

Abstract
The present study investigated the relationships among oxidative stress, β-amyloid and cognitive abilities in the APP/PSEN1 double-transgenic mouse model of Alzheimer’s disease. In two experiments, long-term dietary supplements were given to aged APP/PSEN1 mice containing vitamin C alone (1g/kg diet, Expt. 1) or in combination with a high (750 IU/kg diet, Expts. 1 and 2) or lower (400 IU/kg diet, Expt. 2) dose of vitamin E. Oxidative stress, measured by F4-neuroprostanes or malondialdehyde, was elevated in cortex of control-fed APP/PSEN1 mice and reduced to wild-type levels by vitamin supplementation. High dose vitamin E with C was less effective at reducing oxidative stress than vitamin C alone or the low EC diet combination. The high-dose combination also impaired water maze performance in mice of both genotypes. In Experiment 2 the lower vitamin EC treatment attenuated spatial memory deficits in APP/PSEN1 mice and improved performance in wild-type mice in the water maze. Amyloid deposition was not reduced by antioxidant supplementation in either experiment.
Keywords: Vitamin C, Vitamin E, oxidative stress, Alzheimer’s disease, cognition
1. Introduction
The causes of most cases of Alzheimer’s disease are unknown, but likely involve a combination of genetic predisposition and environmental factors. Mutations in amyloid precursor protein (APP) and the presenilins (PSEN1 and PSEN2) lead to increased production of the 42-amino-acid variant of the β-amyloid (Aβ) peptide. Aβ1-42 is more neurotoxic and more likely to aggregate into plaques than other Aβ species. Although only a small fraction of Alzheimer’s cases are familial (FAD), mutations in APP or one of the presenilins have been identified in each of the cases [1]. Progression of the disease can be influenced by factors such as diet, education and exercise [2, 3]. There is mixed evidence as to the efficacy of an antioxidant-vitamin-rich diet in reducing the risk of developing Alzheimer’s disease or its severity. Although positive relationships have been demonstrated between supplement use and dietary intake of vitamins C and E and reduced incidence of Alzheimer’s disease [4–6], these beneficial results are not universal [7–9].
Oxidative stress arises from an imbalance between the production of free radicals and physiological antioxidant capability [10]. In addition to β-amyloid (Aβ) oligomerization and hyperphosphorylation of tau, oxidative stress is thought to play a key role in the pathogenesis and neuropathological changes of Alzheimer’s disease [11]. The brain is particularly susceptible to oxidative stress due to its large oxygen turnover, which leads to the generation of oxygen-related free radicals. The brain is also rich in polyunsaturated fatty acids and thus relies heavily on the antioxidant properties of the lipid-soluble vitamin E (α-tocopherol), which can cross the blood-brain barrier and accumulate in brain tissues. In addition to lipid peroxidation, the brain suffers from oxidative damage to both proteins and DNA. Thus antioxidants such as water-soluble vitamin C (ascorbic acid) may also reduce oxidative stress in the brain. A synergistic relationship exists between vitamins C and E in that vitamin C recycles vitamin E radicals and can regenerate its antioxidant properties [12].
Transgenic mice carrying mutant versions of APP and/or PSEN1 exhibit increased vulnerability to both protein oxidation and lipid peroxidation [13]. Oxidative stress has been shown to increase β-secretase activity, resulting in the increased production of pathogenic Aβ variants [14]. Increased lipid peroxidation was observed in brain of aged transgenic mice carrying an FAD-linked PSEN1 mutation, relative to controls [15]. Even young mice carrying PSEN1 mutations showed increased lipid peroxidation and reduced activity of the antioxidant enzymes Cu/Zn superoxide dismutase and glutathione reductase [16]. Fibroblasts cultured from brains from AD patients carry PSEN1 mutations had reduced antioxidant capabilities compared to controls [17]. Mice lacking the α-tocopherol transfer protein have diminished brain vitamin E and high levels of lipid peroxidation. When crossed with Tg2576 APP-overexpressing mice they show increased Aβ deposition that is reversed by vitamin E supplementation [18]. Furthermore, Aβ-like structures were observed in the CA1 hippocampal subfield of rats maintained on a vitamin E-deficient diet or exposed to hyperoxic conditions to induce oxidative stress [19]. Aβ is toxic and induces oxidative damage secondary to sustained oxidative stress [20–22]. This neurotoxicity is attenuated by vitamins C and E in rat primary hippocampal neurons [23, 24]. In addition to its antioxidant activity, vitamin C also reduces Aβ secretion in SHS-Y5Y cells [25].
Converging evidence shows that oxidative stress is a critical mediator of cognitive impairments such as those found in Alzheimer’s disease, and that treatment with dietary antioxidants can reverse these deficits. In Long-Evans rats trained on a spatial reference-memory water-maze task, greater oxidative stress-related changes were observed in the hippocampus of rats with a spatial-learning impairment, compared to unimpaired animals of the same age [26]. Oxidative stress induced by hyperoxia significantly impaired water maze and radial maze performance of 3-month-old rats, effects that were partially attenuated by dietary vitamin E supplementation [19, 27]. Long-term feeding of an antioxidant blueberry extract rescued a Y-maze alternation deficit in transgenic mice carrying mutations of APP and PSEN1, although treatments did not reduce plaque deposition [28]. The antioxidant curcumin has been shown to reduce markers of oxidative stress and amyloid deposition in Tg2576 APP-transgenic mice, although memory was not assessed in this study [29].
Although a broad range of antioxidants alone and in combination have been shown to be effective cognition enhancers [30–34], there has been no study of the role of vitamins C and E in preventing the deterioration of learning and memory in a transgenic mouse model of the oxidative stress, plaque formation, and cognitive deficits associated with Alzheimer’s disease. To this end, APPSWE/PSEN1ΔE9 transgenic mice were administered diets containing different amounts of vitamins C and E. A battery of cognitive and control tasks was conducted, after which mice were sacrificed for assessment of oxidative stress and amyloid plaque load. We expected that the antioxidant treatments would normalize oxidative stress and reduce amyloid plaque and cognitive impairments in the transgenic animals.
2. Methods
2.1 Animals
APPSwe/PSEN1ΔE9 bigenic mice were obtained from Jackson Laboratory (Bar Harbor, ME, USA; stock no. 004462) and maintained as double hemizygotes by crossing with wild-type individuals on a B6C3F1/J background strain (Jackson Laboratories stock no. 100010).
This mouse model harbors two separate FAD-linked mutations, the Swedish double-mutation (K595N/M596L) in the APP gene, and a deletion in exon 9 of the PSEN1 gene. The animals express several of the key features of Alzheimer’s disease, including amyloid plaque deposition, elevated oxidative stress and learning and memory deficits. Although these mutations have not been found to be co-expressed in humans, bigenic mice have accelerated development of neuropathological changes relative to single-mutant mice [35], and cognitive deficits from 7 months of age [36]. The mice exhibit amyloid aggregation and amyloid-related neuropathology and cognitive decline from 4–6 months that worsen with increasing age [36–39]. Genotypes were confirmed by polymerase chain reaction analysis of tail biopsies at 21 days of age and again at the end of the experiment when DNA was obtained post-mortem from a new sample of tail tissue. All mice were group-housed by gender in standard tub cages (26.5 × 17 × 12 cm) with fiber bedding under a 12/12-h light/dark cycle (lights on at 0600 h).
Mice were housed in a colony room in the same suite as the behavioral testing rooms. Mice had free access to food and water except in Expt. 1 during testing in operant chambers (4 – 8 months of age) when access to food was restricted to 4 hours per day. Mice were regularly monitored to ensure that weights did not fall to less than 85% of their free-feeding weight. All procedures were approved by the Vanderbilt University Institutional Animal Care and Use Committee and were conducted in accordance with the NIH Guide for the Care and Use of Laboratory Animals. Approximately equal numbers of male and female mice were used in each group.
2.2 Antioxidant treatments
Treatment diets were obtained from Research Diets Inc. (New Brunswick, NJ, USA). The control diet (D10001) contained a standard vitamin mix (V1001) that included no vitamin C but did contain vitamin E (50 IU / kg diet; vitamin E acetate). Mice in Expt. 1 were maintained either on this control diet, or on the same diet supplemented with vitamin C alone (C-alone group: D04101101) or both vitamins C and E (High EC group: D04101102) in addition to control diet levels. A third test diet used in Expt. 2 combined vitamin C and a lower dose of vitamin E (Low EC: D04101103). Details of supplemented diets are given in Table 1. Based on the average daily food intake of the mice, the High EC diet delivered approximately 50 mg/kg body weight/day vitamin E, and 100 mg/kg vitamin C in both the C-alone and High EC diets. Animals were maintained on standard lab chow (#5001, Purina Mills, St. Louis, MO) until 6 months of age, at which time they were randomly allocated to one of the three diet groups in approximately equal numbers (n = 11–17 per group).
Table 1.
Vitamin supplements added to the experimental diets in Experiments 1 and 2.
| Group | Vitamin C - ascorbic acid (g/kg diet)*
|
Vitamin E acetate (IU/kg diet)
|
||||
|---|---|---|---|---|---|---|
| Chow | Supplement | Total | Chow | Supplement | Total | |
| Experiment 1 | ||||||
| Control (D10001) | 0 | 0 | 0 | 50 | 0 | 50 |
| C-alone (D04101101) | 0 | 3 | 3 | 50 | 0 | 50 |
| High EC (D04101102) | 0 | 3 | 3 | 50 | 700 | 750 |
| Experiment 2 | ||||||
| Control (D10001) | 0 | 0 | 0 | 50 | 0 | 50 |
| Low EC (D04101103) | 0 | 3 | 3 | 50 | 350 | 400 |
| High EC (D04101102) | 0 | 3 | 3 | 50 | 700 | 750 |
Bioavailable ascorbic acid is only 33% of that added so actual dose is 1g/kg diet
Diets provided by Research Diets Inc. (New Brunswick, NJ, USA).
