

Summary
Na+/I− symporter (NIS)-mediated active accumulation of I− in thyrocytes is a key step in the biosynthesis of the iodine-containing thyroid hormones T3 and T4. Several NIS mutants have been identified as a cause of congenital I− transport defect (ITD), and their investigation has yielded valuable mechanistic information on NIS. Here we report novel findings derived from the thorough characterization of the ITD-causing mutation R124H, located in the second intracellular loop (IL-2). R124H NIS is incompletely glycosylated and colocalizes with endoplasmic reticulum (ER)-resident protein markers. As a result, R124H NIS is not targeted to the plasma membrane and therefore does not mediate any I− transport in transfected COS-7 cells. Strikingly, however, the mutant is intrinsically active, as revealed by its ability to mediate I− transport in membrane vesicles. Of all the amino acid substitutions we carried out at position 124 (K, D, E, A, W, N and Q), only Gln restored targeting of NIS to the plasma membrane and NIS activity, suggesting a key structural role for the δ-amino group of R124 in the transporter's maturation and cell surface targeting. Using our NIS homology model based on the structure of the Vibrio parahaemolyticus Na+/galactose symporter, we propose an interaction between the δ-amino group of either R or Q124 and the thiol group of C440, located in IL-6. We conclude that the interaction between IL-2 and IL-6 is critical for the local folding required for NIS maturation and plasma membrane trafficking.
Key words: Congenital iodide transport defect, Na+/I− symporter (NIS), Plasma membrane targeting, Impaired intracellular trafficking, Membrane vesicles
Introduction
The Na+/I− symporter (NIS) is a plasma membrane glycoprotein that mediates the active transport of I− into the thyroid gland and other tissues, such as salivary glands, gastric mucosa, small intestine and lactating breast (Altorjay et al., 2007; Nicola et al., 2009; Nicola et al., 2012; Tazebay et al., 2000; Wapnir et al., 2003). In the thyroid, I− accumulation is the first step in the biosynthesis of the iodine-containing hormones T3 and T4 (triiodothyronine and thyroxine, respectively).
Our group cloned NIS and has since characterized it extensively (Dai et al., 1996; Eskandari et al., 1997; Levy et al., 1997; Levy et al., 1998a; Riedel et al., 2001). Our experimentally tested secondary structure model for NIS shows a protein with 13 transmembrane segments (TMS) (Fig. 1A), an extracellularly facing N-terminus, and an intracellularly facing carboxy terminus (Levy et al., 1997; Levy et al., 1998a). Although NIS is N-glycosylated at three positions (Fig. 1A), glycosylation is not essential for stability, targeting or activity of NIS (Levy et al., 1998a). NIS activity results from coupling the inward translocation of Na+ down its electrochemical gradient to the inward transport of I− against its electrochemical gradient, with an electrogenic 2 Na+:1 I− stoichiometry (Eskandari et al., 1997). Significantly, NIS translocates different substrates with different stoichiometries, as NIS-mediated transport of perrhenate (ReO4−) or the environmental pollutant perchlorate (ClO4−) is electroneutral (1 Na+:1 ReO4−/ClO4−) (Dohán et al., 2007).
Fig. 1.

Characterization of expression and activity of R124H Na+/I− symporter. (A) Na+/I− symporter (NIS) secondary structure model. Cylinders represent the 13 transmembrane segments (TMS), in Roman numerals; branches indicate N-linked glycosylation sites (N225, N489 and N502). The N-terminus faces the extracellular milieu and the C-terminus the cytosol. The iodide transport defect (ITD)-causing NIS mutant R124H is indicated in intracellular loop (IL)-2. IL-6 is also labeled. (B) Steady-state I− transport assays in non-transfected (NT), wild-type (WT) or R124H NIS cDNA transiently transfected COS-7 cells. Cells were incubated with 20 µM I− in the absence or presence of 80 µM ClO4−. The results are expressed in pmol I−/µg of DNA ± s.d. Values are representative of at least five different experiments; each experiment was performed in triplicate. (C) Immunofluorescence analysis of permeabilized WT and R124H NIS-transfected COS-7 cells. Immunostaining was performed with anti-human NIS and anti-Na+/K+ ATPase antibodies (Abs), followed by anti-rabbit Alexa 488- and anti-mouse Alexa 555-conjugated Abs. Nuclei were stained with DRAQ5 dye (blue). The overlay of the two images is shown (Merge). Scale bars, 20 µm. (D) Flow cytometry analysis of total and plasma membrane (PM) NIS expression in permeabilized (P) and non-permeabilized (NP) COS-7 cells non-transfected or transfected with WT or R124H NIS. Cells were stained with anti-NIS VJ1 Ab, followed by Alexa-488-conjugated anti-mouse Ab. (E) Immunoblot analysis of cell surface-biotinylated proteins from non-transfected and WT- or R124H NIS-transfected COS-7 cells, performed with anti-human NIS Ab (upper panel). The plasma membrane marker Na+/K+ ATPase was used as a positive control (lower panel). NS, non-specific. (F) Immunoblot of membrane fractions from COS-7 cells transfected with WT or R124H NIS cDNA with anti-human NIS Ab. Letters on the right side of the blot indicate the relative electrophoretic mobilities of the corresponding NIS bands [A: non-glycosylated, B: partially glycosylated, C: fully glycosylated (mature), BB: dimer of the partially glycosylated, and CC: dimer of the mature polypeptide]. α-Tubulin was used as a loading control.
Iodide deficiency disorders (IDDs) remain a major health issue around the world as a direct result of insufficient dietary I− intake. Indeed, they are the most common preventable cause of mental retardation. Because I− is very scarce in the environment, normal thyroid physiology relies on adequate dietary I− intake and on proper NIS functioning (Dohán et al., 2003; Zimmermann, 2009). Fourteen cases of congenital hypothyroidism due to an I− transport defect (ITD) have been reported to date (Nicola et al., 2011; Spitzweg and Morris, 2010). ITD is an uncommon autosomal-recessive condition caused by naturally occurring mutations in the SLC5A5 gene, which encodes NIS.
Studies of different ITD-causing NIS mutations have uncovered key structure/function information on NIS (De la Vieja et al., 2004; De la Vieja et al., 2005; De la Vieja et al., 2007; Dohán et al., 2002; Levy et al., 1998b; Paroder-Belenitsky et al., 2011; Reed-Tsur et al., 2008). For instance, the analysis of the T354P mutation revealed that the hydroxyl group at the β-carbon of the residue at position 354 is essential for NIS function and that other β-hydroxyl–containing residues in TMS IX are involved in Na+ binding/translocation (De la Vieja et al., 2007; Levy et al., 1998b). Similarly, the investigation of the G93R mutation led to the discovery that position 93 in TMS III is critical for substrate selectivity and stoichiometry. Furthermore, changes from an outwardly to an inwardly open conformation during the transport cycle use G93 as a pivot (Paroder-Belenitsky et al., 2011).
Szinnai et al. identified the ITD-causing mutant R124H NIS in a newborn carrying a homozygous transition at nucleotide 718 in exon 2 and reported that, when transiently transfected into COS-7 cells, the R124H NIS protein was inactive, even though it was properly targeted to the plasma membrane (Szinnai et al., 2006). Here, however, we conducted a thorough molecular analysis of the R124H NIS mutant, in which we observed, by immunofluorescence, flow cytometry and cell surface biotinylation, that R124H NIS is retained intracellularly and therefore does not reach the cell surface. Strikingly, R124H NIS is intrinsically active, as determined by I− transport assays in membrane vesicles. Of Lys, Ala, Trp, Asn, Asp, Glu or Gln, the only amino acid that restored NIS function when engineered at position 124 was Gln, suggesting that the presence of the δ-amino group at position 124 is critical for proper NIS maturation and plasma membrane trafficking. Using our NIS homology model based on the structure of the Vibrio parahaemolyticus Na+/galactose symporter (Paroder-Belenitsky et al., 2011), we propose an interaction between the δ-amino group of either R- or Q124 and the thiol group of C440. These data underscore the structural role played by residue R124 in bridging together IL-2 and 6, allowing a local folding required for NIS maturation and trafficking to the plasma membrane.
