of all possible ≤10-mers approximates (4/3) × 106 [=∼(4/3) × 410]. To reduce the possibility of selecting false-positive motifs from ≤10-mers, we performed multiple hypothesis testing using the Bonferroni correction to check each motif at a significance level of 5% divided by (4/3) × 106 (=3.75 × 10−8).
Figure 4.
Conserved sequence motifs upstream of TSSs. (A) List of known motifs identified for outron- and exon-TSSs, and a list of novel motifs. (B) Frequency distribution of motif occurrences and their conservation around representative TSSs. (The upper histogram) The frequency distribution of motifs around outron-TSSs with downstream SL1 sites; (the lower histogram) the motif distribution around exon-TSSs without SL1 sites. The conservation rate of a motif occurrence is the average conservation rate of all positions in a given instance, where the conservation rate in the worm genome was calculated using six allied species (see Methods). Conservation rates ranging from 0 to 1 were divided into five levels. Darker colors indicate higher conservation rate; for example, the darkest color denotes a range from 0.8 to 1. In the figure, an SP1 motif (Xi et al. 2007) was observed around outron-TSSs and exon-TSSs. The SP1 motif was significantly conserved around a strong peak ∼50 bp upstream of the TSS, and P-values were calculated for representative TSSs from the Wilcoxon rank-sum test (Methods). (C) The SP1 motif is orientation independent in the sense that its reverse complement was observed and conserved around outron/exon-TSSs. Motifs were examined upstream of TSSs collected from two samples, embryos and adults. The distributions from the two samples tend to be similar; thus, the graphs for embryos are displayed exclusively hereafter unless significant differences were observed between the distributions of the two samples. (D) Distribution of TATAA-box occurrences around representative TSSs. TATAA-box occurrences were enriched significantly upstream of both types of TSSs according to the rank-sum test (Methods). (E) Distribution of TATAA-box around Gaussian TSSs. (F) A TATAA-box occurs upstream of the representative outron-TSS for NM_058327 (taf-4) that is encoded in the plus strand. The outron is displayed as a yellow box. The information is available at http://wormtss.utgenome.org/ by making a query of taf-4.
Of note, the SP1 motif was “orientation independent” in the sense that occurrences of its reverse complement were enriched and conserved around both outron-TSSs and exon-TSSs (Fig. 4C). Other examples of orientation-independent motifs were six variants of the CGCG motif. These motifs were frequently observed in CG-rich regions for both outron-TSSs and exon-TSSs, were preferentially located 20–80 bp upstream of outron-TSSs, and were significantly conserved around outron-TSSs (Supplemental Fig. S3). The evolutionary conservation of a motif and its positional enrichment at a specific location upstream of TSSs are both potential indicators of the functional importance of these motifs. Although C. elegans has several SP1 homologs, it is not clear if the promoter-proximal motif represents binding of one of these factors or of another unrelated factor with similar sequence specificity.
Another well-known motif found to be enriched in putative promoters was the TATAA-box (Lifton et al. 1978; Goldberg 1979; Basehoar et al. 2004). TATAA motifs are highly conserved promoter elements shared by all eukaryotes, are typically found closely upstream of TSSs, and are correlated with low-CG-content promoters, narrow distributions of TSSs, and a larger variability in expression (Blake et al. 2003; Raser and O'Shea 2004; Carninci et al. 2006; Landry et al. 2007; Yang et al. 2007). We found (Fig. 4D) that TATAA-box motifs showed high conservation across the six nematode species and were significantly enriched 30–40 bp upstream of both representative outron-TSSs (P = 9.26 × 10−16) and exon-TSSs (P = 6.83 × 10−29). The degree of TATAA-box enrichment appears to differ between representative outron-TSSs and representative exon-TSSs in Figure 4D, leading to a concern that TATAA-box occurrences around the full collection of Gaussian outron-TSSs might have been overlooked. Indeed, more TATAA-box occurrences were seen around the full collection of Gaussian outron-TSSs as well as around the full collection of Gaussian exon-TSSs (Fig. 4E), although most of them were less conserved. Figure 4F presents the promoter region for the taf-4 gene (TBP-associated transcription factor, refseq ID NM_058327). taf-4 features a 240-bp outron, indicated by the region between the representative TSS and the SL1 site, and has a TATAA-box motif located 31 bp upstream of its TSS. The TSS data add to previous analysis indicating an upstream TATAA in C. elegans (Grishkevich et al. 2011), providing a demonstration of TATAA-box preference at a specific location upstream.
Supplemental Figure S4 shows several other previously described motifs that we identified upstream of C. elegans TSSs. T-blocks (stretches of TTT or more Ts) are known to occur frequently at 50–100 bp upstream of a start codon, with longer T-block occurrences correlated with lower nucleosome occupancy and a higher expression level (Grishkevich et al. 2011). Using our TSS information, we found that T-blocks and A-blocks, the reverse complement of T-blocks, were significantly conserved around outron-TSSs, while they were not conserved around exon-TSSs. The motif associated with Initiator (Itr) binding ([CT][CT]A.[AT][CT][CT]) (Smale and Baltimore 1989) occurred frequently and was enriched around outron-TSSs. As expected, splice acceptor enrichment was observed proximal to outron-TSS sites.
