

Abstract
Understanding the molecular mechanisms of insulin resistance remains a major medical challenge of the twenty-first century. Over the last half-century, many hypotheses have been proposed to explain insulin resistance, and, most recently, inflammation associated with alterations in adipocytokines has become the prevailing hypothesis. Here we discuss diacylglycerol-mediated insulin resistance as an alternative and unifying hypothesis to explain the most common forms of insulin resistance associated with obesity and type 2 diabetes mellitus, as well as lipodystrophy and aging.
Type 2 diabetes mellitus (T2DM) has reached epidemic proportions, currently affecting an estimated 285 million individuals worldwide. At present in the United States, T2DM affects 7% of the population, with associated health care costs exceeding $350 billion per year1. Although it is clear that beta cell dysfunction is the main factor responsible for the progression to hyperglycemia, insulin resistance predates beta cell dysfunction, and reversibility of insulin resistance delays and prevents the development of diabetes. So, understanding the pathogenesis of insulin resistance remains a major medical challenge.
At a physiological level, the liver and skeletal muscle are the two key insulin-responsive organs that are responsible for most of the glucose disposal after a meal. In vivo studies have shown that a defect in insulin-stimulated muscle glycogen synthesis2, due to a defect in insulin-stimulated muscle glucose transport3,4, is crucial for insulin resistance in muscle. In the liver, defects in insulin-stimulated hepatic glycogen synthesis5,6 and increased rates of hepatic gluconeogenesis5,7 are the main factors that contribute to insulin resistance and fasting hyperglycemia.
Ever since Berson and Yalow found high, rather than low, plasma insulin concentrations in ‘maturity-onset diabetes’8, many hypotheses have been proposed to explain insulin resistance. Most recently, inflammation in adipocytes associated with increased adipocytokines9–11, endoplasmic reticulum stress12, increased reactive oxygen species production13 and genetic alterations in insulin signaling14,15 have become the prevailing explanations for insulin resistance associated with obesity and T2DM. Here we discuss recent evidence in support of the hypothesis that increases in intracellular diacylglycerol content, due to an imbalance between fatty acid delivery and intracellular fatty acid oxidation and storage, lead to activation of new protein kinase C (PKC) isoforms that in turn inhibit insulin action in liver and skeletal muscle.
Lipid-induced muscle insulin resistance
Obesity has long been associated with insulin resistance, suggesting a potential role of lipids in mediating this process. More than 50 years ago, Randle et al.16–18 showed that fatty acids can induce insulin resistance in diaphragm and cardiac muscle in vitro. They hypothesized that increased fatty acid oxidation caused an increase in the intramitochondrial acetyl CoA/CoA and NADH/NAD+ ratios, with subsequent inhibition of pyruvate dehydrogenase activity, resulting in increased citrate concentrations that would, in turn, inhibit phosphofructokinase activity. This would lead to increased glucose-6-phosphate concentrations that would subsequently inhibit hexokinase II activity, resulting in increased intracellular glucose concentrations and decreased glucose transport. In contrast to this model, we have observed decreased intramyocellular glucose-6-phosphate and glucose concentrations in humans when plasma fatty acid concentrations are increased19,20. These results showed that fatty acids induce insulin resistance in skeletal muscle, not by inhibition of pyruvate dehydrogenase activity16–18, but by directly inhibiting insulin-stimulated glucose transport activity. Moreover, fatty acid–induced insulin resistance in muscle can be attributed to a reduction in insulin-stimulated phosphatidylinositol-3 kinase (PI3K)20, and increasing plasma fatty acid concentrations can result in increased intramyocellular acyl CoAs and diacylglycerol content. In turn, these conditions would result in activation of PKC-θ21,22 and, ultimately, decreased insulin receptor substrate 1 (IRS-1)-associated PI3K activity (Fig. 1a). Lipid infusion studies in humans23 confirmed these findings, although PKC-βII and PKC-δ, rather than PKC-θ, were found to be the relevant PKC isoforms.
Figure 1.