Results
R124H NIS is intracellularly retained
According to our experimentally tested NIS secondary structure model, Arg-124 is located in IL-2 (Fig. 1A). R124 is conserved in NIS molecules from all species cloned to date and in 11 out of 12 members of solute carrier family 5 (SLC5A) (see Fig. 2A). COS-7 cells were transfected with wild-type (WT) or R124H NIS cDNA and assayed for I− transport activity. At steady state, no perchlorate (ClO4−)-inhibitable I− accumulation was observed in R124H NIS-transfected cells, in contrast to control WT NIS-expressing cells (Fig. 1B) (Dai et al., 1996; De la Vieja et al., 2004; Dohán et al., 2002; Levy et al., 1997; Levy et al., 1998b). In indirect immunofluorescence experiments, R124H NIS-expressing cells showed intracellular staining, which did not co-localize with the plasma membrane marker Na+/K+ ATPase (Fig. 1C), unlike WT NIS, which partially does (Fig. 1C) (De la Vieja et al., 2004; De la Vieja et al., 2005; Paroder-Belenitsky et al., 2011; Reed-Tsur et al., 2008). Additionally, immunofluorescence performed under non-permeabilized conditions to ascertain NIS expression only at the plasma membrane showed immunoreactivity in cells expressing WT NIS but not in R124H NIS-transfected cells (supplementary material Fig. S1). Consistently with our immunofluorescence data, flow cytometry (FACS) analysis showed that R124H NIS was not targeted to the plasma membrane (Fig. 1D). When FACS was performed under permeabilized conditions to assess total NIS protein expression, we observed positive staining in both WT- and R124H-expressing cells. In contrast, when the experiments were performed in non-permeabilized cells, using an anti-NIS VJ1 Ab directed against an extracellularly facing conformational epitope, positive staining was observed exclusively with WT NIS (Fig. 1D). Cell surface biotinylation showed the reported ∼100 kDa mature NIS polypeptide at the plasma membrane (De la Vieja et al., 2005; Reed-Tsur et al., 2008), whereas no biotinylated NIS polypeptides were detected in R124H NIS-transfected cells (Fig. 1E), in agreement with the immunofluorescence and FACS data, indicating that R124H NIS did not reach the plasma membrane. Membrane fractions from cells expressing either WT or R124H NIS were subjected to immunoblot analysis. As reported (De la Vieja et al., 2005), the electrophoretic pattern of WT NIS comprises non-glycosylated (∼55 kDa, band A), partially glycosylated (∼60 kDa, band B), dimer of the partially glycosylated (∼120 kDa, band BB), mature or fully glycosylated (∼100 kDa, band C), and dimer of the mature (∼200 kDa, band CC) polypeptides. The mature ∼100 kDa NIS polypeptide was absent in R124H NIS-expressing cells, suggesting that R124H NIS was not fully processed (Fig. 1F).
Fig. 2.
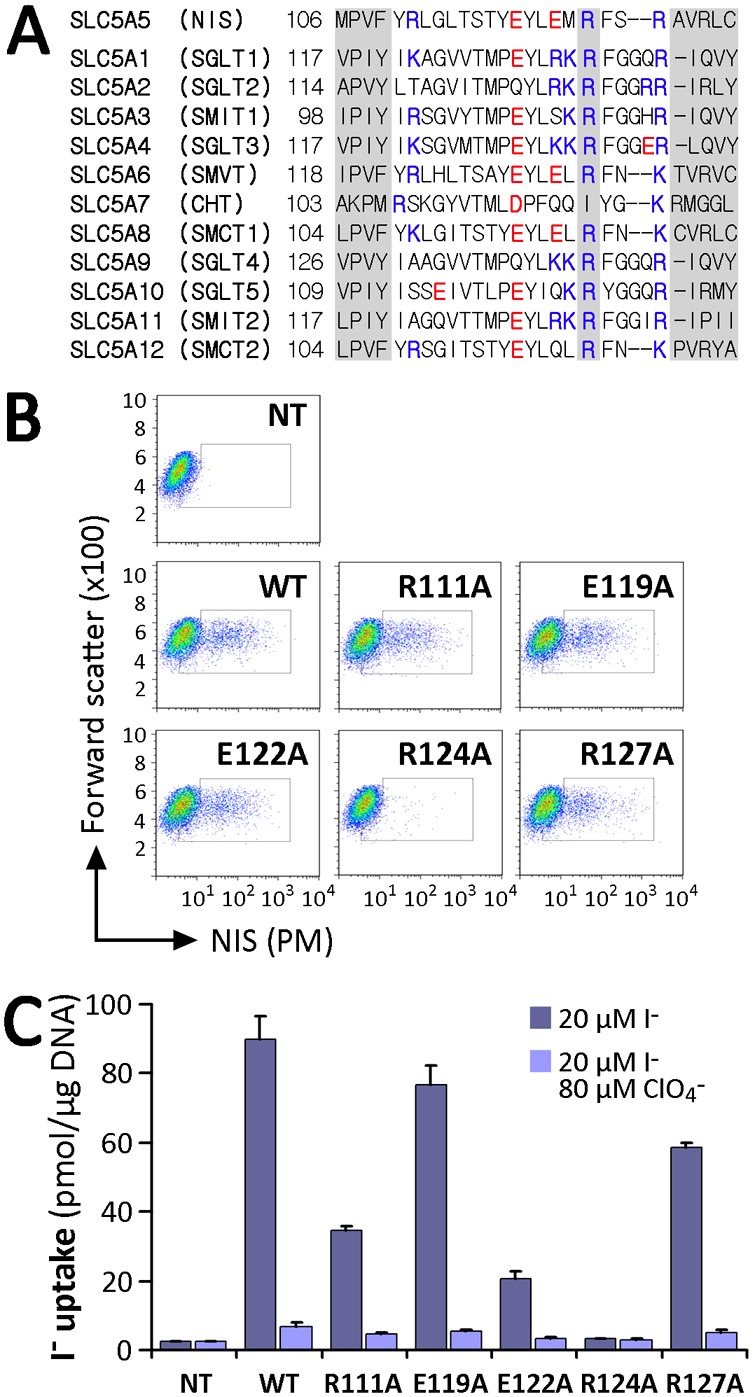
Analysis of charged residues within IL-2 of NIS. (A) Sequence alignment of IL-2 of SLC5A family members. The R124 position in NIS and corresponding residues in other members are indicated. Positively charged residues are indicated in blue and negatively charged ones in red. Left and right shaded sequences represent parts of TMS III and IV, respectively, according to the crystal structure of vSGLT. (B) Flow cytometry of non-transfected (NT) COS-7 cells or COS-7 cells transfected with WT or IL-2 NIS mutants under non-permeabilized conditions to assess NIS expression at the plasma membrane (PM). Staining was done using anti-NIS VJ1 Ab, followed by Alexa-488-conjugated anti-mouse Ab. (C) Steady-state I− transport in WT or R111A, E119A, E122A, R124A and R127A NIS-transfected COS-7 cells. Cells were incubated with 20 µM I− in the absence or presence of 80 µM ClO4−. Values are expressed in pmol I−/µg of DNA ± s.d. and are representative of three different experiments; each experiment was performed in triplicate.