In contrast, Supplemental Figure S5 presents known motifs, most of which were identified in the human or fruit fly genomes but were less evident in the worm genome. BRE, a TFIIB recognition element, was reported to be located immediately upstream of TATAA elements in the human genome (Lagrange et al. 1998); however, this tendency was not evident in the worm genome. BRE occurrences were conserved significantly, but the number of BRE occurrences was fairly small. A DPE (downstream promoter element) was identified in the fruit fly genome (Burke and Kadonaga 1996) but was not enriched as a downstream promoter element in the worm genome (Supplemental Fig. S5). A DCE (downstream core element) (Ohler et al. 2002) was investigated by checking the appearance of one of the three short motifs CTTC, CTGT, and AGC, and was seen ubiquitously (Supplemental Fig. S5). An MTE (Motif Ten Element) that was reported to be conserved from fruit fly to humans (Lim et al. 2004) was not identified as enriched in the worm promoterome.
In addition to these previously identified sequence motifs, we identified eight novel motifs that were statistically enriched proximal to worm TSSs and also showed evidence for evolutionary conservation (Fig. 4A). Six of these motifs may have usage or roles specific to outron-TSSs because they were conserved more significantly around outron-TSSs than around exon-TSSs. The six motifs were each orientation independent and likely to occur in narrow regions, usually ∼50 bp upstream of outron-TSSs (Fig. 5). These motifs were modestly frequent around representative TSSs as well as around Gaussian TSSs. We also noticed that the novel motifs were prevalent in bidirectional promoters (discussed later), suggesting their functional relevance to transcription factor-binding sites. Two less frequent, orientation-dependent motifs were found upstream of exon-TSSs (Supplemental Fig. S6).
Figure 5.

Frequency distributions of six novel orientation-independent motifs around representative and Gaussian outron/exon-TSSs. Individual motifs are given new identifiers BiSL1-A–F. The distributions of the reverse complements of individual motifs are shown in the last two columns. P-values according to the Wilcoxon rank-sum test (see Methods) are shown for the distributions around representative outron/exon-TSSs.
Sequence analysis alone cannot conclusively demonstrate that individual motifs are transcription-factor (TF) binding sites. To this end, the worm modENCODE project (Gerstein et al. 2010) provided an informative list of 12 candidate motifs that were observed in proximity to transcription-factor (TF) binding sites HLH-1, LIN-13, MEP-1, and PHA-4. Of note, the list includes sequences that are highly similar to four of our novel motifs (BiSL1-A, BiSL1-F, reverse complements of BiSL1-D and BiSL1-C in our notation), motivating the experimental validation of other novel motifs as TF-binding sites. We analyzed the frequency distribution of distances between binding sites of each TF and newly determined representative TSSs, observing that binding sites of each TF were enriched 50–100 bp upstream of representative TSSs (Fig. 6A). In addition, we examined the frequency distribution of distances between binding sites of each TF and positions of each sequence motif. We found that the positions of each of four motifs (BiSL1-A, BiSL1-C, BiSL1-D, and BiSL1-F) significantly accorded with those of each TF (Fig. 6B; Supplemental Fig. S7, P < 10−18). They would be good starting points for future functional analysis.
Figure 6.

Motifs associated with transcription factors and tissue-specific genes. (A) For transcription factors (TF), HLH-1, LIN-13, MEP-1, and PHA-4, we obtained TF-binding scores (ChIP-seq signals) from the modENCODE database (Gerstein et al. 2010), calculating the average TF-binding score around the representative TSSs. The TF-binding score is enriched 80–50 bp upstream of representative exon-TSSs as well as outron-TSSs, respectively. (B) Frequency distribution of motif occurrences around positions associated with locally maximum HLH-1 binding scores. The frequency distributions for LIN-13, MEP-1, and PHA-4 can be found in Supplemental Figure S7. Color shade represents conservation rates (see Fig. 4B; Methods). We checked the null hypothesis that two distributions of conservation rates of a motif in proximity to TF-binding sites and in the other nonpromoter genomic regions were equal, by using the Wilcoxon rank-sum test (see Methods). A lower P-value of a motif indicates a higher enrichment of the motif at the binding sites. (C) The rows present sequence motifs, and the columns show tissue types in the LongSAGE data, except for the last column that displays the number of genes associated with each sequence motif among 2342 genes. The table presents P-values of the Wilcoxon rank-sum test for assessing the null hypothesis that two distributions of expression levels of genes with the presence or the absence of the upstream motif were equal in each tissue. To highlight significantly low P-values according to the Bonferroni correction, we colored P-values ≤ 3.75 × 10−8 red, and P-values ranging from 3.75 × 10−8 to 10−3 orange.
Motifs associated with tissue-specific gene expression
Motifs identified upstream of TSSs might have a fundamental role as cis-elements that regulate expression of genes in particular tissue types. We therefore examined the association between each motif and the expression of its associated gene (defined as the nearest downstream gene). As a measure of tissue-specific expression, we used the publicly available LongSAGE data, a comprehensive collection of SAGE data derived from a variety of tissues (e.g., oocytes, embryos, gut, muscle, hypodermal, neurons, neural cells, pharynx cells, and gonad: British Columbia C. elegans Gene Expression Consortium, http://elegans.bcgsc.bc.ca/) (Jones et al. 2001; McKay et al. 2003; Blacque et al. 2005; Khattra et al. 2007; McGhee et al. 2007; Meissner et al. 2009; Wang et al. 2009). Two thousand three hundred and forty-two (2342) genes have SAGE data as well as at least one of our identified motifs upstream of their TSS. To test whether the presence of a particular motif tends to be associated with higher expression (represented by steady-state mRNA levels) in a given tissue compared with genes lacking the motif, we performed Wilcoxon rank-sum tests for assessing the null hypothesis that distributions of expression levels from the two sets of genes were equivalent. Figure 6C and Supplemental Figure S8 show the results of this analysis. We performed multiple hypothesis testing to check each motif at a significance level of 3.75 × 10−8 as we examined the significance of the conservation of a motif. P-values less than the significance level are highlighted in red in Figure 6C. Notably, the presence of the novel BiSL-A motif upstream of a gene's TSS was associated with significantly higher expression in oocytes, embryos, gut, muscle, hypodemal, pharynx, and gonad samples. Genes with CpG motif4 in their putative promoters showed elevated gene expression in neuronal cells. High expression in gonad cells was associated with the presence of several upstream motifs, including AAAA, BiSL1-A, CpG motif 1-2, and SL1. The statistical association of several of our identified motifs with tissue-specific patterns of mRNA expression indicates that these sequences may be functionally important in proper gene expression and are strong candidate transcription factor binding sites.