2.3 Behavioral procedures
The experimental design and order of behavioral tests for experiments 1 and 2 is depicted in Figure 1.
Figure 1.
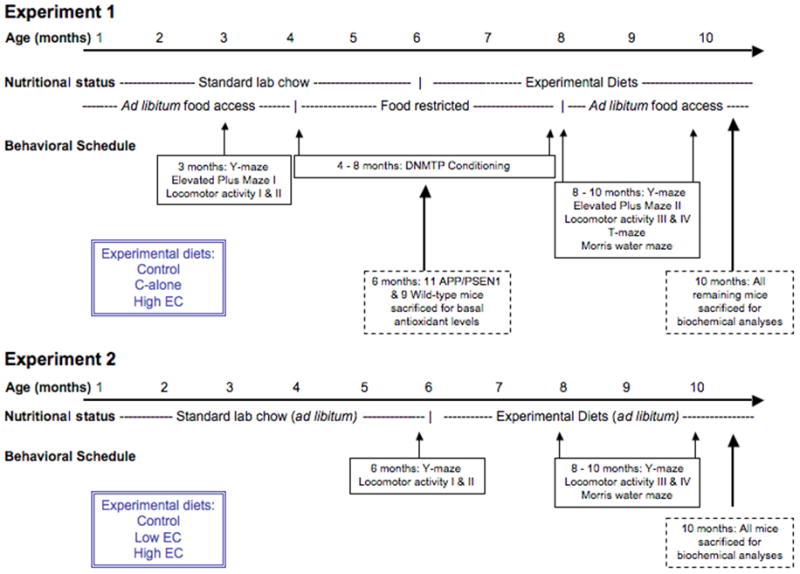
Experimental design and timeline for Experiments 1 and 2
Control tasks
The elevated plus maze was used to assess anxiety via differential exploratory tendencies in enclosed versus open arms, as previously described [36, 37]. Two different standard mazes were used for testing at 3 months (enclosed arms made of translucent gray acrylic) and at 8 months of age (enclosed arms made of clear acrylic), and testing was conducted in a different room each time. The use of separate apparatus ensured that the equipment was novel for both test sessions and limited any effects of habituation during the second session. At the beginning of a session, mice were gently placed in one of the open arms facing the central area, and allowed to explore freely for 5 min. Sessions were recorded and analyzed using NIH Image software on a Macintosh computer, running a macro specially written to collect data about the mouse’s position on the maze throughout the trial [40]. The time spent in closed arms was calculated as the percent of total time on arms, excluding time in the central area. The number of entries into each arm, the amount of time spent in each arm, and total distance traveled were also recorded. Additional control measures of anxiety and locomotor activity were obtained from the cognitive tests described below.
Learning and memory
Spontaneous alternation was tested in a standard Y-maze made of clear acrylic tubing, as previously described [36]. The maze had three identical arms 32 cm long, radiating symmetrically from the center of the maze. The number and sequence of arm entries made during a 5-min. session were recorded. Alternations were defined as an entry into each arm within three consecutive arm choices (e.g. ABC or BAC).
Locomotor activity was assessed in commercially-available activity monitors (ENV-510; MED Associates, Georgia, VT), as previously described [41, 42]. Activity was automatically recorded by the breaking of infrared beams as the mouse explored the chamber. A single session lasted 10 min., and the chambers were cleaned with a 10% alcohol solution between each session. Two sessions were conducted on consecutive days when mice were 3 months old, followed by two sessions on consecutive days at 7 months of age. Implicit memory for the testing context can be measured by comparing performance between sequential test sessions [43, 44]. Typically, activity will decrease from one test session to the next until a baseline level of activity is reached (habituation). Memory savings between the second and third test sessions, which were conducted four months apart, were calculated using the distance traveled (d) during the first three sessions: [100 · (d1−d3)/ (d1−d2)]. An increase in activity between the second and third sessions indicates a ‘loss’ of the initial habituation and is reflected in a savings score of <100%. Inversely, equivalent or reduced activity on the third session (savings score of >100%) indicates that all of the habituation processes have been ‘saved,’ i.e., that the habituation process is complete and that baseline activity levels have been reached. Using this formula memory savings can be compared between groups regardless of initial activity levels.
A delayed non-matching-to-position (DNMTP) version of delayed conditional discrimination task was conducted in commercially-available operant chambers (MED Associates, Georgia VT) as previously described [45, 46]. Responses were recorded, and stimuli presented, using MED-PC software. Mice were trained to respond to a sample stimulus, which was the illumination of a nose-poke response hole on the front wall (either left or right of a dipper used for the delivery of a liquid reinforcer to the inside of the chamber), and then to respond at a third nose-poke hole on the rear wall for 1, 4 or 8 s (delay phase). In the choice phase that followed, both left and right nose poke holes were lit and the mouse was required to respond in the nose-poke hole that was not the sample stimulus in order to earn a reinforcer (4 s access to 0.1 ml 30 % sucrose solution). Incorrect choices were not reinforced and resulted in immediate darkness for 4 s, followed by the 20-s inter-trial interval (ITI). Each daily session consisted of 60 trials or 60 min., whichever came first. A number of procedural variables were also collected during the sessions to assess potential non-cognitive influences such as visual discriminative ability, behavioral slowing, perseveration, etc., as previously described [45–48].
The T-maze provided an additional means to test spatial non-matching-to-position [49, 50]. The maze consisted of a central stem and two choice arms on a white base, and clear acrylic walls. Each area of the maze could be manually closed by means of sliding guillotine doors. Two test sessions were conducted, each comprising one forced-choice trial followed by 12 free-choice trials, with an ITI of 90 s. Each trial began with only one arm open containing a few drops of a 30% sucrose solution. Upon entering the arm with all four paws the mouse was confined for 60 s and allowed to consume the sucrose reinforcer. On each of the 12 free-choice trials the mouse was free to choose either arm, but only the arm not entered on the previous trial contained the reinforcer. Once the mouse had crossed into either arm, the sliding door was closed and the mouse confined for 60 s. During the ITI the mouse was confined in the starting area while the maze was quickly wiped clean with a 10% ethanol solution.
Spatial reference memory was tested in a Morris water maze. Hidden-platform testing was conducted as previously described [37, 45] in a 105-cm diam. pool with a circular acrylic platform (10 cm diam.) submerged 1 cm below the surface of the water with clearly visible cues fixed around the perimeter of the test room. Sessions were captured by an overhead camera and analyzed in real time using an NIH Image macro on a Macintosh computer [51]. Mice received four trials per day for 10 days to learn the location of the escape platform. To assess memory for the platform location, a no-platform probe trial was conducted 24 hours following the final acquisition session. Latency and path length to reach the hidden platform were the variables of interest during task acquisition. Preference for swimming in the target quadrant over non-target quadrants and search accuracy measured as time spent swimming within 20 cm of the platform location (Annulus 20) were the two primary dependent measures derived from the probe trial. Swim speed and peripheral swim time (within 8 cm of the edge) were also assessed in the water maze, to determine whether differences in performance could be attributed to non-cognitive factors.
2.4 Neurochemistry
Mice were anaesthetized using isoflurane and sacrificed by decapitation. Brains were quickly removed, cut along the midline, and stored following one of the methods described below, depending on the subsequent assay.
Vitamin C (ascorbic acid) and Vitamin E (α-tocopherol) levels were measured in cortex and liver. Samples were quickly dissected on ice from fresh tissue, frozen in liquid nitrogen and stored at −80 °C until assay. Vitamin C concentrations were measured by ion pair HPLC [52] and electrochemical detection as previously described [43, 53], except that tetrapentyl ammonium bromide was used as the ion pair reagent. Vitamin E in tissue samples was measured by HPLC as previously described [54].
Lipid peroxidation as F4-neuroprostanes was measured in cortical tissue using methods previously described by Roberts et al. [55]. A small piece of cortical tissue was dissected on ice and immediately frozen in liquid nitrogen and stored at −80 °C until measurements were made. Briefly, esterified F4-neuroprostanes were quantified by stable isotope dilution negative ion chemical ionization gas chromatography mass spectrometry (GC/MS) using [2H4]-15-F2t-isoprostane as an internal standard. Malondialdehyde was measured by homogenizing small amounts of tissue in a 5% TCA solution and reacting the samples with 0.02 M thiobarbituric acid for 35 minutes at 95 °C followed by 10 minutes at 4 °C. Malondialdehyde was then specifically measured using HPLC with inline fluorescence detection of the malondialdehyde-thiobarbituric acid adduct [56]. Neuroprostanes are specific markers for non-enzymatic lipid peroxidation. Malondialdehyde levels are also an important indicator of lipid peroxidation and oxidative stress, and are similarly elevated in Alzheimer’s disease [57].
In order to assess β-amyloid aggregates, hemi-brains were placed in a 10% formalin solution for three days as a fixative and then stored in phosphate buffered saline. Sections 30-μm thick were cut using a freezing microtome, and mounted on slides for histology. Thioflavin S staining was conducted and analyzed as described previously [37, 45]. Digital images of the hippocampus and overlying cortex were taken using a fluorescent imaging microscope with an Axiocam high-resolution camera (Carl Zeiss Microimaging Inc., Thornwood, NY, USA) at a magnification of 40X. The area of the hippocampus and overlying cortical areas occupied by amyloid plaques were determined using the freely-available Image J software (National Institute of Health, Bethesda, MD, USA). Quantification was performed by an experienced experimenter who was blind to the treatment condition of mice. Plaque coverage was calculated as percent of total region measured in pixels. Three sections were assayed per mouse for hippocampus and cortex, and the mean value used to calculate percent coverage for each area.