R124 is the only charged residue in IL-2 that is essential for NIS targeting to the cell surface
Intracellularly facing charged residues have been shown to be important in trafficking of membrane proteins (Kasahara et al., 2004; Li et al., 2012; Wolff et al., 2010). IL-2 contains three basic (R111, R124, R127) and two acid residues (E119, E122). Besides R124, which is highly conserved, the other four of these charged residues are conserved to different extents among members of the SLC5 family (Fig. 2A). To examine the role of these residues, we engineered Ala substitutions at each position. In contrast to the results obtained with R124A, we observed that all of the other NIS mutants were properly targeted to the plasma membrane (Fig. 2B) and active, although to different extents than WT NIS (Fig. 2C). Therefore, R124 is the only charged residue in IL-2 that is critical for proper NIS maturation and plasma membrane expression.
R124H NIS exhibits incomplete maturation
We generated permanently transfected MDCK-II cells expressing either WT or R124H NIS. Cells expressing R124H NIS did not transport I− (supplementary material Fig. S2A), in contrast to those expressing WT NIS. R124H NIS was expressed at levels comparable to those of WT NIS, even though fewer cells expressed R124H than WT NIS (supplementary material Fig. S2B, total). Clearly, R124H NIS did not reach the plasma membrane, as shown by FACS under non-permeabilized conditions (supplementary material Fig. 2B, PM). Therefore, the results in MDCK-II cells recapitulate those obtained in COS-7 cells (Fig. 1B,D). To investigate the impaired plasma membrane targeting of R124H NIS, we used endoglycosidase H (Endo H) treatment. As proteins that have matured beyond the medial Golgi are Endo H resistant, sensitivity to Endo H treatment indicates that a protein has not been processed past the medial-Golgi. Membrane fractions from MDCK-II cells expressing WT or R124H NIS were digested with Endo H and subjected to western blot analysis. The electrophoretic pattern of WT NIS comprises partially glycosylated (∼60 kDa, band A) and mature or fully glycosylated (∼100 kDa, band B) polypeptides (Fig. 3A) (Levy et al., 1997; Riedel et al., 2001). As observed in COS-7 cells (Fig. 1F), the mature NIS polypeptide was also absent in MDCK-II cells expressing R124H NIS. Endo H treatment caused the disappearance of partially glycosylated polypeptides from both WT and R124H NIS. However, as reported, the mature WT NIS polypeptide was Endo H resistant (Fig. 3A) (De la Vieja et al., 2005), whereas PNGase F treatment of WT NIS resulted in the disappearance of both the fully and partially glycosylated polypeptides, with a concomitant increase in the non-glycosylated polypeptide. Similarly, PNGase F digestion of R124H NIS caused the disappearance of the partially glycosylated band and an increase in the non-glycosylated polypeptide (supplementary material Fig. S3).
Fig. 3.
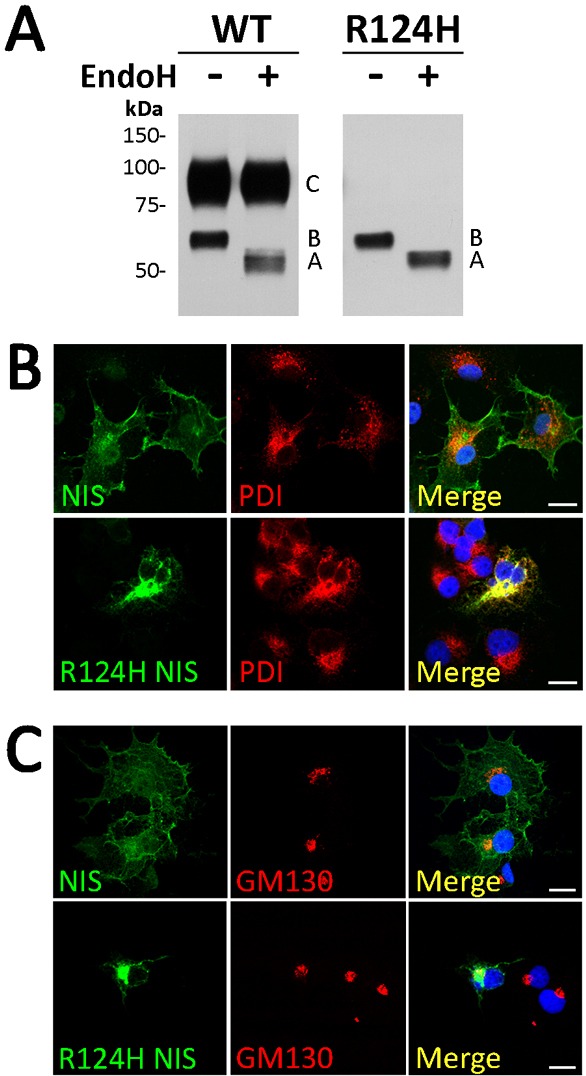
R124H NIS is incompletely glycosylated and intracellularly retained. (A) Immunoblot of membrane fractions treated with Endo H. Membrane proteins (5 µg) from MDCK-II cells permanently expressing WT or R124H NIS were treated (+) or not (−) with Endo H. Samples were resolved by SDS-PAGE and immunoblotted with polyclonal anti-human NIS Ab. Letters on the right side of the blot indicate the relative electrophoretic mobility of the corresponding NIS polypeptides [A: non-glycosylated, B: partially glycosylated, and C: fully glycosylated (mature)]. The blot shown is representative of two independent experiments. (B,C) Immunofluorescence colocalization experiments. Permeabilized WT or R124H NIS-transfected COS-7 cells were incubated with polyclonal anti-human NIS Ab and either anti-protein disulfide isomerase (PDI) or anti-GM130 Abs, followed by Alexa 488- and Alexa 555-conjugated goat anti-rabbit and anti-mouse Abs. Nuclei stained with DRAQ5 are shown in blue. Overlay images (Merge) are shown. Scale bars, 20 µm.
These findings show that R124H NIS is retained in intracellular compartments prior to medial-Golgi. To extend these results, we performed co-localization experiments of NIS expression with endoplasmic reticulum (ER) and Golgi resident markers. R124H NIS co-localized with the ER-residents proteins protein disulfide isomerase (PDI) (Fig. 3B) and calnexin (data not shown), but not with the cis-Golgi marker GM130 (Fig. 3C). These results, taken together, support the conclusion that R124H NIS is retained in the ER, that none of it matures beyond the medial-Golgi, and that the protein's intracellular retention and its consequent failure to reach the cell surface account for the absence of NIS activity in R124H NIS-expressing cells.
Intracellularly retained R124H NIS is intrinsically active
In intact cells, I− transport is only observed when NIS is properly targeted to the plasma membrane (Kaminsky et al., 1994). R124H NIS is not active in transfected COS-7 cells ultimately because it is not targeted to the cell surface. However, the lack of I− transport activity in these cells does not necessarily mean that R124H NIS is intrinsically inactive. To examine whether or not R124H NIS is intrinsically functional, we prepared membrane vesicles (MVs) from non-transfected cells or cells expressing either WT or R124H NIS. As expected, MVs from WT NIS-expressing cells exhibited ClO4−-inhibited I− accumulation (De la Vieja et al., 2005). Strikingly, MVs from R124H NIS-expressing cells also displayed ClO4−-sensitive I− transport, albeit at a modest level (Fig. 4), indicating that, although trafficking of R124H NIS to the cell surface is impaired, the protein is functional.
Fig. 4.