Potential bidirectional promoters
Bidirectional promoters provide a compact mechanism for the joint transcriptional control of two coding regions. A comprehensive catalog of potential bidirectional promoters has not been available for C. elegans, due to limits in our knowledge of TSSs. Our comprehensive collection of TSSs together with information about gene expression levels provides a means of identifying candidate bidirectional promoters. Examining the candidate bidirectional promoters (those associated with TSSs 60–160 bp of each other) revealed that many of the identified motifs were frequently observed in these regions (Fig. 7; Supplemental Fig. S9). This class of C. elegans promoter should be of great interest for future experimental analysis.
Figure 7.
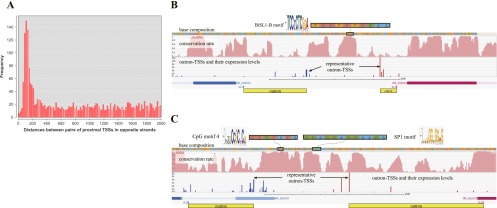
Potential bidirectional promoters. (A) Distribution of distances between paired proximal TSSs in opposite strands. The remarkable number of loci (total instances more than 600) with paired divergent TSSs at a distance of 60–160 bp suggests an abundance of potential bidirectional promoters. This range (60–160 bp) is consistent with the observation that motif sequences (candidate cis-elements) are typically enriched ∼50 bp upstream of the TSSs. In reality, many motifs, including novel ones, were observed frequently in those potential regions. (B) Example of candidate bidirectional promoter region. Two expressed representative outron-TSSs in the opposite strands were separated by 115 bp. A novel motif (TTCTCTCGGA), named “BiSL1-B” in Figure 5 (respectively, its reverse complement), was identified ∼50 bp (∼70 bp) upstream of the plus (minus) strand representative TSS. This is in accordance with the enrichment of the motif ∼30–50 bp (70–100 bp) upstream of the plus (minus) strand TSS obtained from our genome-wide analysis (see BiSL1-B in Fig. 5). Of note, the motif was highly conserved among the six allied species. The two outrons from the representative TSSs to their nearest downstream SL1 sites were different in length, demonstrating the difficulty of estimating outrons and TSSs solely from SL1 sites and the genomic sequence. (C) Another example of a bidirectional promoter. Two common motifs (SP1 and CpG motif 4) were highly conserved between two expressed outron-TSSs. Multiple highly expressed and isolated TSSs were seen in both strands, and the wide distribution of the occurrences of these motifs may account for the multiple outron-TSSs (SP1 in Fig. 4B and CpG motif 4 in Supplemental Fig. S3). Many more examples of regions harboring conserved motifs between expressed outron-TSSs were seen (Supplemental Fig. S9), which could function as potential bidirectional promoters (696 candidates in Supplemental Table S4). Expression levels between the pairs of genes have a very small similarity (Pearson's correlation coefficient <0.066).
Discussion
The study provides a comprehensive collection of TSSs for 7351 trans-spliced genes in C. elegans such that their TSSs and gene bodies were linked by substantial paired-end reads, confirming the validity of the TSSs for the corresponding trans-spliced genes.
Transcription of many genes initiates predominantly at a single site. This could be due to selection for a single, regulatable promoter or for a single defined 5′ UTR. If the selection is at the level of the UTR, we might expect that trans-splicing would eliminate that pressure, since the upstream region is replaced by a uniform 22-nt UTR. To address this question with the current data, we analyzed the average numbers of Gaussian outron/exon-TSSs in the regions of the same length, 3000 bp, upstream of SL1 sites for outron-TSSs and upstream of genes for exon-TSSs. We observed that the outron-TSS average is significantly larger than the exon-TSS average (Supplemental Fig. 10A) according to the z-test (P-value < 10−116). In addition, the analysis of the distributions of the numbers of outron- and exon-TSSs per gene also showed that the outron-TSS distribution was significantly greater than the exon-TSS distribution (Supplemental Fig. 10B) according to the Wilcoxon rank-sum test (P-value < 10−117).
These observations are consistent with models in which the presence of trans-splicing might remove selective pressure for having a single TSS. In addition to potential differences in selective pressure, several conceivable mechanistic differences might serve as a basis for the observed difference in dispersion between exon- and outron-TSS distributions (e.g., subtle differences in sequence composition or chromatin structure between outron and upstream exon regions). The combination of mechanistic and selective differences in the two 5′ regions should certainly become clearer as additional analytic and experimental tools are applied to C. elegans promoters. While sequence composition and technical influences cannot be ruled out, these observations are intriguingly consistent with the possibility that trans-splicing may afford an advantage to the organism in permitting flexibility of 5′ gene structures (Blumenthal 2005).