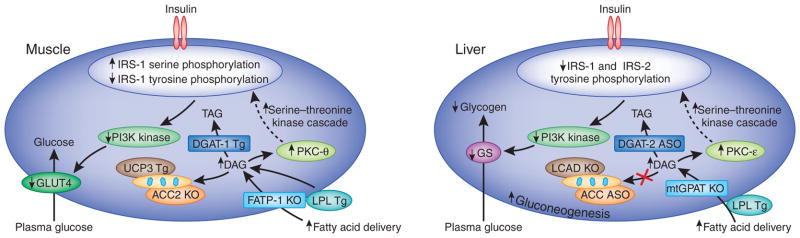
Molecular mechanisms of cellular insulin resistance in muscle and liver. Increased intracellular diacyglycerols lead to activation of PKC-θ and PKC-ε in skeletal muscle and liver, respectively, which, in turn, decreases insulin-stimulated IRS-1/IRS-2 tyrosine phosphorylation, PI3K activation and downstream insulin signaling. (a) In the muscle, this results in decreased muscle glycogen synthesis, owing to reduced insulin-stimulated GLUT4 translocation to the plasma membrane. (b) In the liver, this results in decreased hepatic glycogen synthesis, owing to decreased activation of glycogen synthase, and increased hepatic gluconeogenesis. The use of transgenic and knockout mice, as well as antisense oligonucleotides to knock down specific proteins, has allowed the validation of the diacylglycerol-mediated insulin hypothesis of insulin resistance through the manipulation of key proteins in the pathway. KO, knockout; GLUT4, glucose transporter type 4; TAG, triacylglycerol; LPL, lipoproteinlipase; Tg, transgenic; FATP-1, fatty acid transport protein-1; DGAT, diacylglycerol O-acyltransferase; UCP3, uncoupling protein-3; GS, glycogen synthase; LCAD, long-chain acyl-CoA dehydrogenase; mtGPAT, mitochondrial glycerol-3-phosphate acyltransferase; ASO, antisense oligonucleotide.
Genetic studies with transgenic and knockout mice24–29 have confirmed the above findings and have provided further insight into the mechanisms that link intracellular diacylglycerol concentration to insulin resistance. Of particular interest is the observation that acetyl CoA carboxylase-2 (Acc2) knockout in mice resulted in decreases in intramyocellular diacylglycerol content, PKC-θ activation and muscle insulin resistance28. Although a recent study using another Acc2 knockout mouse strain did not observe protection from fat-induced insulin resistance30, these workers did not measure intramyocellular diacylglcyerol content or PKC-θ activation. Although the reason for this discrepancy is unknown, the more recent work did not report intramyocellular diacylglycerol content or PKC-θ activation. Taken together, these findings suggest that solely inhibiting ACC2 without concomitant increases in mitochondrial uncoupling or some other process to decrease metabolic efficiency would not result in net reduction of intracellular diacylglycerol content and protection from fat-induced insulin resistance.
Lipid-induced liver insulin resistance
A similar mechanism for diacylglycerol-induced insulin resistance operates in the liver (Fig. 1b). Short-term high-fat feeding of rats caused high hepatic diacylglycerol content, which is associated with PKC-ε activation and decreased IRS-2 tyrosine phosphorylation31. The key role of PKC-ε for fat-induced hepatic insulin resistance was demonstrated with antisense oligionucleotide–mediated knockdown of PKC-ε32. PKC-ε–specific antisense oligionucleotide–treated rats were protected from fat-induced hepatic insulin resistance despite similar levels of hepatic diacylglycerol and triacylglycerol content as the control antisense oligionucleotide–treated rats. Likewise, fructose feeding increased hepatic diacylglycerol content, PKC-ε activation and hepatic insulin resistance, which was prevented by knockdown of PGC-1β expression in liver33. This evidence supports the existence of distinct roles of PKC-θ and PKC-ε in fat-induced insulin resistance in skeletal muscle and liver, respectively.