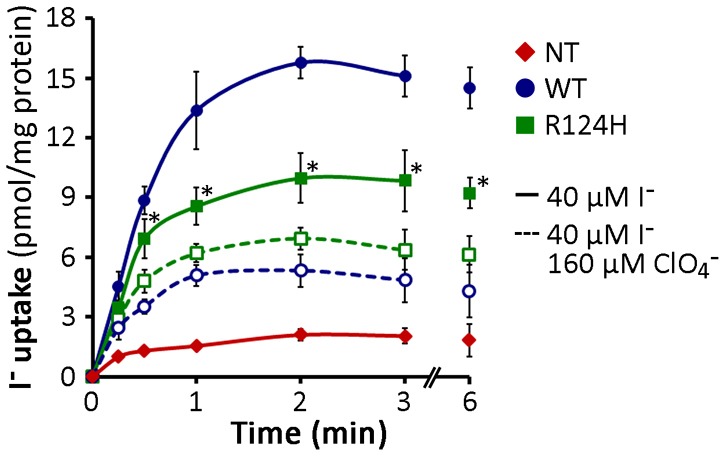
R124H NIS is intrinsically active. I− uptake in membrane vesicles (MV) prepared from non-transfected (NT) and either WT- or R124H NIS-expressing COS-7 cells. Membrane aliquots (100 µg) were assayed for I− uptake at the indicated time points by incubation at room temperature in an equal volume of uptake buffer containing 40 µM I−. Incubations were performed in the presence or absence of 160 µM ClO4−. The results are expressed as pmol I−/mg protein ± s.d.; each experimental point was performed in triplicate. The experiment shown is representative of three independent experiments. *P<0.05 versus same condition in the presence of ClO4− (Student's t-test).
Gln at position 124 restores NIS activity
To investigate whether R124H NIS was inactive because of the absence of the permanent positive charge provided by Arg, we engineered R124K NIS. Steady-state I− transport assays performed in transiently transfected COS-7 cells showed that R124K was inactive (Fig. 5A). Like R124H NIS, R124K NIS was well expressed and intracellularly retained, as was evident from FACS analysis (Fig. 5B). Thus, the lack of the positive charge of Arg was not the cause of the impaired maturation of R124H NIS. We further tested whether negatively charged residues (i.e., Asp and Glu) could restore NIS plasma membrane targeting. R124D and E did not display any significant I− uptake (Fig. 5A) and were not targeted to the plasma membrane (Fig. 5B). In addition, the substitution of a large (Trp) neutral residue at position 124 resulted in the absence of I− transport and no detectable plasma membrane expression, as assessed by FACS (Fig. 5A,B).
Fig. 5.
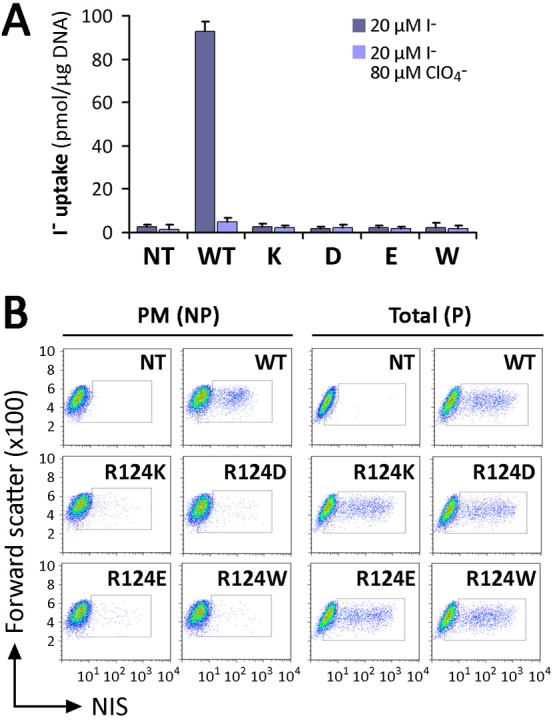
Substitutions with charged residues at position 124 did not recover NIS plasma membrane targeting. (A) Steady-state I− transport in WT and R124K, D, E or W NIS-transfected COS-7 cells. Cells were incubated with 20 µM I− in the absence or presence of 80 µM ClO4−. The results are expressed in pmol I−/µg of DNA ± s.d. Values are representative of at least five different experiments; each experiment was performed in triplicate. (B) Analysis of NIS expression (total) and plasma membrane (PM) targeting by flow cytometry. Non-transfected (NT) or WT and R124K, D, E or W NIS-transfected COS-7 cells were stained under non-permeabilized (NP) or permeabilized (P) conditions with anti-NIS VJ1 Ab, followed by Alexa-488-conjugated anti-mouse Ab.
An important feature of the Arg side chain is its terminal δ-guanidinium group. The nitrogen atoms of the guanidinium group are capable of forming hydrogen bonds or salt bridges, which might be important for the proper secondary and/or tertiary structure of NIS (Borders et al., 1994). Therefore, we replaced R124 with Asn or Gln. Like R124H, R124N NIS did not mature or reach the cell surface. In contrast, R124Q NIS, remarkably, did (Fig. 6A; supplementary material Fig. S4). Furthermore, in steady-state I− uptake assays, R124Q displayed ClO4−-sensitive I− transport, although to a lesser extent than WT NIS (Fig. 6B). In kinetic experiments, no significant variation in the Km for I− was observed in cells expressing R124Q with respect to WT NIS [Km(R124Q): 7.2±2.3 µM; Km(WT): 8.9±1.6 µM] (Fig. 6C), whereas the Vmax was lower in R124Q than in WT NIS [Vmax(R124Q): 9.9±0.7 µM; Vmax(WT): 35.0±2.9 µM] (Fig. 6C). These results suggest that the δ-amino group of Arg and Gln may play a role in NIS folding. Because the replacement of Arg by Gln preserved NIS targeting to the cell surface, we conclude that a guanidinium group at position 124, rather than a positive charge, is required.
Fig. 6.
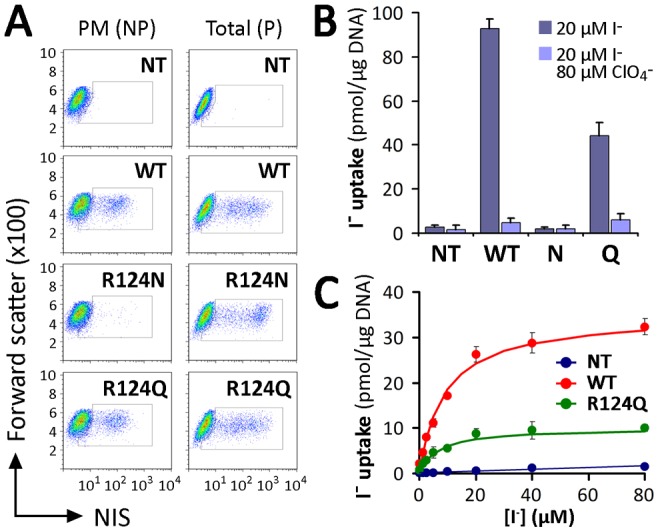
Substitution with Gln at position 124 restores NIS plasma membrane targeting. (A) Flow cytometry analysis of NIS expression in non-transfected (NT), WT-, R124N or Q NIS-transfected COS-7 cells. Staining was performed under non-permeabilized (NP) or permeabilized (P) conditions with anti-NIS VJ1 Ab, followed by Alexa-488-conjugated anti-mouse Ab. (B) Steady-state I− transport in WT or R124Q and N NIS-transfected COS-7 cells. Cells were incubated with 20 µM I− in the absence or presence of 80 µM ClO4−. The results are expressed in pmol I−/µg of DNA ± s.d. Values are representative of three different experiments; each experiment was performed in triplicate. (C) Initial rates (2 min time points) of I− uptake were determined at the indicated concentrations of I−. Calculated curves (smooth lines) were generated using the equation v = (Vmax*[I−])/(Km+[I−]) adjusted to consider background data obtained with non-transfected cells. Data shown are from a representative experiment out of three independent experiments.