2.5 Statistical analyses
Y-maze spontaneous alternation, elevated plus maze, water maze probe measures (except quadrant preference), open field memory savings, and neurochemical measures were analyzed using univariate analysis of variance (ANOVA) with genotype and treatment as between-subjects factors. Locomotor activity, DNMTP, T-maze delayed alternation, and water maze training data were analyzed using repeated-measures ANOVA (RMANOVA) with genotype and treatment as the between-subjects factors and session as the repeated measure. Preference for the target quadrant during water maze probe trials was also assessed by RMANOVA within each group separately, comparing percent time spent swimming in the target quadrant versus the mean time spent in the non-target quadrants. To protect against a Type I error, follow-up tests were restricted to comparison of the groups of interest, i.e., wild-type control vs. each APP/PSEN1 group to assess the effects of the mutant transgenes and to establish the effects of treatments on behavior, amyloid aggregation, and neurochemistry in the transgenic mice. Each treatment was compared to the control diet within genotype to assess the effects of antioxidant supplementation. Gender differences were not observed on any measure, so data for male and female mice were combined.
3. Results
3.1 Experiment 1
Vitamin C and vitamin E levels at 6 months of age
In order to establish that there were no baseline differences between genotypes in antioxidant levels, vitamins C and E were measured in 11 APP/PSEN1 and nine wild-type mice before starting the experimental diets. Neither liver nor brain content of vitamin C differed across genotypes [Fs <1.22, Ps>.28] or vitamin E [Fs <.11, Ps>.74] (data not shown).
Behavioral data
Mice were tested continuously on the DNMTP task from 3 months until 7 months of age, before and during administration of test diets. Response accuracy was measured for all trials, at each delay that was imposed between sample and choice response (1, 4 & 8 s). Representative data are presented from the final week of testing (Fig. 2a). A main effect for delay indicated that mice made fewer correct responses when required to remember the sample stimulus for longer periods of time [F(2, 112) = 57.41, P <.001]. Contrary to expectation however, APP/PSEN1 mice made more correct responses than wild-type mice overall [F(1, 56) = 7.24, P <.01]. There was no effect of treatment group on response accuracy and no significant interactions between factors. Furthermore, there were no genotype or treatment effects on non-cognitive performance control measures of choice latency, sample latency, discrimination ratio, perseveration (errors after errors), or number of completed trials (data not shown). Accuracy in the T-maze was also similar between genotypes and treatment groups on both days of testing [Fs<2.15, Ps>.13].
Figure 2. Behavioral data - Experiment 1.

All mice received continuous training on the DNMTP task from 3 to 7 months of age. (a) During the final 5 days of DNMTP testing APP/PSEN1 mice (solid shapes) made more correct responses than wild-type mice (open shapes; P<.01). At 8 months of age mice were trained on a hidden-platform version of the water maze followed 24 hours later by a 60-s no-platform probe trial. (b) Although the pattern of swim-time in each quadrant varied among groups, all groups showed a significant preference for the target quadrant over the mean time spent in non-target quadrants during the probe trial (Ps<.05). (c) High EC-treated mice were less accurate than control-fed animals, with less time spent within 20 cm of the platform (P<.05). + P <.05 APP/PSEN1 mice different from wild-type mice and * P <0.05 vitamin-supplemented group different from control-treated mice.
Locomotor activity was assessed in four 10-minute sessions — two sessions 3 months before initiation of the experimental diets, and two sessions after 1 month on the diets (data not shown). All mice showed the expected decrease in activity across the four sessions [F(3, 204) = 30.89, P<.001] with significant habituation between the first two (P<.001) and the second two test sessions (P<.001) and no differences between genotypes or subsequent treatment groups at either time point [Fs < 2.81, Ps>.059]. The 4-month gap between testing sessions was long enough so that a proportion (45%-65%) of mice in each group had savings scores below 100%; however, savings scores did not vary among the groups [Fs < .82, Ps>.37]. Percent alternation in the Y-maze also did not differ among treatment groups or genotypes [Fs <1.575, Ps>.215]. In the elevated plus maze neither genotype nor dietary treatment had any effect on performance on measures of exploration (path length and total number of arm entries) or anxiety (percent entries into closed arms and percent time spent in closed arms; Fs <2.69, Ps >.11).
During water-maze acquisition at 8 months of age all mice learned to swim to the escape platform in a direct manner as shown by a decrease in escape latency [F(10, 640) = 64.78, P<.001] and path length [F(10, 640) = 50.40, P <.001] across test sessions. Neither measure was affected by genotype or diet [Fs<1.47, Ps>.15]. During the 60-s probe trial, memory for the target location is inferred by a preference for swimming in the quadrant that previously contained the platform. RMANOVA revealed a preference for target over non-target quadrants, suggesting that mice remembered the location of the hidden platform [wild-type: Control F(1, 10) = 20.61, P<.001; C-alone F(1, 10) = 16.86, P <.01; High EC F(1, 11) = 4.99, P <.05; APP/PSEN1: Control F(1, 13) = 32.04, P <.001; C-alone F(1, 10) = 13.50, P <.01; High EC F(1, 10) = 10.11, P <.01, Fig. 2b]. Selective search may also be assessed by examination of time swimming close to the former platform location (e.g. Annulus 20 [45, 58]). APP/PSEN1 and wild-type mice overall spent the same amount of time swimming within 20 cm of the platform location [Genotype F(1, 64) = .006, P=.94; Genotype x Treatment F(2, 64) = .016, P =.96]. Although the main treatment effect was not significant [F(2, 64) = 3.13, P =.051], planned follow-up comparisons showed that High EC-treated mice performed less well than controls (P<.05; Fig. 2c). C-alone groups performed at an intermediate level but did not differ significantly from control or High-EC groups (P =.16). Treatment groups did not differ on the distance traveled during the probe trial, swim speed, or time spent swimming in the periphery of the maze [Fs<2.06, Ps>.14].
Neurochemistry data at 10 months of age
Vitamin C supplementation led to an increase in liver vitamin C compared to control-treated animals [F(2, 29) = 34.20, P<.001; for C-alone and High EC groups separately, Ps < .001; Fig. 3a]. Antioxidant treatments had no effect on vitamin C levels in the cortex [F(2, 24) = .74, P =.49; Fig. 3b]. Vitamin C did not vary according to genotype in either organ assayed [Fs<2.20, Ps>.13]. Treatment had a large effect on vitamin E in the liver [F(2, 29) = 12.61, P =.001, Fig. 3c]; mice that received High EC supplements had greater amounts of vitamin E in the liver than control-fed mice (P<.001). Overall, transgenic animals had higher liver vitamin E than wild-type mice [F(1, 29) =6.23, P <.05]; there was no Genotype x Treatment interaction [F(2, 29) = 2.662, P=.087]. There was no main effect of genotype on cortical vitamin E [F(1, 16) = 2.531, P=.131], but levels varied among treatment groups [F(1, 16) = 12.70, P < .001; Fig. 3d]. A significant Treatment x Genotype interaction [F(2, 16) = 5.72, P<.05] revealed that increased vitamin E levels in the High EC-treated animals over control-fed mice were evident in the APP/PSEN1 mice (P<.001) but not in wild-type mice (P=1.0), and only APP/PSEN1 mice on the High EC treatment had higher vitamin E levels than wild-type mice on the same dietary regimen (P<.001). C-alone and control-fed animals did not differ by genotype (Ps>.44).
Figure 3. Neurochemical data - Experiment 1.

Vitamins C and E were measured in cortex and liver of mice following antioxidant supplementation and behavioral testing. Treatment diets led to increased vitamin C (a) in the liver (P<.01) (b) but not cortex (P=.49) of all supplemented mice. (c) Vitamin E levels were elevated in the liver of mice that received the High EC treatment (P<.001) but not in mice that received vitamin C alone. (d) Vitamin E was also elevated in the cortex of mice fed the High EC-supplemented diet (P<.001) with the highest levels observed in APP/PSEN1 mice. F4-neuroprostanes were measured in the cortex as a marker of oxidative stress in response to dietary supplementation with vitamins. (e) F4-neuroprostanes were significantly elevated in APP/PSEN1 control-treated mice compared to wild-type mice (P<.001). Both the vitamin C-alone and the High EC diets decreased transgenic F4-neuroprostanes to wild-type levels (P<.05). (f) Aβ plaque deposition was higher in hippocampus of Vitamin C-alone-treated mice compared to control and High EC-treated mice (P<.01), but plaque deposition in the cortex did not differ among treatment groups (P=.798). * P <0.05 supplemented groups different from control-treated mice and + P <.05 APP/PSEN1 mice different from wild-type mice
Oxidative stress in the form of F4-neuroprostanes was assessed in the cortex from three to seven mice per group. As shown in Figure 3e, F4-neuroprostane levels were elevated in the brains of transgenic animals but decreased by supplementation of dietary antioxidants. There were significant overall effects of genotype [F(1, 26) = 6.78, P<.05] and treatment [F(2, 26) = 6.29, P <.01] and a Genotype x Treatment interaction [F(2, 26) = 5.08, P <.05]. Follow up analyses confirmed that oxidative stress was elevated in APP/PSEN1 mice on the control diet compared to control-treated wild-type mice (P<.001). There were no genotype differences in either of the supplemented groups (Ps>.64), and there were significant effects of antioxidant treatment in the APP/PSEN1 mice (Ps<.001) but not the wild-type mice (Ps=1.0). Amyloid levels were assessed in four to six APP/PSEN1 transgenic mice per treatment group. In the hippocampus plaque load varied among the treatment groups [F(2, 12) = 10.37, P<.01; Fig. 3f], with a higher amyloid load in C-alone treatment mice than control-fed animals (P<.01). High EC-fed mice did not differ from control-fed mice (P=.71). Plaque load in the cortex did not differ among groups [F(2, 13) = .230, P=.798].