Inhibiting hepatic diacylglycerol synthesis by knocking out the gene encoding mitochondrial glycerol-3-phosphate acyltransferase protects mice from fat-induced hepatic insulin resistance34. Unexpectedly, knockdown of the gene encoding diacylglycerol O-acyltransferase-2 also lowers hepatocellular diacylglycerol content, owing to downregulation of sterol regulatory element–binding transcription factor-1 expression, decreased PKC-ε activation and protection from fat-induced hepatic insulin resistance35. Last, promoting increased fatty acid oxidation and mitochondrial uncoupling by knockdown of Acc1 and Acc2 or the use of 2,4-dinitrophenol resulted in reduced diacylglycerol content, decreased PKC-ε activation and protection from fat-induced hepatic insulin resistance31,36. Conversely, decreased β-oxidation of long-chain acyl CoAs can result in insulin resistance by increasing the intracellular diacylglycerol content37.
Causes of increased diacylglycerol content
There are multiple pathological causes for high net intracellular diacylglycerol content in the liver and in skeletal muscle (Fig. 2). The most common is overnutrition, which increases the rate of fatty acid delivery to these tissues, exceeding the rates of intracellular fat oxidation and triglyceride synthesis and storage. This causes a net increase in intramyocellular and intrahepatocellular lipid content, leading to liver and muscle insulin resistance38. Lipodystrophy, inherited39 or acquired40, can also result in redistribution of fat into the liver and the skeletal muscle owing to a lack of peripheral adipocytes. Moreover, low rates of mitochondrial fatty acid oxidation in healthy, lean, insulin-resistant offspring of parents with T2DM41,42 and in healthy, lean, elderly individuals43 are associated with increased intramyocellular lipid content and muscle insulin resistance.
Figure 2.
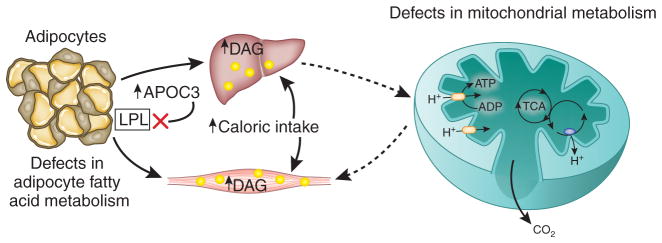
Mechanisms for intracellular diacylglycerol (DAG) accumulation in muscle and liver. DAGs accumulate in skeletal muscle and liver when the rate of fatty acid delivery to these tissues exceeds the rates of intracellular fat oxidation and/or conversion to neutral lipid. The four main causes of net DAG accumulation are excess caloric intake, defects in adipocyte metabolism (which would include lipid storage and lipolysis), defects in mitochondrial fatty acid oxidation and gene variation in apolipoprotein C3, leading to inhibition of lipoprotein lipase activity. TCA, tricarboxylic acid.
Whether this reduced mitochondrial activity is a primary or secondary factor in the development of insulin resistance remains to be determined but, given the key role of intracellular diacylglycerols, reductions in mitochondrial fatty acid oxidation would certainly be expected to exacerbate diacylglycerol-induced muscle insulin resistance. These results suggest that increased intramyocellular diacylglycerol content may also be responsible for the insulin resistance caused by glucose toxicity associated with poorly controlled type 1 and 2 diabetes. Notably, high intramuscular diacylglycerol content resulting from reduced activity of diacylglycerol kinase-δ has also been associated with insulin resistance in patients with poorly controlled T2DM44. Last, a common gene variant in apolipoprotein C-III (APOC3) results in increased plasma concentrations of APOC3, leading to inhibition of lipoprotein lipase activity and increased postprandial hypertriglyceridemia; thus, this variant predisposes carriers to nonalcoholic fatty liver disease and insulin resistance45.