The interaction between R-124 and C-440 may be key for proper NIS folding
The only protein belonging to the SLC5 family whose crystal structure has been determined is the Vibrio parahaemolyticus Na+/galactose symporter (vSGLT) (Faham et al., 2008). Using the structure of this protein as a template, we have generated a homology model for NIS (Paroder-Belenitsky et al., 2011). To understand why a δ-amino group is required at position 124 of NIS, we searched for potential interaction partners in our NIS homology model. We observed a possible interaction between the guanidinium group of R124 and the backbones of G434 and L437, and identified C440 as a residue that potentially interacts with the δ-amino group of R- or Q124 NIS (Fig. 7A). To test this hypothesis, we engineered a C440A mutant in the background of WT or R124Q NIS and functionally tested these proteins in COS-7 cells. In steady-state I− uptake assays, C440A NIS displayed ClO4−-sensitive I− transport at the same level as WT NIS (Fig. 7B). However, in contrast to R124Q NIS, the double mutant R124Q/C440A did not exhibit any significant I− transport. Unlike C440A, which matured and reached the cell surface, the double mutant R124Q/C440A NIS did not (Fig. 7C). Additionally, MVs generated from transiently transfected COS-7 cells expressing R124Q/C440A did not display detectable I− uptake (not shown), indicating that either the protein was intrinsically inactive or its activity was so low as to be undetectable.
Fig. 7.

Proposed intramolecular interactions between IL-2 and IL-6. (A) Close-up of NIS homology model based on the structure of vSGLT. C440 interacts with the δ-amino group present in Arg and Gln. R124, but not Q124, interacts with the backbones of G434 and L437. IL-2 and IL-6 are represented in pink and green, respectively, as in Fig. 1A. Potential interactions are indicated by dotted lines. Structural representations were generated using PyMol (Schrödinger, Portland, OR, USA). (B) Steady-state I− transport in WT and NIS mutant-transfected COS-7 cells. Cells were incubated with 20 µM I− in the absence or presence of 80 µM ClO4−. The results are expressed in pmol I−/µg of DNA ± s.d. Values are representative of three different experiments; each experiment was performed in triplicate. (C) Analysis of plasma membrane (PM) and total (P) NIS expression by FACS. COS-7 cells transiently expressing WT and mutant NIS constructs were stained under permeabilized (P) or non-permeabilized (NP) conditions with anti-NIS VJ1 Ab, followed by Alexa-488-conjugated anti-mouse Ab.
Having identified a mutant NIS protein that is intrinsically active but is retained intracellularly (R124H) and a mutant that is both inactive and retained intracellularly (R124Q/C440A), we sought to investigate whether other alterations in the IL-2/IL-6 interaction might result in molecules that are inactive but properly targeted to the plasma membrane. R124Q/C440S NIS turned out to be just such a molecule (Fig. 7B,C), suggesting that the hydrogen bond formed by the thiol group of C440 may be mimicked by S440 to achieve the local conformation required for targeting to the cell surface, but not mimicked closely enough to preserve all the conformations involved in the transport cycle.
Discussion
The detailed molecular analysis of several ITD-causing NIS mutants has yielded key mechanistic information on NIS structure/function, such as the following findings: a β-hydroxyl group at residue 354 is essential for NIS function (De la Vieja et al., 2007; Levy et al., 1998b); the presence of a charge or a large side-chain at position 395 interferes with NIS function (Dohán et al., 2002); any charged residue other than Glu at position 267 renders NIS inactive (De la Vieja et al., 2004), and a β-branched residue is required at position 59 for NIS to be active (Reed-Tsur et al., 2008). Furthermore, we have recently reported that position 93 is critical for substrate selectivity and stoichiometry (Paroder-Belenitsky et al., 2011). Five NIS mutants (V59E, G93R, T354P, G395R, and Q267E) have been shown to be fully glycosylated and properly targeted to the plasma membrane (De la Vieja et al., 2004; De la Vieja et al., 2007; Dohán et al., 2002; Levy et al., 1998b; Paroder-Belenitsky et al., 2011; Reed-Tsur et al., 2008). G543E, by contrast, was the first NIS mutant reported to mature only partially and be retained intracellularly rather than being targeted to the cell surface (De la Vieja et al., 2005).
Whereas Szinnai et al. (Szinnai et al., 2006) reported that R124H NIS, when transfected into COS-7 cells, reached the plasma membrane but was inactive, we here conclusively show, instead, that R124H NIS is not expressed at the plasma membrane. Only the immaturely glycosylated ∼60 kDa R124H NIS and its ∼120 kDa dimer were detected by immunoblot (Fig. 1F, Fig. 3A), species that, upon Endo H treatment, disappeared with a simultaneous increase in the non-glycosylated ∼50 kDa species (Fig. 3A). The subcellular localization of R124H was thoroughly investigated by immunofluorescence and FACS analysis. We showed that R124H NIS was detectable as intracellular staining only in permeabilized cells but not in non-permeabilized cells (Fig. 1C,D; supplementary material Figs S1, S2). In addition, cell surface biotinylation experiments demonstrated that no R124H NIS molecules reached the plasma membrane (Fig. 1E). Furthermore, as R124H NIS is Endo H sensitive, this mutant is retained at some point before the medial-Golgi, the site where Endo H resistance is conferred. Colocalization studies conducted by confocal microscopy with intracellular organelle markers established that R124H NIS is primarily retained in the ER (Fig. 3B). Incomplete glycosylation of R124H NIS is unlikely to be the direct cause of the protein's intracellular retention, given our previous demonstration that glycosylation is not essential for NIS plasma membrane localization or transport activity (Levy et al., 1998a). When all three glycosylated Asp residues were replaced with Gln, non-glycosylated NIS was still targeted to the cell surface and transported I− with a Km for I− very similar to that of WT NIS (Levy et al., 1998a). Strikingly, when assayed in membrane vesicles, R124H NIS displayed ClO4−-sensitive I− transport, albeit at lower levels than WT NIS, indicating that R124H NIS, while retained intracellularly in intact cells, remains intrinsically active.
During biosynthesis, integral plasma membrane proteins associate with molecular chaperones that facilitate their folding and keep them in the ER until they reach their proper conformation (Hartl et al., 2011). This quality control mechanism ensures that only properly folded proteins are delivered to their final destinations, thereby retaining and degrading misfolded polypeptides without preventing the export of properly folded proteins. However, several human disease–related mutant proteins with varying degrees of activity are retained in the ER, such as Duox2, CFTR, aquaporin-2, angiotensin I–converting enzyme, the V2 vasopressin receptor, the sulfonylurea receptor, the HERG K+ channel, the melanocortin-4 receptor and the luteinizing hormone G protein-coupled receptor (Danilov et al., 2010; Grasberger et al., 2007; Li et al., 1993; Meimaridou et al., 2011; Morello et al., 2000; Newton et al., 2011; Partridge et al., 2001; Tamarappoo and Verkman, 1998; Zhou et al., 1999). Most ITD-causing NIS mutants are, like WT NIS, targeted to the plasma membrane. R124H NIS, on the other hand, is the first NIS mutant to be identified that, despite being active, does not leave the ER, causing congenital hypothyroidism.
R124 is located in IL-2 between TMS III and IV (Fig. 1A). Interestingly, R124 is highly conserved among members of solute carrier family 5 (SLC5A), unlike the many other charged residues in this loop, which are conserved to different extents (Fig. 2A). The only charged residue in this loop that proved crucial for targeting of NIS to the plasma membrane was R124 (Fig. 2B). However, a charged residue at position 124 is not a requirement for proper NIS maturation, as Lys, Glu, or Asp at this position did not restore plasma membrane NIS targeting (Fig. 5B). In addition, of all the amino acid substitutions at position 124 (K, D, E, A, W, N and Q), only Gln rescued NIS cell surface targeting and activity (Fig. 6), suggesting a critical role for the δ-amino group of R124. Gln has a δ-amino group that can be oriented to form a hydrogen bond equivalent to that formed by Arg. Indeed, analysis of Arg substitutions in the protein database indicates that Gln is often found in place of Arg, as frequently as Ile in place of Met, Trp in place of Val or Phe, and Thr or Ala in place of Ser. Only Lys in place of Arg is found more often (Henikoff and Henikoff, 1992).