Based on data from ours and other laboratories [36, 37, 59], we expected to find cognitive impairments in the APP/PSEN1 mice from 7 months of age. A primary difference between previous studies and the present experiment is the extensive daily testing in operant chambers: mice were trained for at least 6 days per week between the ages of 3 and 7 months. DNMTP acquisition was accompanied by dietary restriction to motivate subjects to perform tasks for food reinforcement. These mice therefore had an existence that comprised dietary restriction, an antioxidant-rich diet (except in the case of the control animals), high levels of cognitive activity, and moderate physical activity, which are the four recommendations made by Mattson [2] to reduce the risk of Alzheimer’s disease and other age-related neurodegeneration. We also identified a detrimental effect of the combined E and C supplementation in spatial memory retention in the water maze. Poorer accuracy was observed in both wild-type and transgenic mice on the High EC diet during the probe trial (Figure 2b & c).
In Experiment 1 we showed that the High EC treatment increased the amount of vitamin E detected in the liver and brain but did not reduce oxidative stress, and impaired, or at best did not improve cognitive function. Given the evidence in support of combination antioxidant therapies we decided to investigate the possibility that vitamin E supplements were too high and had negative impact on oxidative stress and cognition. Therefore, in Experiment 2 we retained the same High EC treatment as in Experiment 1, and added an additional treatment group receiving the same amount of vitamin C as the High EC treatment with approximately half the amount of vitamin E. Furthermore, we dramatically reduced the amount cognitive testing that mice underwent in order to avoid the memory-improving effects of environmental enrichment and maintained cognitive activity that were responsible for masking/attenuating the expected deficits in the transgenic mice.
3.2 Experiment 2
Behavioral data
All mice showed normal habituation processes in the locomotor activity monitors, with a decrease in activity across sessions [F(1, 53) = 23.93, P<.001; data not shown]. The change was significant between sessions one and two at 6 months of age (P <.001) but not between sessions three and four at 8 months (P=.086), suggesting that most mice had reached a baseline level of exploration by the third trial. The 2-month delay between test sessions was shorter than that used in Experiment 1, and most mice retained some memory of the testing environment and thus exhibited memory savings scores of greater than 100%. Savings scores did not differ according among the groups [Fs <1.93, Ps >.15]. Percent alternation in the Y-maze did not differ between the groups [F(1, 58) =.27, P=.61].
Overall, mice learned to locate the hidden platform in the water maze, demonstrating improvements across test sessions in both latency and path length [F(9, 702) = 64.27, P <.001; F(9, 702) = 56.45, P<.001; Fig. 4a]. There were no significant omnibus effects of genotype or treatment during acquisition, and no Genotype x Treatment interaction [Fs <3.565, Ps>.063]. However, planned comparisons revealed that both escape latencies and path lengths were significantly longer in APP/PSEN1 mice on the control diet, compared to control-fed wild-type mice (Ps<.05). This spatial-memory deficit is identical to that previously reported in these bigenic mice [36, 37, 59], and suggests that the unimpaired performance in control-fed transgenic mice in Experiment 1 was indeed attributable to their long-term food restriction and cognitive “exercise”. Neither of the supplemented APP/PSEN1 groups differed from the control-fed wild-type mice on either acquisition measure (Ps>.17, Fig. 4a). Differences during acquisition were not due to swim speed or peripheral swimming, which did not differ among groups [Fs<.43, Ps>.45].
Figure 4. Behavioral data - Experiment 2.

At 8 months of age mice were trained for 10 days on a hidden-platform version of the water maze. (a) Neither of the two vitamin-supplemented wild-type groups differed from the control-fed wild-type group, and were excluded from the graph for clarity. Control fed APP/PSEN1 mice were slower at locating the hidden platform than wild-type control-fed mice (Ps<.05). There was no such impairment in vitamin-treated APP/PSEN1 mice (Ps>.17). For clarity, data are collapsed into 2-day time bins. (b) During the 60-s probe trial all wild-type mice showed a preference for swimming in the target quadrant over the three non-target quadrants (Ps<.01). Only APP/PSEN1 treated with the Low EC-supplemented diet showed the same preference for the platform quadrant (Control and High EC Ps>.082; Low EC P<.001). (c) Both APP/PSEN1 and wild-type mice fed the vitamin Low EC diet spent a greater portion of the time swimming within 20 cm of the platform location than control-fed mice (P<.05), indicating a better memory for the platform location. * P <0.05 different from control-treated mice.
During the probe trial, all wild-type mice spent a greater percentage of time swimming in the target quadrant compared to non-target quadrants [Fig. 4b; Control F(1, 13) = 9.14, P<.01; Low EC F(1, 14) = 29.59, P <.001; High EC F(1, 16) = 19.45, P <.001]. Consistent with their poor performance during water maze acquisition, APP/PSEN1 mice on the control diet did not show selective search for the former platform location [F(1, 14) = 3.52, P=.082]; transgenic mice fed the High EC diet had similarly poor spatial memory [F(1, 10) = 1.54, P =.243]. In contrast, APP/PSEN1 mice on the Low EC diet exhibited good spatial memory, spending nearly 50% of the probe trial in the target quadrant [F(1, 15) = 22.97, P <.001]. Overall, APP/PSEN1 mice spent less time swimming in the target quadrant than wild-type mice [F1, 78 = 5.41, P =.023]. Diet also had a significant effect on spatial memory [F2, 78 = 5.79, P = .005], due to the Low EC mice spending more time swimming in the target quadrant than either control or High EC-fed mice (Ps<.015). Genotype was not a significant predictor of swim accuracy on the annulus 20 measure [F(1, 82) = 2.71, P =.10; Genotype x Diet F(2, 82) = .25, P =.78; Fig. 4c]. However, there was a significant main effect of diet. Both wild-type and transgenic mice receiving the Low EC-treatment spent more time swimming closer to the platform than mice in the control group [F(2, 82) = 3.10, P <.05; Fig. 4c]. Groups did not differ in the amount of time spent swimming in the periphery of the maze [Fs<1.21, Ps>.28], and differences in swim speed were not significant across groups [Fs<3.88, Ps>.053]. Mice in all groups performed equally well on the cued-platform control version of the task when the platform was located in a different location on each trial but was marked by a conspicuous black flag (data not shown). Latency and path length improved significantly over the course of 8 days of cued-platform testing [Fs(7, 539) >11.93, Ps<.001], and there were no main effects of genotype or diet or interaction between the factors [Fs<.52, Ps>.56].
Neurochemical data
Both experimental diets contained the same high level of vitamin C but different amounts of vitamin E. As expected, the treatments significantly increased liver vitamin C levels [F(2, 17) = 7.16, P<.01]. Higher levels of vitamin C were detected in livers of both Low EC and High EC (Ps<.05) groups relative to untreated controls (Fig. 5a), but transgenic and wild-type mice had similar values [F(1, 17) = .23, P=.64]. Cortical vitamin C levels were similar among the three treatment groups and in both genotypes [Fs <1.76, Ps >.20; Fig. 5b]. As in Experiment 1, there were significant effects of treatment on vitamin E levels in the liver [Fig. 5c; F(2, 23) = 31.01, P<.001]. High EC-treated mice had elevated levels of vitamin E compared to control mice (P<.001), whereas the Low EC-fed animals were not significantly different from controls (P=1.0). Vitamin E in the liver did not vary according to genotype [F(1, 23) = 2.01, P=.17]. Dietary treatments also led to differences in vitamin E in cortex [F(2, 26) = 5.42, P<.05; Fig. 5d]. Cortical vitamin E was elevated in High EC-(P<.05) but not Low EC-treated mice (P=.91) compared to control-fed mice.
Figure 5. Neurochemical data – Experiment 2.
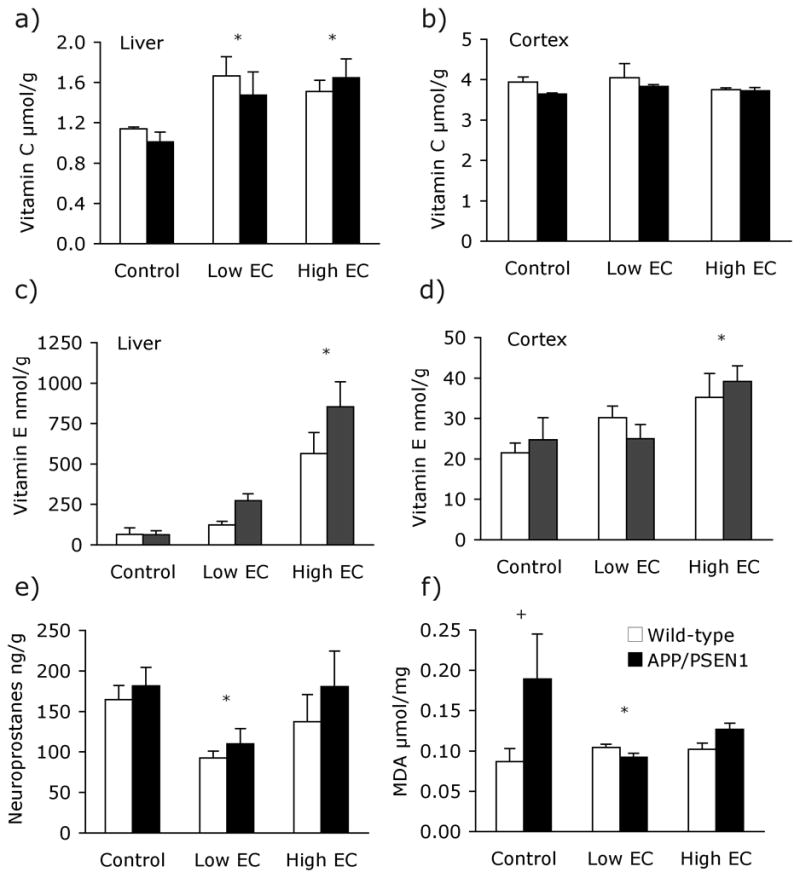
Vitamins C and E were measured in the cortex and liver of mice following treatment and behavioral testing. (a) Both vitamin treatents led to increases in vitamin C in the liver (P<.01). (b) Vitamin C levels in the cortex were unaffected by vitamin treatments (P>.20). The High EC treatment led to elevated vitamin E levels (c) in the liver (P<.001) (d) and cortex (P<.05). Liver and brain vitamin E levels in mice fed the Low EC treatment did not differ from those of control-fed mice (Ps>.91). (e) F4-Neuroprostane levels did not differ significantly between APP/PSEN1 (black bars) and wild-type mice (white bars) in Experiment 2; however, supplementation with the Low EC treatment led to a significant decrease in F4-neuroprostanes in the cortex (P<.05). Neuroprostane measurements in the brains of mice supplemented with High EC treatment did not differ from control-treated animals (P=.82). (f) Malondialdehyde levels in the cortex of APP/PSEN1 on the control diet were elevated relative to control-fed wild-type mice (P<.05). Genotype differences were not seen in either of the supplemented groups however Low EC-fed APP/PSEN1 mice had lower malondialdehyde levels than control-fed transgenic animals. * P <0.05 Low EC mice different from control-treated mice and + P <.05 APP/PSEN1 mice different from wild-type mice.