Inflammation and insulin resistance
Obesity leads to the recruitment of macrophages to fat cells. The macrcophages subsequently produce cytokines, causing a low-grade inflammatory response46,47. The mechanisms that lead to this proinflammatory state are still being investigated, but overnutrition undoubtedly has a role in elevating levels of circulating cytokines. Over the past decade, inflammation has become the prevailing view to explain insulin resistance associated with T2DM9–11,48. However, several lines of evidence suggest that inflammation may not be the primary cause. First, as discussed above, young, lean, insulin-resistant offspring of parents with T2DM49 or healthy, lean, insulin-resistant elderly individuals43 can manifest severe muscle insulin resistance, independent of any alterations in circulating adipocytokine concentrations.
Second, individuals with severe lipodystrophy have severe liver and muscle insulin resistance despite lacking visceral and subcutaneous fat and having low concentrations of circulating adipocytokines39. Leptin-replacement therapy reverses insulin resistance in these patients, and this improvement is associated with reductions in liver and intramyocellular lipid content39. Similar observations have been made in a mouse model of severe lipodystrophy in which liver and muscle insulin resistance were associated with increased hepatocellular and intramyocellular fatty acyl CoA content and reductions in insulin activation of IRS-1–associated and IRS-2–associated PI3K activity50. These changes were all reversed by transplanting fat from wild-type littermates into the lipodystrophic mice.
Last, moderate (approximately 8-kg) reductions in body weight normalized fasting hyperglycemia in obese patients with T2DM51. These changes were associated with reductions in hepatic triglyceride content but were independent of changes in circulating plasma concentrations of tumor necrosis factor-α (TNF-α), interleukin-6 (IL-6), retinol-binding protein-4 and leptin51. TNF-α and IL-6 are often cited as crucial cytokines that modulate insulin resistance and signaling in T2DM. However, the roles of these cytokines remain uncertain52,53. It is clear that the high concentrations of TNF-α that are observed in individuals affected by burns or cancer can result in insulin resistance, but the levels typically observed in people with T2DM is more characteristic of a lower-grade inflammation. Furthermore, antibodies targeted to neutralize TNF-α in individuals with T2DM do not reverse insulin resistance54.
In addition to what we have discussed so far, the diacylglycerol hypothesis might also explain the insulin-sensitization effects of thiazoladinediones55,56 and omega fatty acids57. By activating peroxisome proliferator-activated receptor-γ (PPAR-γ) in adipocytes, thiazoladinediones redistribute fat from the liver and muscle to the subcutaneous fat55,58. This redistribution lowers intracellular diacylglycerol concentrations in the liver and in muscle and restores insulin sensitivity in these organs. This would explain the paradoxical improvement in insulin sensitivity that is often associated with weight gain in patients with T2DM who are treated with thiazoladinedione. In addition to activating PPAR-γ, omega fatty acids activate PPAR-α, promote peroxisomal and mitochondrial biogenesis and increase fatty acid oxidation.
Conclusion
Given the crucial role of insulin resistance in the pathogenesis of T2DM, understanding the molecular mechanism of insulin resistance is vital for the development of new therapies. Although diacylglycerol-induced insulin resistance and the inflammatory causes of insulin resistance are not mutually exclusive, they point to different sets of targets to reverse insulin resistance. Sorting out the relative roles of these pathways in the pathogenesis of liver and muscle insulin resistance is crucial for the development of new and more effective therapies for T2DM.
Footnotes
COMPETING FINANCIAL INTERESTS
The authors declare no competing financial interests.