Mutations in other members of the SLC5A family of transporters, such as the Na+/glucose symporters SLC5A1 (SGLT1) and SLC5A2 (SGLT2), also affect protein trafficking (Martín et al., 1997; Martín et al., 1996). However, the mechanisms that regulate the targeting of these transporters to the plasma membrane remain unknown (van den Heuvel et al., 2002; Wright et al., 2011). As previously mentioned, the R124 residue of NIS protein is well conserved among the members of the SLC5A family. Underscoring the importance of this Arg residue, autosomal recessive glucose-galactose malabsorption and renal glucosuria are caused by the inactivating mutations R135W in SGLT1 and R132H in SGLT2, respectively (see Fig. 2A) (Calado et al., 2006; Martín et al., 1996), pointing to a key role for this residue in SLC5A transporters. As in our own observations with R124H NIS, R135W SGLT1 is expressed to levels similar to those of WT SGLT1, although the mutant protein is retained in the ER and only exhibits core glycosylation (Wright et al., 2002).
Chemical chaperones have been shown to reverse the cellular mislocalization or misfolding of certain plasma membrane proteins, such as ΔF508 CFTR, the most common cystic fibrosis-causing mutant, and aquaporin-2, a molecule associated with nephrogenic diabetes insipidus (Fischer et al., 2001; Sato et al., 1996; Tamarappoo and Verkman, 1998; Zeitlin, 1999). Therefore, we examined the effect on R124H NIS of several treatments that have been effective at promoting cell surface targeting of other intracellularly retained plasma membrane proteins. None of these treatments [glycerol, DMSO, trimethylamine-N-oxide (TMAO), thapsigargin, the proteasome inhibitor MG101, or low temperature (27°C)] increased the targeting of R124H NIS to the plasma membrane (data not shown). The inhibition of protein synthesis has been reported to increase plasma membrane targeting of pendrin, a protein retained in the ER in some patients with Pendred syndrome (Shepshelovich et al., 2005). However, treatment of R124H NIS-expressing COS-7 cells with cycloheximide or puromycin did not increase cell surface expression of R124H NIS either (data not shown). All these treatments also failed to target G543E NIS to the plasma membrane (De la Vieja et al., 2005), suggesting that the mechanism for maturation and trafficking of NIS may be different from those of other proteins.
Our NIS homology model based on the structure of the bacterial Na+/galactose symporter (vSGLT) (Paroder-Belenitsky et al., 2011), predicts a hydrogen bond between the δ-amino group of R124 in IL-2 and the thiol group of C440 in IL-6. If this amino group is involved in a hydrogen bond necessary for the correct folding of NIS, it should be possible to replace R124 with a residue that contains a hydrogen-bond-donor group at the δ-position of its side-chain, without losing plasma membrane targeting or functionality of the protein. As predicted, R124Q was the only substitution at position 124 that resulted in NIS molecules that were properly targeted to the cell surface and functional (Fig. 6). We therefore conclude that the δ-amino group at position 124, rather than the positive charge, is required for NIS activity. The model also explains why Lys and Asn cannot functionally replace Arg: Lys is too long and Asn too short to interact with C440, which is conserved among most species. One possible interpretation of our results is that local differences in folding in the IL2/IL6 region are recognized by ER chaperones, blocking the trafficking of NIS from the ER, and that an interaction between the δ-amino group of either Arg or Gln and Cys-440 plays a critical role in NIS folding.
Materials and methods
Cell culture and transfections
COS-7 and MDCK-II cell lines obtained from the American Type Culture Collection was cultured in DMEM media (Life Technologies, Grand Island, NY, USA) supplemented with 10% fetal bovine serum (Gemini BioProducts, Woodland, CA, USA), 10 mM glutamine, 100 U/ml penicillin and 100 µg/ml streptomycin (Cellgro, Herndon, VA, USA). Cells were transiently transfected with 4 µg of NIS cDNAs per 10 cm plate using Lipofectamine/Plus reagent (Life Technologies) following the manufacturer's instructions. Cells were split 24 h after transfection and analyzed by FACS to determine the transfection efficiency (De la Vieja et al., 2005). I− uptake activity, immunoblot, and surface biotinylation were assayed 48 h post-transfection.
MDCK-II polyclonal populations expressing WT or R124H NIS were selected and propagated in growth medium containing 1 g/L G418 (Mediatech, Herndon, VA, USA) as described (Paroder et al., 2006).
Site-directed mutagenesis
Generation of R124H, K, D, E, A, W, Q and N mutants in human NIS cDNA cloned into pSVSport was as described (De la Vieja et al., 2004; De la Vieja et al., 2005; Dohán et al., 2002). Mutagenesis of R111A, R124A and H, R127A, E119A, E122A in human NIS cloned into pcDNA3.1, or R124Q, C440A, S and T in hemagglutinin-tagged human NIS cloned into pcDNA3.1 were introduced using QuikChange™ Site-Directed Mutagenesis Kit (Strategene, La Jolla, CA, USA) as reported (Nicola et al., 2010). All constructs were sequenced to verify specific nucleotide substitutions.
Deglycosylation assays and immunoblotting
Protein deglycosylation was performed as previously described (De la Vieja et al., 2005; Levy et al., 1998a). Briefly, membrane fractions (5 µg) were incubated in glycoprotein denaturing buffer (New England Biolabs, Ipswich, MI, USA) containing 0.5% (w/v) sodium dodecyl sulfate and 40 mM dithiothreitol at 37°C (30 min). Deglycosylation reaction was performed with endo-β-acetylglucosaminidase H (Endo H) (New England Biolabs) in 50 mM sodium citrate (pH 5.5) at 37°C for 2 h. As negative control, membrane vesicles were incubated in the same buffer without enzyme. SDS-PAGE, electrotransference to nitrocellulose membranes and immunoblotting was as described (Levy et al., 1997). Membranes were incubated with 4 nM of an affinity-purified polyclonal anti-human NIS Ab directed against the last 13 residues of the cytosolic NIS carboxyl terminus (Tazebay et al., 2000). Equal loading or purification controls were assessed by stripping and reprobing the same blot with 0.25 µg/ml monoclonal anti-α-tubulin Ab (Sigma-Aldrich, St. Louis, MO, USA) or 0.5 µg/ml monoclonal anti-Na+/K+ ATPase α-subunit Ab (Affinity BioReagents, Golden, CO, USA). Horseradish peroxidase (HRP)-linked secondary anti-mouse and anti-rabbit Abs were from Jackson ImmunoResearch (West Grove, PA, USA) and Amersham Biosciences (Piscataway, NJ, USA), respectively. Proteins were visualized using the enhanced chemiluminescence western blot detection system (Amersham Biosciences). Band intensities were measured densitometrically using the ImageJ Image Software (National Institutes of Health, Bethesda, MD, USA).
Cell surface biotinylation
Biotinylation of cell surface proteins was performed as described (De la Vieja et al., 2004; De la Vieja et al., 2007). Briefly, transfected cells were incubated with 1 mg/mL of the membrane-impermeable biotin reagent sulfo-NHS-SS-biotin (Pierce Chemical, Rockford, IL, USA), which covalently interacts with extracellular primary amines. Cells were lysed and biotinylated proteins were precipitated overnight with streptavidin-coated beads (Pierce Chemical). Beads were washed and adsorbed proteins were eluted with sample buffer containing 10 mM dithiothreitol at 75°C for 5 min, and analyzed by immunoblot.