F4-neuroprostanes were measured in five to 13 mice per group. As shown in Figure 5e, F4-neuroprostane levels varied among treatment groups [F(2, 46)= 3.61, P<.05]. Low-EC-supplemented animals had lower neuroprostane levels than control animals (P<05), but control- and High EC-treated mice did not differ (P=.82). There was no main effect of genotype and no Treatment x Genotype interaction [Fs < 1.35, Ps>.25], suggesting that the Low EC treatment had had an equally beneficial effect for both genotypes. In contrast, malondialdehyde levels were elevated in control-fed APP/PSEN1 mice relative to wild-type controls. Although there was no main effects of diet or genotype and no interaction (Fs <1.66, Ps>.21), planned follow up comparisons revealed a pattern of effects similar to that observed in F4-neuroprostane levels in Experiment 1. Control-fed APP/PSEN1 mice had significantly elevated malondialdehyde (P<.05), and both vitamin treatments returned it to normal wild-type levels (Ps>.64; Fig. 5f). The low EC diet significantly reduced malondialdehyde levels in APP/PSEN1 mice (P<.05). High EC-fed mice also had lower malondialdehyde levels than control-fed mice, but the difference was not statistically significant (P>.12). Diet did not affect malondialdehyde levels in wild-type mice (Ps>.71). β-amyloid plaque deposition was analyzed in the hippocampus and overlying cortex in a subset of the APP/PSEN1 mice. Plaque deposition was similar among treatment groups [Fs(2, 23) <1.363, Ps>.28] in both brain areas assayed.
4. Discussion
To our knowledge this is the first long-term assessment of dietary administration of vitamins C and E in a mouse model of Alzheimer’s disease. The use of animal models allows us to control all of the features that make human population studies so variable, including past experience, health and diet, and compliance with the treatment schedule. Although our sample provides results that show the importance of such factors on the effects of oxidative stress and cognition, they are regulated between the groups. We show here that antioxidant supplements, administered over the course of several months, can have dramatic effects on lipid peroxidation in the brain and improve cognitive function in APP/PSEN1 and wild-type mice. We have also shown that a high dose of vitamin E has adverse effects in wild-type and transgenic mice under some conditions.
Oxidative stress and antioxidant levels
At 6 months of age, vitamin C and E levels did not differ between control-fed genotypes. Feeding of the antioxidant diets from 6 to 10 months of age increased vitamin C levels in the liver of all supplemented mice. Transport of vitamin C across the blood-brain barrier and into neurons is tightly controlled [60], and levels in the cortex did not differ from control-fed animals. 10-month-old mice treated with the High EC diet showed increased vitamin E levels in both liver and brain. In contrast, mice treated with the Low EC diet had increased vitamin E levels in the liver but not brain. Consistent with our previous data [37], oxidative stress was increased in APP/PSEN1 mice on the control diet compared to wild-type mice in Experiment 1 (F4-neuroprostanes) and Experiment 2 (malondialdehyde). In keeping with our experimental hypothesis, oxidative stress markers were reduced following vitamin supplementation, even in wild-type mice, although effects were more pronounced in transgenics.
It is probable that the additional circulating antioxidants resulting from supplemented diets led to the reduction of lipid peroxidation in treated transgenic animals. An interesting caveat to this result is that oxidative stress levels were not decreased further by the combined High EC treatment compared to the Low EC treatment or Vitamin C alone. In fact, the High EC combination diet appeared to be the least effective of the treatments in reducing oxidative stress in both APP/PSEN1 and wild-type mice. Long-term administration of higher doses of vitamin E than were used in the present study (up to 500 mg/kg mouse weight/day) have been shown to confer protection against markers of oxidative stress [30, 31, 34], but this beneficial effect is not universally observed [61, 62]. Nor does there appear to be a direct relationship between the dose administered and the effect on oxidative stress. A synergy exists between vitamins C and E [63], such that when the α-tocopherol (vitamin E) radical is generated by donation of an electron to a radical species, it is subsequently recycled by vitamin C. Supplementation with high doses of vitamin C reduced oxidation of low-density lipoprotein in humans, with the action presumed to be through recycling of tocopherol radicals [64]. However, vitamin E may be antioxidant, neutral, or pro-oxidant, depending on conditions [65]. In the absence of sufficient availability of vitamin C, the tocopherol radicals would remain in a reactive, pro-oxidant state or could assume another thus-far undefined role, but one that is not in keeping with the usual antioxidant function of vitamin E [65, 66].
Higher vitamin E levels were found in the liver and cortex of APP/PSEN1 than wild-type animals in Experiment 1 and to a lesser extent in Experiment 2. These divergent levels may reflect a difference in vitamin E storage and use between the genotypes. Either stored levels were greater in transgenic animals because less vitamin E was utilized in antioxidant processes (an inefficiency which might explain higher oxidative stress in these mice), or greater levels may be stored by transgenic animals as a response to the increased oxidative stress.
F4-neuroprostane data differed between the two experiments suggesting that oxidative stress may be influenced by a number of ‘lifestyle’ factors, including diet and cognitive activity. Even in wild-type mice F4-neuroprostane levels were higher in Experiment 1 than 2. Although each data set revealed a beneficial role of antioxidant supplementation in reducing this marker of oxidative stress, there was only a marginal increase in F4-neuroprostane levels in APP/PSEN1 in Experiment 2. The greatest benefits of antioxidant treatment were seen in the highly trained mice in Experiment 1, where the increase in oxidative stress in the transgenic mice was greatest. Oxidative stress levels in APP/PSEN1 mice may have been exacerbated by extensive training in Experiment 1, or conversely, the extensive cognitive activity may have allowed wild-type mice some form of protection that differed for APP/PSEN1 mice. The data highlight the fact that oxidative stress is not a unitary measure and thus it is important to consider more than one measure at a time. Thus, in Experiment 2, increased malondialdehyde levels confirmed the oxidant injury in APP/PSEN1 mice as well as the benefit of vitamin treatments despite the absence of a significant increase in APP/PSEN1 F4-neuroprostane levels.
Cognitive ability
A number of novel results are evident in the behavioral data presented here. First, transgene-dependent deficits were only seen in mice without extensive cognitive training (Expt. 2 but not Expt. 1), in contrast to our previously-published results with these double-transgenics [36, 37, 59]. This failure to replicate the cognitive deficit occurred despite significant oxidant damage as indicated by F4-neuroprostane levels. As noted above, important factors that differed between the two experiments, specifically dietary restriction, an antioxidant-rich diet (except in the case of the control animals), high levels of cognitive activity (environmental enrichment), and moderate physical activity have been shown to reduce age-related increases in oxidative stress and memory loss, and to reduce amyloid load and protect against cognitive deficits in wild-type mice and mouse models of Alzheimer’s disease [67–72]. Thus we are confident that the good cognitive performance of APP/PSEN1 mice in Experiment 1 was attributable to the daily regimen associated with the performing the DNMTP task.
Low EC-treated wild-type mice spent aprox. 20% more time swimming in the target quadrant and within 20 cm of the platform location (annulus 20) than control-fed mice. Nevertheless, Low-EC treatment improved water maze performance in both wild-type and transgenic mice, whereas the High-EC treatment did not. High levels of oxidative stress may impair cognition by a variety of means, for example, by altering membrane fluidity or synaptic function [73, 74]. Nevertheless, it appears that the associated effects on learning and memory can be attenuated by antioxidant supplementation. Very old (18 months) APP/PSEN1 bigenic mice are more cognitively impaired than those in the present study [58], and thus using older mice may have allowed us to observe an even greater effect of dietary supplementation. However, a general aim of disease prevention is to alleviate suffering in the early stages of the disease when there is still time to protect the relatively healthy brain. Despite the varied behavioral, environmental, and dietary conditions, no changes were observed in plaque load in the transgenic groups. Amyloid aggregation also did not change in conjunction with brain vitamin or oxidative stress levels. This is in accordance with a growing literature suggesting that plaque formation is a consequence and not a cause of the disease process [75, 76]. Thus we show that it is possible to enhance cognition with dietary antioxidant supplements without affecting the overt neuropathology that defines Alzheimer’s disease. Combined supplementation of vitamins C and E along with non-steroidal anti-inflammatory drugs (NSAIDs) helped to preserve cognitive abilities across an 8-year period in Alzheimer’s patients [77]. The protective effect was greatest in those who carried at least one copy of the ApoE E4 allele. These data are consistent with our results in that they support a role for antioxidant therapies in those who carry a strong genetic predisposition to the Alzheimer’s disease and in individuals who are most heavily compromised.