References
- 1.International Diabetes Federation. 2009 http://www.idf.org/latest-diabetes-figures-paint-grim-global-picture/
- 2.Shulman GI, et al. N Engl J Med. 1990;322:223–228. doi: 10.1056/NEJM199001253220403. [DOI] [PubMed] [Google Scholar]
- 3.Cline GW, et al. N Engl J Med. 1999;341:240–246. doi: 10.1056/NEJM199907223410404. [DOI] [PubMed] [Google Scholar]
- 4.Rothman DL, Shulman RG, Shulman GI. J Clin Invest. 1992;89:1069–1075. doi: 10.1172/JCI115686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Magnusson I, Rothman DL, Katz LD, Shulman RG, Shulman GI. J Clin Invest. 1992;90:1323–1327. doi: 10.1172/JCI115997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Roden M, et al. Diabetes. 2000;49:701–707. doi: 10.2337/diabetes.49.5.701. [DOI] [PubMed] [Google Scholar]
- 7.Landau BR, et al. J Clin Invest. 1996;98:378–385. doi: 10.1172/JCI118803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Yalow RS, Berson SA. J Clin Invest. 1960;39:1157–1175. doi: 10.1172/JCI104130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Schenk S, Saberi M, Olefsky JM. J Clin Invest. 2008;118:2992–3002. doi: 10.1172/JCI34260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Shoelson SE, Lee J, Goldfine AB. J Clin Invest. 2006;116:1793–1801. doi: 10.1172/JCI29069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hotamisligil GS. Nature. 2006;444:860–867. doi: 10.1038/nature05485. [DOI] [PubMed] [Google Scholar]
- 12.Hotamisligil GS. Int J Obes (Lond) 2008;32(Suppl 7):S52–S54. doi: 10.1038/ijo.2008.238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Evans JL, Goldfine ID, Maddux BA, Grodsky GM. Endocr Rev. 2002;23:599–622. doi: 10.1210/er.2001-0039. [DOI] [PubMed] [Google Scholar]
- 14.Kulkarni RN, et al. Cell. 1999;96:329–339. doi: 10.1016/s0092-8674(00)80546-2. [DOI] [PubMed] [Google Scholar]
- 15.Brüning JC, et al. Cell. 1997;88:561–572. doi: 10.1016/s0092-8674(00)81896-6. [DOI] [PubMed] [Google Scholar]
- 16.Randle PJ, Garland PB, Hales CN, Newsholme EA. Lancet. 1963;1:785–789. doi: 10.1016/s0140-6736(63)91500-9. [DOI] [PubMed] [Google Scholar]
- 17.Randle PJ, Newsholme EA, Garland PB. Biochem J. 1964;93:652–665. doi: 10.1042/bj0930652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Randle PJ, Garland PB, Newsholme EA, Hales CN. Ann NY Acad Sci. 1965;131:324–333. doi: 10.1111/j.1749-6632.1965.tb34800.x. [DOI] [PubMed] [Google Scholar]
- 19.Roden M, et al. J Clin Invest. 1996;97:2859–2865. doi: 10.1172/JCI118742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Dresner A, et al. J Clin Invest. 1999;103:253–259. doi: 10.1172/JCI5001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yu C, et al. J Biol Chem. 2002;277:50230–50236. doi: 10.1074/jbc.M200958200. [DOI] [PubMed] [Google Scholar]
- 22.Griffin ME, et al. Diabetes. 1999;48:1270–1274. doi: 10.2337/diabetes.48.6.1270. [DOI] [PubMed] [Google Scholar]
- 23.Itani SI, Ruderman NB, Schmieder F, Boden G. Diabetes. 2002;51:2005–2011. doi: 10.2337/diabetes.51.7.2005. [DOI] [PubMed] [Google Scholar]
- 24.Kim JK, et al. Proc Natl Acad Sci USA. 2001;98:7522–7527. doi: 10.1073/pnas.121164498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kim JK, et al. J Clin Invest. 2004;113:756–763. doi: 10.1172/JCI18917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Liu L, et al. J Clin Invest. 2007;117:1679–1689. doi: 10.1172/JCI30565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Choi CS, et al. J Clin Invest. 2007;117:1995–2003. doi: 10.1172/JCI13579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Choi CS, et al. Proc Natl Acad Sci USA. 2007;104:16480–16485. doi: 10.1073/pnas.0706794104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kim JK, et al. J Clin Invest. 2004;114:823–827. doi: 10.1172/JCI22230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hoehn KL, et al. Cell Metab. 2010;11:70–76. doi: 10.1016/j.cmet.2009.11.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Samuel VT, et al. J Biol Chem. 2004;279:32345–32353. doi: 10.1074/jbc.M313478200. [DOI] [PubMed] [Google Scholar]