Flow cytometry
Paraformaldehyde-fixed cells were incubated in PBS containing 0.2% BSA for non-permeabilized conditions or an additional 0.2% saponin for permeabilized conditions with anti-NIS VJ1 mouse monoclonal Ab (dilution 1∶50) directed against an undefined extracellular conformational epitope of NIS (Pohlenz et al., 2000). After washing, cells were incubated with 50 nM of Alexa-488-conjugated goat anti-mouse antibody (Life Technologies). The fluorescence of 104 cells per tube was assayed in FACSCalibur flow cytometer (BD Bio-sciences, San Jose, CA, USA).
Immunofluorescence and confocal microscopy
Cells were stained using indirect immunofluorescence procedures (Paroder et al., 2006). Briefly, cells seeded onto glass coverslips were fixed in 2% paraformaldehyde in PBS. Cells were immunostained with 8 nM polyclonal anti-human NIS Ab, 0.2 µg/ml anti-HA rat monoclonal Ab (Roche Applied Science, Indianapolis, IN, USA), 1∶50 anti-human NIS VJ1 mouse monoclonal Ab, 1 µg/ml monoclonal anti-Na+/K+ ATPase α-subunit Ab, 1 µg/ml monoclonal anti-Calnexin Ab (BD Transduction, San Jose, CA, USA), 2 µg/ml monoclonal anti-protein disulfide-isomerase (Enzo Life Sciences, Farmingdale, NY, USA), or 1 µg/ml monoclonal anti-GM130 (BD Transduction) in PBS containing 0.2% BSA (Sigma-Aldrich) and 0.1% Triton-100. For analysis under non-permeabilized conditions, Triton-100 was omitted. Secondary staining proceeded was carried out with 50 nM anti-rabbit Alexa-488 and anti-mouse Alexa-555 Abs (Life Technologies). Nuclei were stained with DRAQ5 (Cell Signaling Technologies, Danvers, MA, USA). Coverslips were mounted with ProLong Gold Antifade reagent (Cell Signaling Technologies) and images were acquired on a Zeiss LSM 510 laser-scanning confocal microscope (Carl Zeiss, Germany).
I− transport in whole cells
Transfected cells were washed twice with Hanks' balanced salt solution (HBSS) [140 mM NaCl, 5.4 mM KCl, 1.3 mM CaCl2, 0.4 mM MgSO4, 0.5 mM MgCl2, 0.4 mM Na2HPO4, 0.44 mM KH2PO4, 5.55 mM glucose and 10 mM HEPES (pH 7.5)] as described (Purtell et al., 2012). For steady-state experiments, cells were incubated with HBSS containing 20 µM KI supplemented with carrier-free Na125I (specific activity 50 µCi/µmol). Incubations proceeded at 37°C for 45 min in a humidified atmosphere. For I−-dependent kinetic analysis, cells were incubated in HBSS buffer containing the indicated I− concentrations (1.2–160 µM) for 2 min. Accumulated 125I− was released with ice-cold ethanol and then quantified in a Wizard gamma-counter (Perkin Elmer, Boston, MA, USA). DNA was determined by the diphenylamine method after trichloroacetic acid precipitation (Nicola et al., 2009). Iodide uptake was expressed as picomoles of I− per µg DNA (pmol/µg DNA). In all flux experiments, the activity was standardized by NIS expression at the cell surface analyzed by FACS under non-permeabilized conditions. Initial-rate data were analyzed by non-linear regression using the following equation: v = (Vmax*[I−])/(Km+[I−]). Background obtained in non-transfected cells was subtracted. Data were analyzed with Gnuplot software (www.gnuplot.info). The indicated kinetic parameters Km and Vmax are expressed as mean ± s.d. of three independent experiments.
I− transport in membrane vesicles
MVs were prepared from transiently transfected cells as described elsewhere (De la Vieja et al., 2005; Levy et al., 1997). Briefly, transfected COS-7 cells were resuspended in ice-cold 250 mM sucrose, 1 mM EGTA, and 10 mM HEPES (pH 7.5) supplemented with protease inhibitors. Cells were disrupted with a motor-driven Teflon-glass homogenizer. The homogenate was centrifuged twice at 500×g for 15 min at 4°C, and the resulting supernatant was centrifuged at 100,000×g for 1 h at 4°C. Pellets were resuspended in ice-cold 250 mM sucrose, 1 mM MgCl2, 10 mM HEPES (pH 7.5). Aliquots containing 100 µg of protein were assayed for 125I− uptake by incubating at room temperature with an equal volume of a solution containing 80 µM 125I− (specific activity 100 µCi/µmol), 1 mM MgCl2, 10 mM HEPES (pH 7.5), 2 mM methimazole and 200 mM NaCl. Reactions were terminated at the indicated time points by the addition of 4 ml of ice-cold quenching solution containing 1 mM Tris-HCl (pH 7.5), 250 mM KCl, 1 mM methimazole followed by rapid filtration through wet nitrocellulose filters (0.22-µm pore diameter). Radioactivity retained by MVs was determined as described above. I− uptake was expressed as picomoles of I− per mg of protein (pmol/mg protein). Background values obtained at time 0 were subtracted from each condition.
Supplementary Material
Acknowledgments
We thank Dr Sabine Costagliola (Institute of Interdisciplinary Research, Free University of Brussels, Brussels, Belgium) for providing the anti-human NIS VJ1 monoclonal antibody. We are grateful to Dr L. Mario Amzel for insightful discussions, and to the members of the Carrasco laboratory for suggestions and critical reading of the manuscript. The authors have nothing to disclose.
Footnotes
Author contributions
V.P., J.P.N. and N.C. conceived, designed and performed the experiments. C.S.G. designed and generated plasmids. V.P., J.P.N. and N.C. analyzed data and wrote the manuscript. All authors read and approved the final version of the manuscript.
Funding
V.P. was supported in part by Medical Scientist Training Program Grant T32 JM007288 from National Institutes of Health (NIH). J.P.N. was supported by a Brown-Coxe postdoctoral fellowship from Yale University School of Medicine. This work was supported by NIH grant DK-41544 (N.C.). Deposited in PMC for release after 12 months.