Finally, we identified a detrimental effect of the combined E and C supplementation in spatial memory retention in the water maze. In contrast to the Low EC supplementation, High EC treatments impaired water-maze performance of both wild-type and transgenic mice in Experiment 1, with approximately 25% less time spent in annulus 20 than controls, but did not impair or improve performance of either group in Experiment 2. In Experiment 1, control-fed APP/PSEN1 mice were not only unimpaired but performed better than wild-type mice normally do. Control-fed wild-type mice spent nearly 50% of the probe trial time within annulus 20 (Fig. 2c). Under normal conditions, wild-type mice of this age spend only 35–40% of the time within annulus 20 (Fig. 4c). A similar phenomenon was evident in the APP/PSEN1 mice that are normally impaired on tests of spatial cognition [36, 37, 59]. The effect of any drug is in part a function of the baseline level of performance [78] and it is likely that the continued cognitive training on the DNMTP task in Experiment 1 led to improved spatial learning in mice of both genotypes. Although the DNMTP task uses a discrete visual cue, it requires a left-right spatial discrimination and this spatial practice may have generalized to spatial learning in the water maze [45, 50]. Under the conditions of enhanced spatial cognition in Experiment 1, the High EC treatment impaired performance in mice of both genotypes; however, when performance was in its normal range for each respective genotype in Experiment 2, the High EC treatment did not affect water-maze performance. Many studies have shown improved cognition, and in some cases improved sensorimotor abilities, under different experimental conditions following administration of a broad range of doses vitamin E [30–34]. Furthermore, other studies reveal no improvement, or in some cases impairments, in cognition and sensorimotor skills following vitamin E administration [61, 66, 79]. Although it is difficult to demonstrate a clear relationship between dose and effect given the range of experimental methods, in general higher vitamin E doses were used in the studies that failed to find beneficial effects of supplementation.
β-Amyloid
We found no difference in the levels of Aβ plaque deposits between treatment groups. Similar results have been reported in other transgenic APP and APP/PSEN1 mouse models of AD [28, 45, 80]. The relationship between amyloid deposition, oxidative stress and cognition is not fully understood, and reports in the literature are inconsistent with respect to whether or not amyloid load correlates with cognition [58, 81, 82]. Indeed, considerable evidence shows that soluble Aβ can have an acute, drug-like amnestic effect in the absence of amyloid aggregation [83, 84] and it is possible that antioxidant treatments may have lowered levels of soluble Aβ without a measurable effect on deposited amyloid [30].
5. Conclusion
The behavioral and biochemical data presented here show that oxidative stress and memory can be improved by nutritional changes without significantly impacting aggregated amyloid proteins in the brain. If these data can be extended to humans, one may conclude that a vitamin-rich diet, or supplements, could help to reduce the overt cognitive deficits caused by Alzheimer’s disease as well as a number of disorders in which oxidative stress and cognitive impairment are factors, such as Parkinson’s disease and amyotrophic lateral sclerosis. Furthermore, our data suggest that the therapeutic window for the benefits of vitamin supplementation may be narrower than previously thought or may be restricted by benefits afforded by additional interventions.
Acknowledgments
This work was supported by grants from the National Institute of Health (AG023138 to James May & AG022439 to Mike McDonald). We thank Dr. Sean Davies for help with neuroprostane measurements.
Footnotes
The authors have no actual or potential conflicts of interest to report.
References
- 1.Tandon A, Rogaeva E, Mullan M, St George-Hyslop PH. Molecular genetics of Alzheimer’s disease: the role of beta-amyloid and the presenilins. Curr Opin Neurol. 2000;13:377–84. doi: 10.1097/00019052-200008000-00003. [DOI] [PubMed] [Google Scholar]
- 2.Mattson MP. Existing data suggest that Alzheimer’s disease is preventable. Ann N Y Acad Sci. 2000;924:153–9. doi: 10.1111/j.1749-6632.2000.tb05573.x. [DOI] [PubMed] [Google Scholar]
- 3.Weih M, Wiltfang J, Kornhuber J. Non-pharmacologic prevention of Alzheimer’s disease: nutritional and life-style risk factors. J Neural Transm. 2007 doi: 10.1007/s00702-007-0704-x. [DOI] [PubMed] [Google Scholar]
- 4.Engelhart MJ, Geerlings MI, Ruitenberg A, et al. Dietary intake of antioxidants and risk of Alzheimer disease. Jama. 2002;287:3223–9. doi: 10.1001/jama.287.24.3223. [DOI] [PubMed] [Google Scholar]
- 5.Morris MC, Beckett LA, Scherr PA, et al. Vitamin E and vitamin C supplement use and risk of incident Alzheimer disease. Alzheimer Dis Assoc Disord. 1998;12:121–6. doi: 10.1097/00002093-199809000-00001. [DOI] [PubMed] [Google Scholar]
- 6.Zandi PP, Anthony JC, Khachaturian AS, et al. Reduced risk of Alzheimer disease in users of antioxidant vitamin supplements: the Cache County Study. Arch Neurol. 2004;61:82–8. doi: 10.1001/archneur.61.1.82. [DOI] [PubMed] [Google Scholar]
- 7.Fillenbaum GG, Kuchibhatla MN, Hanlon JT, et al. Dementia and Alzheimer’s disease in community-dwelling elders taking vitamin C and/or vitamin E. Ann Pharmacother. 2005;39:2009–14. doi: 10.1345/aph.1G280. [DOI] [PubMed] [Google Scholar]
- 8.Laurin D, Masaki KH, Foley DJ, White LR, Launer LJ. Midlife dietary intake of antioxidants and risk of late-life incident dementia: the Honolulu-Asia Aging Study. Am J Epidemiol. 2004;159:959–67. doi: 10.1093/aje/kwh124. [DOI] [PubMed] [Google Scholar]
- 9.Luchsinger JA, Tang MX, Shea S, Mayeux R. Antioxidant vitamin intake and risk of Alzheimer disease. Arch Neurol. 2003;60:203–8. doi: 10.1001/archneur.60.2.203. [DOI] [PubMed] [Google Scholar]
- 10.Andersen JK. Oxidative stress in neurodegeneration: cause or consequence? Nat Med. 2004;10 (Suppl):S18–25. doi: 10.1038/nrn1434. [DOI] [PubMed] [Google Scholar]
- 11.Christen Y. Oxidative stress and Alzheimer disease. Am J Clin Nutr. 2000;71:621S–629S. doi: 10.1093/ajcn/71.2.621s. [DOI] [PubMed] [Google Scholar]
- 12.Niki E. Interaction of ascorbate and alpha-tocopherol. Ann N Y Acad Sci. 1987;498:186–99. doi: 10.1111/j.1749-6632.1987.tb23761.x. [DOI] [PubMed] [Google Scholar]
- 13.Mohmmad Abdul H, Wenk GL, Gramling M, Hauss-Wegrzyniak B, Butterfield DA. APP and PS-1 mutations induce brain oxidative stress independent of dietary cholesterol: implications for Alzheimer’s disease. Neurosci Lett. 2004;368:148–50. doi: 10.1016/j.neulet.2004.06.077. [DOI] [PubMed] [Google Scholar]
- 14.Tamagno E, Guglielmotto M, Aragno M, et al. Oxidative stress activates a positive feedback between the gamma- and beta-secretase cleavages of the beta-amyloid precursor protein. J Neurochem. 2008;104:683–95. doi: 10.1111/j.1471-4159.2007.05072.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Schuessel K, Frey C, Jourdan C, et al. Aging sensitizes toward ROS formation and lipid peroxidation in PS1M146L transgenic mice. Free Radic Biol Med. 2006;40:850–62. doi: 10.1016/j.freeradbiomed.2005.10.041. [DOI] [PubMed] [Google Scholar]
- 16.Leutner S, Czech C, Schindowski K, Touchet N, Eckert A, Muller WE. Reduced antioxidant enzyme activity in brains of mice transgenic for human presenilin-1 with single or multiple mutations. Neurosci Lett. 2000;292:87–90. doi: 10.1016/s0304-3940(00)01449-x. [DOI] [PubMed] [Google Scholar]
- 17.Gibson GE, Zhang H, Sheu KR, Park LC. Differential alterations in antioxidant capacity in cells from Alzheimer patients. Biochim Biophys Acta. 2000;1502:319–29. doi: 10.1016/s0925-4439(00)00057-0. [DOI] [PubMed] [Google Scholar]
- 18.Nishida Y, Yokota T, Takahashi T, Uchihara T, Jishage K, Mizusawa H. Deletion of vitamin E enhances phenotype of Alzheimer disease model mouse. Biochem Biophys Res Commun. 2006;350:530–6. doi: 10.1016/j.bbrc.2006.09.083. [DOI] [PubMed] [Google Scholar]
- 19.Fukui K, Takatsu H, Shinkai T, Suzuki S, Abe K, Urano S. Appearance of amyloid beta-like substances and delayed-type apoptosis in rat hippocampus CA1 region through aging and oxidative stress. J Alzheimers Dis. 2005;8:299–309. doi: 10.3233/jad-2005-8309. [DOI] [PubMed] [Google Scholar]