Arrays of positioned and phased nucleosomes have been seen around TSSs in mammals and other eukaryotes (Mavrich et al. 2008; Kaplan et al. 2009; Sasaki et al. 2009; Weiner et al. 2010; Valouev et al. 2011), with this study confirming this characteristic in C. elegans. In addition, nucleosome positioning and Pol II enrichment downstream from SL1 sites suggested a distinctive association between signals for nucleosome positioning and signals for trans-splicing. In the regions upstream of newly identified TSSs, several evolutionarily conserved motifs were uncovered, including known motifs (e.g., TATAA-box and SP1) found in other species as well as six novel TSS-specific motifs with downstream SL1 sites. These discoveries led us to reveal 694 potential bidirectional promoters of ∼100 bp in length that harbor orientation-independent motifs such as CpG, SP1, and novel motifs. We hope that this data set will serve as a valuable resource in functional genomics studies for C. elegans.
Methods
5′-End library construction
Figure 1A illustrates the process of 5′-end library construction (Hashimoto et al. 2009). C. elegans nuclei were prepared by disruption of animals and sedimentation of lysates through a 2 M sucrose cushion (lysis and gradient reagents from Sigma-Aldrich, catalog no. NUC-101). We started with ∼2 mL of worm grind (adult or embryo) for each nuclear preparation, with purified nuclei stored in 200 μL of the NUC-101 storage buffer. We used TRIzol for the isolation of total RNA, with subsequent cleaning using the RNeasy Mini (QIAGEN). We then replaced the cap structure of mRNA with an Illumina RNA linker according to the oligo-cap method (Maruyama and Sugano 1994) as follows: The purified total RNA (starting with 5 μg) was treated with bacterial alkaline phosphatase (TaKaRa). The RNA was extracted twice with phenol:chloroform (1:1), ethanol-precipitated, and treated with tobacco acid pyrophosphatase (TAP). Then a RNA linker was ligated using RNA ligase (TaKaRa): a 5′-oligo (5′-ACAGGUUCAGAGUUCUACAGUCCAGCAG-3′) linker. After linker ligation, the RNA was depleted of ribosomal RNA using the RiboMinus Eukaryote Kit (for RNA-seq, Invitrogen, catalog no. 10837-08). After removing ribosomal RNA, a cDNA library was produced from 10 pmol of random hexamer primer (5′-CAAGCAGAAGACGGCATACGACAGCAGNNNNNNC-3′) using SuperScript III (Invitrogen) by incubating for 10 min at 26°C, 90 min at 50°C, and 15 min at 70°C. After first-strand synthesis, RNA was no longer needed and was degraded in 15 mM NaOH for 1 h at 65°C. cDNA was amplified in a volume of 100 μL using PCR with 16 pmol of 5′ (5′-AATGATACGGCGACCACCGACAGGTTCAGAGTTCTACAGTCC-3′) and 3′ (5′-CAAGCAGAAGACGGCATACGA-3′) PCR primers. The cDNA was produced using 15–20 cycles of 1 min at 94°C, 1 min at 58°C, and 10 min at 72°C. The PCR fragments were size-fractionated by 8% polyacrylamide gel electrophoresis, and the fraction of 150–300 bp was recovered. The quality and quantity of the obtained single-stranded first-strand cDNAs were assessed using BioAnalyzer (Agilent).
Read alignment
To map the short reads of Illumina GAII to the reference genome of C. elegans, we used BWA with the default setting (4% of mismatches of read lengths were allowed). Many reads were eliminated unless they contributed to the TSS identification. For example, several trans-spliced reads failed to be mapped because of splice-reader sequences (SL1, SL2, etc.) attached to the head of the sequences after transcription. These unmapped reads were useless in identifying TSSs and were therefore discarded. Despite application of the RiboMinus Eukaryote Kit, we found that one-third of the reads were fragments of rRNA. These numerous rRNA reads simply reconfirmed rRNA regions that were well annotated in WormBase.
TSS peak calling
After mapping of the short-read sequences to the reference genome, we sorted reads in the order of the genome coordinate and then computed the number of reads mapped to the same position to collect candidate TSS peaks. The distributions of the peaks showed various shapes; single-peak, multiple peaks, broadly distributed peaks, etc. To classify these peak types, we attempted to fit a Gaussian mixture model to our data. One issue was to determine the number of Gaussian distributions. We used X-means clustering (Pelleg and Moore 2000) of peaks that used Gaussian model and penalty score to avoid overfitting (Bayesian-information criteria, BIC), so that the result of X-means clustering is expected to have a nice fit to a Gaussian-mixture model representing the observed peaks. The observed number of clusters before genes ranged from 1 to 70. These clusters contain noisy peaks that may inadequately distort the Gaussian distributions. Thus, for each cluster, we trimmed marginal peaks (less than 5 tag counts) and applied the Gaussian distribution so as to compute N (gene expression level) and σ (distribution shape) values. TSSs within 10 bp from the SL1 sites were removed to avoid confusion with the trans-spliced tags. We selected clusters with the highest expression level within 3000 bp before the gene start sites as representative TSSs. Supplemental Table S1 gives a gene-by-gene list of representative TSSs as well as Gaussian TSSs. In the table, we used the gene annotations in WormBase WS220. We merged gene models that overlapped in individual regions of the genome into unique groups (choosing the longest model in each case) so as to associate each TSS with a single neighboring entity.