- 32.Samuel VT, et al. J Clin Invest. 2007;117:739–745. doi: 10.1172/JCI30400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Nagai Y, et al. Cell Metab. 2009;9:252–264. doi: 10.1016/j.cmet.2009.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Neschen S, et al. Cell Metab. 2005;2:55–65. doi: 10.1016/j.cmet.2005.06.006. [DOI] [PubMed] [Google Scholar]
- 35.Choi CS, et al. J Biol Chem. 2007;282:22678–22688. doi: 10.1074/jbc.M704213200. [DOI] [PubMed] [Google Scholar]
- 36.Savage DB, et al. J Clin Invest. 2006;116:817–824. doi: 10.1172/JCI27300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zhang D, et al. Proc Natl Acad Sci USA. 2007;104:17075–17080. doi: 10.1073/pnas.0707060104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Weiss R, et al. Lancet. 2003;362:951–957. doi: 10.1016/S0140-6736(03)14364-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Petersen KF, et al. J Clin Invest. 2002;109:1345–1350. doi: 10.1172/JCI15001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Luzi L, et al. Am J Physiol Endocrinol Metab. 2003;284:E274–E280. doi: 10.1152/ajpendo.00391.2001. [DOI] [PubMed] [Google Scholar]
- 41.Morino K, et al. J Clin Invest. 2005;115:3587–3593. doi: 10.1172/JCI25151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Befroy DE, et al. Diabetes. 2007;56:1376–1381. doi: 10.2337/db06-0783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Petersen KF, et al. Science. 2003;300:1140–1142. doi: 10.1126/science.1082889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Chibalin AV, et al. Cell. 2008;132:375–386. doi: 10.1016/j.cell.2007.12.035. [DOI] [PubMed] [Google Scholar]
- 45.Petersen KF, et al. N Engl J Med. 2009;362:1082–1089. doi: 10.1056/NEJMoa0907295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Weisberg SP, et al. J Clin Invest. 2003;112:1796–1808. doi: 10.1172/JCI19246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Xu H, et al. J Clin Invest. 2003;112:1821–1830. doi: 10.1172/JCI19451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Taubes G. Science. 2009;325:256–260. doi: 10.1126/science.325_256. [DOI] [PubMed] [Google Scholar]
- 49.Petersen KF, Dufour S, Befroy D, Garcia R, Shulman GI. N Engl J Med. 2004;350:664–671. doi: 10.1056/NEJMoa031314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kim JK, Gavrilova O, Chen Y, Reitman ML, Shulman GI. J Biol Chem. 2000;275:8456–8460. doi: 10.1074/jbc.275.12.8456. [DOI] [PubMed] [Google Scholar]
- 51.Petersen KF, et al. Diabetes. 2005;54:603–608. doi: 10.2337/diabetes.54.3.603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Pedersen BK, Febbraio MA. J Appl Physiol. 2007;102:814–816. doi: 10.1152/japplphysiol.01208.2006. [DOI] [PubMed] [Google Scholar]
- 53.Shoelson SE. J Appl Physiol. 2007;102:820. doi: 10.1152/japplphysiol.01353.2006. [DOI] [PubMed] [Google Scholar]
- 54.Ofei F, Hurel S, Newkirk J, Sopwith M, Taylor R. Diabetes. 1996;45:881–885. doi: 10.2337/diab.45.7.881. [DOI] [PubMed] [Google Scholar]
- 55.Mayerson AB, et al. Diabetes. 2002;51:797–802. doi: 10.2337/diabetes.51.3.797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Belfort R, et al. N Engl J Med. 2006;355:2297–2307. doi: 10.1056/NEJMoa060326. [DOI] [PubMed] [Google Scholar]
- 57.Neschen S, et al. Diabetes. 2007;56:1034–1041. doi: 10.2337/db06-1206. [DOI] [PubMed] [Google Scholar]
- 58.Kim JK, et al. Diabetes. 2003;52:1311–1318. doi: 10.2337/diabetes.52.6.1311. [DOI] [PubMed] [Google Scholar]