- 20.Mark RJ, Blanc EM, Mattson MP. Amyloid beta-peptide and oxidative cellular injury in Alzheimer’s disease. Mol Neurobiol. 1996;12:211–24. doi: 10.1007/BF02755589. [DOI] [PubMed] [Google Scholar]
- 21.Pappolla MA, Chyan YJ, Omar RA, et al. Evidence of oxidative stress and in vivo neurotoxicity of beta-amyloid in a transgenic mouse model of Alzheimer’s disease: a chronic oxidative paradigm for testing antioxidant therapies in vivo. Am J Pathol. 1998;152:871–7. [PMC free article] [PubMed] [Google Scholar]
- 22.Butterfield DA. Amyloid beta-peptide (1-42)-induced oxidative stress and neurotoxicity: implications for neurodegeneration in Alzheimer’s disease brain. A review. Free Radic Res. 2002;36:1307–13. doi: 10.1080/1071576021000049890. [DOI] [PubMed] [Google Scholar]
- 23.Goodman Y, Mattson MP. Secreted forms of beta-amyloid precursor protein protect hippocampal neurons against amyloid beta-peptide-induced oxidative injury. Exp Neurol. 1994;128:1–12. doi: 10.1006/exnr.1994.1107. [DOI] [PubMed] [Google Scholar]
- 24.Prehn JH, Bindokas VP, Jordan J, et al. Protective effect of transforming growth factor-beta 1 on beta-amyloid neurotoxicity in rat hippocampal neurons. Mol Pharmacol. 1996;49:319–28. [PubMed] [Google Scholar]
- 25.Huang J, May JM. Ascorbic acid protects SH-SY5Y neuroblastoma cells from apoptosis and death induced by beta-amyloid. Brain Res. 2006;1097:52–8. doi: 10.1016/j.brainres.2006.04.047. [DOI] [PubMed] [Google Scholar]
- 26.Nicolle MM, Gonzalez J, Sugaya K, et al. Signatures of hippocampal oxidative stress in aged spatial learning-impaired rodents. Neuroscience. 2001;107:415–31. doi: 10.1016/s0306-4522(01)00374-8. [DOI] [PubMed] [Google Scholar]
- 27.Fukui K, Omoi NO, Hayasaka T, et al. Cognitive impairment of rats caused by oxidative stress and aging, and its prevention by vitamin E. Ann N Y Acad Sci. 2002;959:275–84. doi: 10.1111/j.1749-6632.2002.tb02099.x. [DOI] [PubMed] [Google Scholar]
- 28.Joseph JA, Denisova NA, Arendash G, et al. Blueberry supplementation enhances signaling and prevents behavioral deficits in an Alzheimer disease model. Nutr Neurosci. 2003;6:153–62. doi: 10.1080/1028415031000111282. [DOI] [PubMed] [Google Scholar]
- 29.Lim GP, Chu T, Yang F, Beech W, Frautschy SA, Cole GM. The curry spice curcumin reduces oxidative damage and amyloid pathology in an Alzheimer transgenic mouse. J Neurosci. 2001;21:8370–7. doi: 10.1523/JNEUROSCI.21-21-08370.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Conte V, Uryu K, Fujimoto S, et al. Vitamin E reduces amyloidosis and improves cognitive function in Tg2576 mice following repetitive concussive brain injury. J Neurochem. 2004;90:758–64. doi: 10.1111/j.1471-4159.2004.02560.x. [DOI] [PubMed] [Google Scholar]
- 31.Joseph JA, Shukitt-Hale B, Denisova NA, et al. Long-term dietary strawberry, spinach, or vitamin E supplementation retards the onset of age-related neuronal signal-transduction and cognitive behavioral deficits. J Neurosci. 1998;18:8047–55. doi: 10.1523/JNEUROSCI.18-19-08047.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kolosova NG, Shcheglova TV, Sergeeva SV, Loskutova LV. Long-term antioxidant supplementation attenuates oxidative stress markers and cognitive deficits in senescent-accelerated OXYS rats. Neurobiol Aging. 2006;27:1289–97. doi: 10.1016/j.neurobiolaging.2005.07.022. [DOI] [PubMed] [Google Scholar]
- 33.Navarro A, Gomez C, Sanchez-Pino MJ, et al. Vitamin E at high doses improves survival, neurological performance, and brain mitochondrial function in aging male mice. Am J Physiol Regul Integr Comp Physiol. 2005;289:R1392–9. doi: 10.1152/ajpregu.00834.2004. [DOI] [PubMed] [Google Scholar]
- 34.Veinbergs I, Mallory M, Sagara Y, Masliah E. Vitamin E supplementation prevents spatial learning deficits and dendritic alterations in aged apolipoprotein E-deficient mice. Eur J Neurosci. 2000;12:4541–6. [PubMed] [Google Scholar]
- 35.Price DL, Wong PC, Markowska AL, et al. The value of transgenic models for the study of neurodegenerative diseases. Ann N Y Acad Sci. 2000;920:179–91. doi: 10.1111/j.1749-6632.2000.tb06920.x. [DOI] [PubMed] [Google Scholar]
- 36.Reiserer RS, Harrison FE, Syverud DC, McDonald MP. Impaired spatial learning in the APPSwe + PSEN1DeltaE9 bigenic mouse model of Alzheimer’s disease. Genes Brain Behav. 2007;6:54–65. doi: 10.1111/j.1601-183X.2006.00221.x. [DOI] [PubMed] [Google Scholar]
- 37.Bernardo A, Harrison FE, McCord M, et al. Elimination of GD3 synthase improves memory and reduces amyloid-beta plaque load in transgenic mice. Neurobiol Aging. 2008 doi: 10.1016/j.neurobiolaging.2007.12.022. [DOI] [PubMed] [Google Scholar]
- 38.Lalonde R, Kim HD, Maxwell JA, Fukuchi K. Exploratory activity and spatial learning in 12-month-old APP(695)SWE/co+PS1/DeltaE9 mice with amyloid plaques. Neurosci Lett. 2005;390:87–92. doi: 10.1016/j.neulet.2005.08.028. [DOI] [PubMed] [Google Scholar]
- 39.Garcia-Alloza M, Robbins EM, Zhang-Nunes SX, et al. Characterization of amyloid deposition in the APPswe/PS1dE9 mouse model of Alzheimer disease. Neurobiol Dis. 2006;24:516–24. doi: 10.1016/j.nbd.2006.08.017. [DOI] [PubMed] [Google Scholar]
- 40.Miyakawa T, Yamada M, Duttaroy A, Wess J. Hyperactivity and intact hippocampus-dependent learning in mice lacking the M1 muscarinic acetylcholine receptor. J Neurosci. 2001;21:5239–50. doi: 10.1523/JNEUROSCI.21-14-05239.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.McDonald MP, Wong R, Goldstein G, Weintraub B, Cheng SY, Crawley JN. Hyperactivity and learning deficits in transgenic mice bearing a human mutant thyroid hormone beta1 receptor gene. Learn Mem. 1998;5:289–301. [PMC free article] [PubMed] [Google Scholar]
- 42.Siesser WB, Cheng SY, McDonald MP. Hyperactivity, impaired learning on a vigilance task, and a differential response to methylphenidate in the TRbetaPV knock-in mouse. Psychopharmacology (Berl) 2005;181:653–63. doi: 10.1007/s00213-005-0024-5. [DOI] [PubMed] [Google Scholar]
- 43.Harrison FE, Yu SS, Van Den Bossche KL, Li L, May JM, McDonald MP. Elevated oxidative stress and sensorimotor deficits but normal cognition in mice that cannot synthesize ascorbic acid. J Neurochem. 2008;106:1198–1208. doi: 10.1111/j.1471-4159.2008.05469.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Platel A, Porsolt RD. Habituation of exploratory activity in mice: a screening test for memory enhancing drugs. Psychopharmacology (Berl) 1982;78:346–52. doi: 10.1007/BF00433739. [DOI] [PubMed] [Google Scholar]
- 45.Bernardo A, McCord M, Troen AM, Allison JD, McDonald MP. Impaired spatial memory in APP-overexpressing mice on a homocysteinemia-inducing diet. Neurobiol Aging. 2007;28:1195–205. doi: 10.1016/j.neurobiolaging.2006.05.035. [DOI] [PubMed] [Google Scholar]
- 46.McDonald MP, Willard LB, Wenk GL, Crawley JN. Coadministration of galanin antagonist M40 with a muscarinic M1 agonist improves delayed nonmatching to position choice accuracy in rats with cholinergic lesions. J Neurosci. 1998;18:5078–85. doi: 10.1523/JNEUROSCI.18-13-05078.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.McDonald MP, Wenk GL, Crawley JN. Analysis of galanin and the galanin antagonist M40 on delayed non-matching-to-position performance in rats lesioned with the cholinergic immunotoxin 192 IgG-saporin. Behav Neurosci. 1997;111:552–63. doi: 10.1037//0735-7044.111.3.552. [DOI] [PubMed] [Google Scholar]
- 48.McDonald MP, Crawley JN. Galanin receptor antagonist M40 blocks galanin-induced choice accuracy deficits on a delayed-nonmatching-to-position task. Behav Neurosci. 1996;110:1025–32. doi: 10.1037//0735-7044.110.5.1025. [DOI] [PubMed] [Google Scholar]
- 49.Lalonde R. The neurobiological basis of spontaneous alternation. Neurosci Biobehav Rev. 2002;26:91–104. doi: 10.1016/s0149-7634(01)00041-0. [DOI] [PubMed] [Google Scholar]
- 50.McDonald MP, Overmier JB. Present imperfect: a critical review of animal models of the mnemonic impairments in Alzheimer’s disease. Neurosci Biobehav Rev. 1998;22:99–120. doi: 10.1016/s0149-7634(97)00024-9. [DOI] [PubMed] [Google Scholar]
- 51.Miyakawa T, Yared E, Pak JH, Huang FL, Huang KP, Crawley JN. Neurogranin null mutant mice display performance deficits on spatial learning tasks with anxiety related components. Hippocampus. 2001;11:763–75. doi: 10.1002/hipo.1092. [DOI] [PubMed] [Google Scholar]