Finding motifs and their occurrences around TSSs
Finding motif sequences is a computationally expensive task because of numerous combinations of ACGT letters of various lengths. MEME is a widely used program for detecting motif sequences. To speed up the analysis, we used an MPI version of MEME, and it took us ∼1 d to run the program using a cluster of machines equipped with 64 CPUs using the option “-p 64 -maxsize 10000000 -dna -nmotifs 30 -mod zoops -minw 3 -maxw 10.” Each letter in a motif is associated with bits that express a likelihood of the presence of the letter at the position in the motif. In searching all promoter regions (350 bp to −50 bp around TSSs) for a motif, we used letters associated with ≥1 bits in a motif for testing exact matches, while we replaced other letters by ?, a wild card matching any one letter. In this way, we transformed a motif into a regular expression. For example, we converted the SP1 motif in Figure 4B into the regular expression “C?CC?CC” and the TATAA-box in Figure 4D into “TATAAAA.” We then searched all promoter regions for the matches of the regular expression corresponding to a motif. We note that the use of regular expressions to search for many known motifs was preferable over weight-based methods, because the relevant publications lacked bit-significance scores. To be consistent with MEME, for the motifs found by MEME, we also used regular expressions of characters whose bit-significance scores were >1.
Measuring the significance of the conservation of a motif around TSSs
We calculated the conservation rates of motifs at each position from a comparison of the genomes of six related nematode species, C. elegans (The C. elegans Sequencing Consortium 1998), Caenorhabditis brenneri (Stein et al. 2003), Caenorhabditis briggsae, Caenorhabditis remanei, Caenorhabditis japonica, and Pristionchus pacificus (Thomas 2008), with alignments derived from the UCSC Genome Browser resource (Siepel et al. 2005; Fujita et al. 2011).
Subsequently, we tested the significance of the conservation of a motif in promoter regions that range from 300 bp to −50 bp of representative TSSs. If Gaussian TSSs are considered, the same motif occurrence can be counted more than once. To avoid this double counting, we focused on representative TSSs so as to associate any motif occurrence with its downstream representative TSS, uniquely. We checked the null hypothesis that two distributions of conservation rates of a motif in promoter regions and in the other nonpromoter genomic regions were equal, by using the Wilcoxon rank-sum test. The number of all occurrences of a focal motif in nonpromoter regions was huge, and using them all was likely to lead to incorrect calculation of P-values. We therefore selected 10,000 instances at random from all the occurrences, and performed this trial 100 times to calculate P-values. Since some P-values happened to be extremely small or large, the median of the P-values was treated as the representative P-value. Because we considered motifs of ≤10-mers in length, according to the Bonferroni correction, we tested each motif at a significance level of 5% divided by (4/3) × 106 (=3.75 × 10−8), where (4/3) × 106 approximates the number of all possible ≤10-mers.
Data access
All sequence data are deposited at the NCBI Sequence Read Archive (SRA) (http://www.ncbi.nlm.nih.gov/sra) under accession number SRA060670. Our genome browser is available at http://wormtss.utgenome.org/. Both the primary data and analytical tables in BAM and WIG format are also available at http://wormtss.utgenome.org/browser/download.jsp, so that public databases such as WormBase and UCSC can incorporate our data easily into their sites.
Acknowledgments
This work was supported in part by PHS (Grants T32 CA09151), NIGMS (Grants GM37706 [to A.F.], GM42432 [to T.B.]), and MEXT (Grant-in-Aid for Scientific Research A 23241058 [to S.M.], Global COE program “Deciphering Biosphere from Genome Big Bang” [to S.M.]), Innovative Cell Biology by Innovative Technology [to S.M.]). We are grateful to Will Kruesi, Leighton Core, Colin Waters, John Lis, and Barbara Meyer, whose sharing of outron lengths for a representative set of genes from their preliminary GroSeq analysis was useful in our decision to consider multiple and long 5′-outron structures. We thank Karen Artiles, Julia Pak, Cecilia Mello, Poornima Parameswaran, Ayelet Lamm, Huibin Zhang, and Diane Wu for their help and suggestions; Yutaka Suzuki and Sumio Sugano for Illumina sequencing; and Hiroshi Minoshima and Kouji Matsushima for their help.
Footnotes
[Supplemental material is available for this article.]
Article published online before print. Article, supplemental material, and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.151571.112.
Freely available online through the Genome Research Open Access option.