- 52.Pachla LA, Kissinger PT. Analysis of ascorbic acid by liquid chromatography with amperometric detection. Methods Enzymol. 1979;62:15–24. doi: 10.1016/0076-6879(79)62183-3. [DOI] [PubMed] [Google Scholar]
- 53.May JM, Qu ZC, Mendiratta S. Protection and recycling of alpha-tocopherol in human erythrocytes by intracellular ascorbic acid. Arch Biochem Biophys. 1998;349:281–9. doi: 10.1006/abbi.1997.0473. [DOI] [PubMed] [Google Scholar]
- 54.Lang JK, Gohil K, Packer L. Simultaneous determination of tocopherols, ubiquinols, and ubiquinones in blood, plasma, tissue homogenates, and subcellular fractions. Anal Biochem. 1986;157:106–16. doi: 10.1016/0003-2697(86)90203-4. [DOI] [PubMed] [Google Scholar]
- 55.Roberts LJ, 2nd, Montine TJ, Markesbery WR, et al. Formation of isoprostane-like compounds (neuroprostanes) in vivo from docosahexaenoic acid. J Biol Chem. 1998;273:13605–12. doi: 10.1074/jbc.273.22.13605. [DOI] [PubMed] [Google Scholar]
- 56.Sabharwal AK, May JM. alpha-Lipoic acid and ascorbate prevent LDL oxidation and oxidant stress in endothelial cells. Mol Cell Biochem. 2008;309:125–32. doi: 10.1007/s11010-007-9650-z. [DOI] [PubMed] [Google Scholar]
- 57.Casado A, Encarnacion Lopez-Fernandez M, Concepcion Casado M, de La Torre R. Lipid peroxidation and antioxidant enzyme activities in vascular and Alzheimer dementias. Neurochem Res. 2008;33:450–8. doi: 10.1007/s11064-007-9453-3. [DOI] [PubMed] [Google Scholar]
- 58.Savonenko A, Xu GM, Melnikova T, et al. Episodic-like memory deficits in the APPswe/PS1dE9 mouse model of Alzheimer’s disease: relationships to beta-amyloid deposition and neurotransmitter abnormalities. Neurobiol Dis. 2005;18:602–17. doi: 10.1016/j.nbd.2004.10.022. [DOI] [PubMed] [Google Scholar]
- 59.Ding Y, Qiao A, Wang Z, et al. Retinoic acid attenuates beta-amyloid deposition and rescues memory deficits in an Alzheimer’s disease transgenic mouse model. J Neurosci. 2008;28:11622–34. doi: 10.1523/JNEUROSCI.3153-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Agus DB, Gambhir SS, Pardridge WM, et al. Vitamin C crosses the blood-brain barrier in the oxidized form through the glucose transporters. J Clin Invest. 1997;100:2842–8. doi: 10.1172/JCI119832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Socci DJ, Crandall BM, Arendash GW. Chronic antioxidant treatment improves the cognitive performance of aged rats. Brain Res. 1995;693:88–94. doi: 10.1016/0006-8993(95)00707-w. [DOI] [PubMed] [Google Scholar]
- 62.Sumien N, Forster MJ, Sohal RS. Supplementation with vitamin E fails to attenuate oxidative damage in aged mice. Exp Gerontol. 2003;38:699–704. doi: 10.1016/s0531-5565(03)00068-8. [DOI] [PubMed] [Google Scholar]
- 63.Padayatty SJ, Katz A, Wang Y, et al. Vitamin C as an antioxidant: evaluation of its role in disease prevention. J Am Coll Nutr. 2003;22:18–35. doi: 10.1080/07315724.2003.10719272. [DOI] [PubMed] [Google Scholar]
- 64.Harats D, Chevion S, Nahir M, Norman Y, Sagee O, Berry EM. Citrus fruit supplementation reduces lipoprotein oxidation in young men ingesting a diet high in saturated fat: presumptive evidence for an interaction between vitamins C and E in vivo. Am J Clin Nutr. 1998;67:240–5. doi: 10.1093/ajcn/67.2.240. [DOI] [PubMed] [Google Scholar]
- 65.Upston JM, Terentis AC, Stocker R. Tocopherol-mediated peroxidation of lipoproteins: implications for vitamin E as a potential antiatherogenic supplement. Faseb J. 1999;13:977–94. doi: 10.1096/fasebj.13.9.977. [DOI] [PubMed] [Google Scholar]
- 66.Sumien N, Heinrich KR, Sohal RS, Forster MJ. Short-term vitamin E intake fails to improve cognitive or psychomotor performance of aged mice. Free Radic Biol Med. 2004;36:1424–33. doi: 10.1016/j.freeradbiomed.2004.02.081. [DOI] [PubMed] [Google Scholar]
- 67.Halagappa VK, Guo Z, Pearson M, et al. Intermittent fasting and caloric restriction ameliorate age-related behavioral deficits in the triple-transgenic mouse model of Alzheimer’s disease. Neurobiol Dis. 2007;26:212–20. doi: 10.1016/j.nbd.2006.12.019. [DOI] [PubMed] [Google Scholar]
- 68.Hashimoto T, Watanabe S. Chronic food restriction enhances memory in mice--analysis with matched drive levels. Neuroreport. 2005;16:1129–33. doi: 10.1097/00001756-200507130-00019. [DOI] [PubMed] [Google Scholar]
- 69.Ward WF, Qi W, Van Remmen H, Zackert WE, Roberts LJ, 2nd, Richardson A. Effects of age and caloric restriction on lipid peroxidation: measurement of oxidative stress by F2-isoprostane levels. J Gerontol A Biol Sci Med Sci. 2005;60:847–51. doi: 10.1093/gerona/60.7.847. [DOI] [PubMed] [Google Scholar]
- 70.Arendash GW, Garcia MF, Costa DA, Cracchiolo JR, Wefes IM, Potter H. Environmental enrichment improves cognition in aged Alzheimer’s transgenic mice despite stable beta-amyloid deposition. Neuroreport. 2004;15:1751–4. doi: 10.1097/01.wnr.0000137183.68847.4e. [DOI] [PubMed] [Google Scholar]
- 71.Costa DA, Cracchiolo JR, Bachstetter AD, et al. Enrichment improves cognition in AD mice by amyloid-related and unrelated mechanisms. Neurobiol Aging. 2007;28:831–44. doi: 10.1016/j.neurobiolaging.2006.04.009. [DOI] [PubMed] [Google Scholar]
- 72.Jankowsky JL, Melnikova T, Fadale DJ, et al. Environmental enrichment mitigates cognitive deficits in a mouse model of Alzheimer’s disease. J Neurosci. 2005;25:5217–24. doi: 10.1523/JNEUROSCI.5080-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Hong JH, Kim MJ, Park MR, et al. Effects of vitamin E on oxidative stress and membrane fluidity in brain of streptozotocin-induced diabetic rats. Clin Chim Acta. 2004;340:107–15. doi: 10.1016/j.cccn.2003.10.003. [DOI] [PubMed] [Google Scholar]
- 74.Omoi NO, Arai M, Saito M, et al. Influence of oxidative stress on fusion of pre-synaptic plasma membranes of the rat brain with phosphatidyl choline liposomes, and protective effect of vitamin E. J Nutr Sci Vitaminol (Tokyo) 2006;52:248–55. doi: 10.3177/jnsv.52.248. [DOI] [PubMed] [Google Scholar]
- 75.Castellani RJ, Lee HG, Zhu X, Nunomura A, Perry G, Smith MA. Neuropathology of Alzheimer disease: pathognomonic but not pathogenic. Acta Neuropathol. 2006;111:503–9. doi: 10.1007/s00401-006-0071-y. [DOI] [PubMed] [Google Scholar]
- 76.Lee HG, Zhu X, Castellani RJ, Nunomura A, Perry G, Smith MA. Amyloid-beta in Alzheimer disease: the null versus the alternate hypotheses. J Pharmacol Exp Ther. 2007;321:823–9. doi: 10.1124/jpet.106.114009. [DOI] [PubMed] [Google Scholar]
- 77.Fotuhi M, Zandi PP, Hayden KM, et al. Better cognitive performance in elderly taking antioxidant vitamins E and C supplements in combination with nonsteroidal anti-inflammatory drugs: the Cache County Study. Alzheimers Dement. 2008;4:223–7. doi: 10.1016/j.jalz.2008.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Wilder J. Stimulus and Response: The Law Of Initial Value. Bristol, England: Wright; 1967. [Google Scholar]
- 79.McDonald SR, Sohal RS, Forster MJ. Concurrent administration of coenzyme Q10 and alpha-tocopherol improves learning in aged mice. Free Radic Biol Med. 2005;38:729–36. doi: 10.1016/j.freeradbiomed.2004.11.014. [DOI] [PubMed] [Google Scholar]
- 80.Stackman RW, Eckenstein F, Frei B, Kulhanek D, Nowlin J, Quinn JF. Prevention of age-related spatial memory deficits in a transgenic mouse model of Alzheimer’s disease by chronic Ginkgo biloba treatment. Exp Neurol. 2003;184:510–20. doi: 10.1016/s0014-4886(03)00399-6. [DOI] [PubMed] [Google Scholar]
- 81.Jensen MT, Mottin MD, Cracchiolo JR, Leighty RE, Arendash GW. Lifelong immunization with human beta-amyloid (1-42) protects Alzheimer’s transgenic mice against cognitive impairment throughout aging. Neuroscience. 2005;130:667–84. doi: 10.1016/j.neuroscience.2004.09.055. [DOI] [PubMed] [Google Scholar]
- 82.Leighty RE, Nilsson LN, Potter H, et al. Use of multimetric statistical analysis to characterize and discriminate between the performance of four Alzheimer’s transgenic mouse lines differing in Abeta deposition. Behav Brain Res. 2004;153:107–21. doi: 10.1016/j.bbr.2003.11.004. [DOI] [PubMed] [Google Scholar]
- 83.McDonald MP, Dahl EE, Overmier JB, Mantyh P, Cleary J. Effects of an exogenous beta-amyloid peptide on retention for spatial learning. Behav Neural Biol. 1994;62:60–7. doi: 10.1016/s0163-1047(05)80059-7. [DOI] [PubMed] [Google Scholar]
- 84.Sweeney WA, Luedtke J, McDonald MP, Overmier JB. Intrahippocampal injections of exogenous beta-amyloid induce postdelay errors in an eight-arm radial maze. Neurobiol Learn Mem. 1997;68:97–101. doi: 10.1006/nlme.1997.3770. [DOI] [PubMed] [Google Scholar]