References
- Allen MA, Hillier LW, Waterston RH, Blumenthal T 2011. A global analysis of C. elegans trans-splicing. Genome Res 21: 255–264 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bailey TL, Elkan C 1994. Fitting a mixture model by expectation maximization to discover motifs in biopolymers. Proc Int Conf Intell Syst Mol Biol 2: 28–36 [PubMed] [Google Scholar]
- Basehoar AD, Zanton SJ, Pugh BF 2004. Identification and distinct regulation of yeast TATA box-containing genes. Cell 116: 699–709 [DOI] [PubMed] [Google Scholar]
- Baugh LR, Demodena J, Sternberg PW 2009. RNA Pol II accumulates at promoters of growth genes during developmental arrest. Science 324: 92–94 [DOI] [PubMed] [Google Scholar]
- Bernstein BE, Liu CL, Humphrey EL, Perlstein EO, Schreiber SL 2004. Global nucleosome occupancy in yeast. Genome Biol 5: R62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blacque OE, Perens EA, Boroevich KA, Inglis PN, Li C, Warner A, Khattra J, Holt RA, Ou G, Mah AK, et al. 2005. Functional genomics of the cilium, a sensory organelle. Curr Biol 15: 935–941 [DOI] [PubMed] [Google Scholar]
- Blake WJ, Kaern M, Cantor CR, Collins JJ 2003. Noise in eukaryotic gene expression. Nature 422: 633–637 [DOI] [PubMed] [Google Scholar]
- Blumenthal T 2005. Trans-splicing and operons. In WormBook (ed. The C. elegans Research Community), pp. 1–9. http://www.wormbook.org [DOI] [PubMed]
- Boyle AP, Guinney J, Crawford GE, Furey TS 2008. F-Seq: A feature density estimator for high-throughput sequence tags. Bioinformatics 24: 2537–2538 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burke TW, Kadonaga JT 1996. Drosophila TFIID binds to a conserved downstream basal promoter element that is present in many TATA-box-deficient promoters. Genes Dev 10: 711–724 [DOI] [PubMed] [Google Scholar]
- The C. elegans Sequencing Consortium 1998. Genome sequence of the nematode C. elegans: A platform for investigating biology. Science 282: 2012–2018 [DOI] [PubMed] [Google Scholar]
- Carninci P, Sandelin A, Lenhard B, Katayama S, Shimokawa K, Ponjavic J, Semple CA, Taylor MS, Engstrom PG, Frith MC, et al. 2006. Genome-wide analysis of mammalian promoter architecture and evolution. Nat Genet 38: 626–635 [DOI] [PubMed] [Google Scholar]
- Chen Z, Duan X 2011. Ribosomal RNA depletion for massively parallel bacterial RNA-sequencing applications. Methods Mol Biol 733: 93–103 [DOI] [PubMed] [Google Scholar]
- Conrad R, Thomas J, Spieth J, Blumenthal T 1991. Insertion of part of an intron into the 5′ untranslated region of a Caenorhabditis elegans gene converts it into a trans-spliced gene. Mol Cell Biol 11: 1921–1926 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Culetto E, Combes D, Fedon Y, Roig A, Toutant JP, Arpagaus M 1999. Structure and promoter activity of the 5′ flanking region of ace-1, the gene encoding acetylcholinesterase of class A in Caenorhabditis elegans. J Mol Biol 290: 951–966 [DOI] [PubMed] [Google Scholar]
- Dibb NJ, Maruyama IN, Krause M, Karn J 1989. Sequence analysis of the complete Caenorhabditis elegans myosin heavy chain gene family. J Mol Biol 205: 603–613 [DOI] [PubMed] [Google Scholar]
- Field Y, Kaplan N, Fondufe-Mittendorf Y, Moore IK, Sharon E, Lubling Y, Widom J, Segal E 2008. Distinct modes of regulation by chromatin encoded through nucleosome positioning signals. PLoS Comput Biol 4: e1000216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frith MC, Ponjavic J, Fredman D, Kai C, Kawai J, Carninci P, Hayashizaki Y, Sandelin A 2006. Evolutionary turnover of mammalian transcription start sites. Genome Res 16: 713–722 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujita PA, Rhead B, Zweig AS, Hinrichs AS, Karolchik D, Cline MS, Goldman M, Barber GP, Clawson H, Coelho A, et al. 2011. The UCSC Genome Browser database: Update 2011. Nucleic Acids Res 39: D876–D882 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerstein MB, Lu ZJ, Van Nostrand EL, Cheng C, Arshinoff BI, Liu T, Yip KY, Robilotto R, Rechtsteiner A, Ikegami K, et al. 2010. Integrative analysis of the Caenorhabditis elegans genome by the modENCODE project. Science 330: 1775–1787 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldberg ML. 1979. “Sequence analysis of Drosophila histone genes.” PhD thesis, Stanford University, CA. [Google Scholar]
- Grishkevich V, Hashimshony T, Yanai I 2011. Core promoter T-blocks correlate with gene expression levels in C. elegans. Genome Res 21: 707–717 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu SG, Fire A 2010. Partitioning the C. elegans genome by nucleosome modification, occupancy, and positioning. Chromosoma 119: 73–87 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris TW, Antoshechkin I, Bieri T, Blasiar D, Chan J, Chen WJ, De La Cruz N, Davis P, Duesbury M, Fang R, et al. 2010. WormBase: A comprehensive resource for nematode research. Nucleic Acids Res 38: D463–D467 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hashimoto S, Qu W, Ahsan B, Ogoshi K, Sasaki A, Nakatani Y, Lee Y, Ogawa M, Ametani A, Suzuki Y, et al. 2009. High-resolution analysis of the 5′-end transcriptome using a next generation DNA sequencer. PLoS ONE 4: e4108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hillier LW, Reinke V, Green P, Hirst M, Marra MA, Waterston RH 2009. Massively parallel sequencing of the polyadenylated transcriptome of C. elegans. Genome Res 19: 657–666 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huber R, Schlessinger D, Pilia G 1998. Multiple Sp1 sites efficiently drive transcription of the TATA-less promoter of the human glypican 3 (GPC3) gene. Gene 214: 35–44 [DOI] [PubMed] [Google Scholar]
- Hughes A, Rando OJ 2009. Chromatin ‘programming' by sequence—is there more to the nucleosome code than %GC? J Biol 8: 96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hunt-Newbury R, Viveiros R, Johnsen R, Mah A, Anastas D, Fang L, Halfnight E, Lee D, Lin J, Lorch A, et al. 2007. High-throughput in vivo analysis of gene expression in Caenorhabditis elegans. PLoS Biol 5: e237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones SJ, Riddle DL, Pouzyrev AT, Velculescu VE, Hillier L, Eddy SR, Stricklin SL, Baillie DL, Waterston R, Marra MA 2001. Changes in gene expression associated with developmental arrest and longevity in Caenorhabditis elegans. Genome Res 11: 1346–1352 [DOI] [PubMed] [Google Scholar]
- Kaplan N, Moore IK, Fondufe-Mittendorf Y, Gossett AJ, Tillo D, Field Y, LeProust EM, Hughes TR, Lieb JD, Widom J, et al. 2009. The DNA-encoded nucleosome organization of a eukaryotic genome. Nature 458: 362–366 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khattra J, Delaney AD, Zhao Y, Siddiqui A, Asano J, McDonald H, Pandoh P, Dhalla N, Prabhu AL, Ma K, et al. 2007. Large-scale production of SAGE libraries from microdissected tissues, flow-sorted cells, and cell lines. Genome Res 17: 108–116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolasinska-Zwierz P, Down T, Latorre I, Liu T, Liu XS, Ahringer J 2009. Differential chromatin marking of introns and expressed exons by H3K36me3. Nat Genet 41: 376–381 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krause M, Hirsh D 1987. A trans-spliced leader sequence on actin mRNA in C. elegans. Cell 49: 753–761 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lagrange T, Kapanidis AN, Tang H, Reinberg D, Ebright RH 1998. New core promoter element in RNA polymerase II-dependent transcription: Sequence-specific DNA binding by transcription factor IIB. Genes Dev 12: 34–44 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Landry CR, Lemos B, Rifkin SA, Dickinson WJ, Hartl DL 2007. Genetic properties influencing the evolvability of gene expression. Science 317: 118–121 [DOI] [PubMed] [Google Scholar]
- Lasda EL, Blumenthal T 2011. Trans-splicing. Wiley Interdiscip Rev RNA 2: 417–434 [DOI] [PubMed] [Google Scholar]
- Li H, Durbin R 2009. Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics 25: 1754–1760 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lifton RP, Goldberg ML, Karp RW, Hogness DS 1978. The organization of the histone genes in Drosophila melanogaster: Functional and evolutionary implications. Cold Spring Harb Symp Quant Biol 42: 1047–1051 [DOI] [PubMed] [Google Scholar]
- Lim CY, Santoso B, Boulay T, Dong E, Ohler U, Kadonaga JT 2004. The MTE, a new core promoter element for transcription by RNA polymerase II. Genes Dev 18: 1606–1617 [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacMorris M, Kumar M, Lasda E, Larsen A, Kraemer B, Blumenthal T 2007. A novel family of C. elegans snRNPs contains proteins associated with trans-splicing. RNA 13: 511–520 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maruyama K, Sugano S 1994. Oligo-capping: A simple method to replace the cap structure of eukaryotic mRNAs with oligoribonucleotides. Gene 138: 171–174 [DOI] [PubMed] [Google Scholar]
- Mavrich TN, Ioshikhes IP, Venters BJ, Jiang C, Tomsho LP, Qi J, Schuster SC, Albert I, Pugh BF 2008. A barrier nucleosome model for statistical positioning of nucleosomes throughout the yeast genome. Genome Res 18: 1073–1083 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGhee JD, Sleumer MC, Bilenky M, Wong K, McKay SJ, Goszczynski B, Tian H, Krich ND, Khattra J, Holt RA, et al. 2007. The ELT-2 GATA-factor and the global regulation of transcription in the C. elegans intestine. Dev Biol 302: 627–645 [DOI] [PubMed] [Google Scholar]
- McKay SJ, Johnsen R, Khattra J, Asano J, Baillie DL, Chan S, Dube N, Fang L, Goszczynski B, Ha E, et al. 2003. Gene expression profiling of cells, tissues, and developmental stages of the nematode C. elegans. Cold Spring Harb Symp Quant Biol 68: 159–169 [DOI] [PubMed] [Google Scholar]
- Meissner B, Warner A, Wong K, Dube N, Lorch A, McKay SJ, Khattra J, Rogalski T, Somasiri A, Chaudhry I, et al. 2009. An integrated strategy to study muscle development and myofilament structure in Caenorhabditis elegans. PLoS Genet 5: e1000537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morton JJ, Blumenthal T 2011. Identification of transcription start sites of trans-spliced genes: Uncovering unusual operon arrangements. RNA 17: 327–337 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ni T, Corcoran DL, Rach EA, Song S, Spana EP, Gao Y, Ohler U, Zhu J 2010. A paired-end sequencing strategy to map the complex landscape of transcription initiation. Nat Methods 7: 521–527 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohler U, Liao GC, Niemann H, Rubin GM 2002. Computational analysis of core promoters in the Drosophila genome. Genome Biol 3: RESEARCH0087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okkema PG, Harrison SW, Plunger V, Aryana A, Fire A 1993. Sequence requirements for myosin gene expression and regulation in Caenorhabditis elegans. Genetics 135: 385–404 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park YS, Kramer JM 1990. Tandemly duplicated Caenorhabditis elegans collagen genes differ in their modes of splicing. J Mol Biol 211: 395–406 [DOI] [PubMed] [Google Scholar]
- Park YS, Kramer JM 1994. The C. elegans sqt-1 and rol-6 collagen genes are coordinately expressed during development, but not at all stages that display mutant phenotypes. Dev Biol 163: 112–124 [DOI] [PubMed] [Google Scholar]
- Pelleg D, Moore A 2000. X-means: Extended k-means with efficient estimation of the number of clusters. In 17th International Conference on Machine Learning, Stanford, CA, pp. 727–734. Morgan Kaufmann, Stanford, CA
- Rach EA, Winter DR, Benjamin AM, Corcoran DL, Ni T, Zhu J, Ohler U 2011. Transcription initiation patterns indicate divergent strategies for gene regulation at the chromatin level. PLoS Genet 7: e1001274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raser JM, O'Shea EK 2004. Control of stochasticity in eukaryotic gene expression. Science 304: 1811–1814 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruan Y, Le Ber P, Ng HH, Liu ET 2004. Interrogating the transcriptome. Trends Biotechnol 22: 23–30 [DOI] [PubMed] [Google Scholar]
- Sasaki S, Mello CC, Shimada A, Nakatani Y, Hashimoto S, Ogawa M, Matsushima K, Gu SG, Kasahara M, Ahsan B, et al. 2009. Chromatin-associated periodicity in genetic variation downstream of transcriptional start sites. Science 323: 401–404 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saxonov S, Berg P, Brutlag DL 2006. A genome-wide analysis of CpG dinucleotides in the human genome distinguishes two distinct classes of promoters. Proc Natl Acad Sci 103: 1412–1417 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schug J, Schuller WP, Kappen C, Salbaum JM, Bucan M, Stoeckert CJ Jr 2005. Promoter features related to tissue specificity as measured by Shannon entropy. Genome Biol 6: R33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Segal E, Widom J 2009. Poly(dA:dT) tracts: Major determinants of nucleosome organization. Curr Opin Struct Biol 19: 65–71 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siepel A, Bejerano G, Pedersen JS, Hinrichs AS, Hou M, Rosenbloom K, Clawson H, Spieth J, Hillier LW, Richards S, et al. 2005. Evolutionarily conserved elements in vertebrate, insect, worm, and yeast genomes. Genome Res 15: 1034–1050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sleumer MC, Wei G, Wang Y, Chang H, Xu T, Chen R, Zhang MQ 2012. Regulatory elements of Caenorhabditis elegans ribosomal protein genes. BMC Genomics 13: 433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smale ST, Baltimore D 1989. The “initiator” as a transcription control element. Cell 57: 103–113 [DOI] [PubMed] [Google Scholar]
- Spieth J, Brooke G, Kuersten S, Lea K, Blumenthal T 1993. Operons in C. elegans: Polycistronic mRNA precursors are processed by trans-splicing of SL2 to downstream coding regions. Cell 73: 521–532 [DOI] [PubMed] [Google Scholar]
- Stein LD, Bao Z, Blasiar D, Blumenthal T, Brent MR, Chen N, Chinwalla A, Clarke L, Clee C, Coghlan A, et al. 2003. The genome sequence of Caenorhabditis briggsae: A platform for comparative genomics. PLoS Biol 1: e45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strauss EJ, Guthrie C 1991. A cold-sensitive mRNA splicing mutant is a member of the RNA helicase gene family. Genes Dev 5: 629–641 [DOI] [PubMed] [Google Scholar]
- Sutton RE, Boothroyd JC 1986. Evidence for trans splicing in trypanosomes. Cell 47: 527–535 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki Y, Taira H, Tsunoda T, Mizushima-Sugano J, Sese J, Hata H, Ota T, Isogai T, Tanaka T, Morishita S, et al. 2001. Diverse transcriptional initiation revealed by fine, large-scale mapping of mRNA start sites. EMBO Rep 2: 388–393 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas JH 2008. Genome evolution in Caenorhabditis. Brief Funct Genomics Proteomics 7: 211–216 [DOI] [PubMed] [Google Scholar]
- Tillo D, Kaplan N, Moore IK, Fondufe-Mittendorf Y, Gossett AJ, Field Y, Lieb JD, Widom J, Segal E, Hughes TR 2010. High nucleosome occupancy is encoded at human regulatory sequences. PLoS ONE 5: e9129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valouev A, Ichikawa J, Tonthat T, Stuart J, Ranade S, Peckham H, Zeng K, Malek JA, Costa G, McKernan K, et al. 2008. A high-resolution, nucleosome position map of C. elegans reveals a lack of universal sequence-dictated positioning. Genome Res 18: 1051–1063 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valouev A, Johnson SM, Boyd SD, Smith CL, Fire AZ, Sidow A 2011. Determinants of nucleosome organization in primary human cells. Nature 474: 516–520 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Zhao Y, Wong K, Ehlers P, Kohara Y, Jones SJ, Marra MA, Holt RA, Moerman DG, Hansen D 2009. Identification of genes expressed in the hermaphrodite germ line of C. elegans using SAGE. BMC Genomics 10: 213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiner A, Hughes A, Yassour M, Rando OJ, Friedman N 2010. High-resolution nucleosome mapping reveals transcription-dependent promoter packaging. Genome Res 20: 90–100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whittle CM, McClinic KN, Ercan S, Zhang X, Green RD, Kelly WG, Lieb JD 2008. The genomic distribution and function of histone variant HTZ-1 during C. elegans embryogenesis. PLoS Genet 4: e1000187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xi H, Yu Y, Fu Y, Foley J, Halees A, Weng Z 2007. Analysis of overrepresented motifs in human core promoters reveals dual regulatory roles of YY1. Genome Res 17: 798–806 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang C, Bolotin E, Jiang T, Sladek FM, Martinez E 2007. Prevalence of the initiator over the TATA box in human and yeast genes and identification of DNA motifs enriched in human TATA-less core promoters. Gene 389: 52–65 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu J, He F, Hu S, Yu J 2008. On the nature of human housekeeping genes. Trends Genet 24: 481–484 [DOI] [PubMed] [Google Scholar]