Supplementary material available online at http://jcs.biologists.org/lookup/suppl/doi:10.1242/jcs.120246/-/DC1
References
- Altorjay A., Dohán O., Szilágyi A., Paroder M., Wapnir I. L., Carrasco N. (2007). Expression of the Na+/I- symporter (NIS) is markedly decreased or absent in gastric cancer and intestinal metaplastic mucosa of Barrett esophagus. BMC Cancer 7, 5 10.1186/1471-2407-7-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borders C. L., Jr, Broadwater J. A., Bekeny P. A., Salmon J. E., Lee A. S., Eldridge A. M., Pett V. B. (1994). A structural role for arginine in proteins: multiple hydrogen bonds to backbone carbonyl oxygens. Protein Sci. 3, 541–548 10.1002/pro.5560030402 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calado J., Loeffler J., Sakallioglu O., Gok F., Lhotta K., Barata J., Rueff J. (2006). Familial renal glucosuria: SLC5A2 mutation analysis and evidence of salt-wasting. Kidney Int. 69, 852–855 10.1038/sj.ki.5000194 [DOI] [PubMed] [Google Scholar]
- Dai G., Levy O., Carrasco N. (1996). Cloning and characterization of the thyroid iodide transporter. Nature 379, 458–460 10.1038/379458a0 [DOI] [PubMed] [Google Scholar]
- Danilov S. M., Kalinin S., Chen Z., Vinokour E. I., Nesterovitch A. B., Schwartz D. E., Gribouval O., Gubler M. C., Minshall R. D. (2010). Angiotensin I-converting enzyme Gln1069Arg mutation impairs trafficking to the cell surface resulting in selective denaturation of the C-domain. PLoS ONE 5, e10438 10.1371/journal.pone.0010438 [DOI] [PMC free article] [PubMed] [Google Scholar]
- De la Vieja A., Ginter C. S., Carrasco N. (2004). The Q267E mutation in the sodium/iodide symporter (NIS) causes congenital iodide transport defect (ITD) by decreasing the NIS turnover number. J. Cell Sci. 117, 677–687 10.1242/jcs.00898 [DOI] [PubMed] [Google Scholar]
- De la Vieja A., Ginter C. S., Carrasco N. (2005). Molecular analysis of a congenital iodide transport defect: G543E impairs maturation and trafficking of the Na+/I- symporter. Mol. Endocrinol. 19, 2847–2858 10.1210/me.2005-0162 [DOI] [PubMed] [Google Scholar]
- De la Vieja A., Reed M. D., Ginter C. S., Carrasco N. (2007). Amino acid residues in transmembrane segment IX of the Na+/I- symporter play a role in its Na+ dependence and are critical for transport activity. J. Biol. Chem. 282, 25290–25298 10.1074/jbc.M700147200 [DOI] [PubMed] [Google Scholar]
- Dohán O., Gavrielides M. V., Ginter C., Amzel L. M., Carrasco N. (2002). Na(+)/I(-) symporter activity requires a small and uncharged amino acid residue at position 395. Mol. Endocrinol. 16, 1893–1902 10.1210/me.2002-0071 [DOI] [PubMed] [Google Scholar]
- Dohán O., De la Vieja A., Paroder V., Riedel C., Artani M., Reed M., Ginter C. S., Carrasco N. (2003). The sodium/iodide Symporter (NIS): characterization, regulation, and medical significance. Endocr. Rev. 24, 48–77 10.1210/er.2001-0029 [DOI] [PubMed] [Google Scholar]
- Dohán O., Portulano C., Basquin C., Reyna-Neyra A., Amzel L. M., Carrasco N. (2007). The Na+/I symporter (NIS) mediates electroneutral active transport of the environmental pollutant perchlorate. Proc. Natl. Acad. Sci. USA 104, 20250–20255 10.1073/pnas.0707207104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eskandari S., Loo D. D., Dai G., Levy O., Wright E. M., Carrasco N. (1997). Thyroid Na+/I- symporter. Mechanism, stoichiometry, and specificity. J. Biol. Chem. 272, 27230–27238 10.1074/jbc.272.43.27230 [DOI] [PubMed] [Google Scholar]
- Faham S., Watanabe A., Besserer G. M., Cascio D., Specht A., Hirayama B. A., Wright E. M., Abramson J. (2008). The crystal structure of a sodium galactose transporter reveals mechanistic insights into Na+/sugar symport. Science 321, 810–814 10.1126/science.1160406 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischer H., Fukuda N., Barbry P., Illek B., Sartori C., Matthay M. A. (2001). Partial restoration of defective chloride conductance in DeltaF508 CF mice by trimethylamine oxide. Am. J. Physiol. 281, L52–L57 [DOI] [PubMed] [Google Scholar]
- Grasberger H., De Deken X., Miot F., Pohlenz J., Refetoff S. (2007). Missense mutations of dual oxidase 2 (DUOX2) implicated in congenital hypothyroidism have impaired trafficking in cells reconstituted with DUOX2 maturation factor. Mol. Endocrinol. 21, 1408–1421 10.1210/me.2007-0018 [DOI] [PubMed] [Google Scholar]
- Hartl F. U., Bracher A., Hayer-Hartl M. (2011). Molecular chaperones in protein folding and proteostasis. Nature 475, 324–332 10.1038/nature10317 [DOI] [PubMed] [Google Scholar]
- Henikoff S., Henikoff J. G. (1992). Amino acid substitution matrices from protein blocks. Proc. Natl. Acad. Sci. USA 89, 10915–10919 10.1073/pnas.89.22.10915 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaminsky S. M., Levy O., Salvador C., Dai G., Carrasco N. (1994). Na(+)-I- symport activity is present in membrane vesicles from thyrotropin-deprived non-I(-)-transporting cultured thyroid cells. Proc. Natl. Acad. Sci. USA 91, 3789–3793 10.1073/pnas.91.9.3789 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kasahara K., Nakayama Y., Ikeda K., Fukushima Y., Matsuda D., Horimoto S., Yamaguchi N. (2004). Trafficking of Lyn through the Golgi caveolin involves the charged residues on alphaE and alphaI helices in the kinase domain. J. Cell Biol. 165, 641–652 10.1083/jcb.200403011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levy O., Dai G., Riedel C., Ginter C. S., Paul E. M., Lebowitz A. N., Carrasco N. (1997). Characterization of the thyroid Na+/I- symporter with an anti-COOH terminus antibody. Proc. Natl. Acad. Sci. USA 94, 5568–5573 10.1073/pnas.94.11.5568 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levy O., De la Vieja A., Ginter C. S., Riedel C., Dai G., Carrasco N. (1998a). N-linked glycosylation of the thyroid Na+/I- symporter (NIS). Implications for its secondary structure model. J. Biol. Chem. 273, 22657–22663 10.1074/jbc.273.35.22657 [DOI] [PubMed] [Google Scholar]
- Levy O., Ginter C. S., De la Vieja A., Levy D., Carrasco N. (1998b). Identification of a structural requirement for thyroid Na+/I- symporter (NIS) function from analysis of a mutation that causes human congenital hypothyroidism. FEBS Lett. 429, 36–40 10.1016/S0014-5793(98)00522-5 [DOI] [PubMed] [Google Scholar]
- Li C., Ramjeesingh M., Reyes E., Jensen T., Chang X., Rommens J. M., Bear C. E. (1993). The cystic fibrosis mutation (delta F508) does not influence the chloride channel activity of CFTR. Nat. Genet. 3, 311–316 10.1038/ng0493-311 [DOI] [PubMed] [Google Scholar]
- Li H. C., Kucher V., Li E. Y., Conforti L., Zahedi K. A., Soleimani M. (2012). The role of aspartic acid residues 405 and 416 of the kidney isotype of sodium-bicarbonate cotransporter 1 in its targeting to the plasma membrane. Am. J. Physiol. 302, C1713–C1730 10.1152/ajpcell.00147.2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martín M. G., Turk E., Lostao M. P., Kerner C., Wright E. M. (1996). Defects in Na+/glucose cotransporter (SGLT1) trafficking and function cause glucose-galactose malabsorption. Nat. Genet. 12, 216–220 10.1038/ng0296-216 [DOI] [PubMed] [Google Scholar]
- Martín M. G., Lostao M. P., Turk E., Lam J., Kreman M., Wright E. M. (1997). Compound missense mutations in the sodium/D-glucose cotransporter result in trafficking defects. Gastroenterology 112, 1206–1212 10.1016/S0016-5085(97)70132-X [DOI] [PubMed] [Google Scholar]
- Meimaridou E., Gooljar S. B., Ramnarace N., Anthonypillai L., Clark A. J., Chapple J. P. (2011). The cytosolic chaperone Hsc70 promotes traffic to the cell surface of intracellular retained melanocortin-4 receptor mutants. Mol. Endocrinol. 25, 1650–1660 10.1210/me.2011-1020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morello J. P., Salahpour A., Laperrière A., Bernier V., Arthus M. F., Lonergan M., Petäjä-Repo U., Angers S., Morin D., Bichet D. G. et al. (2000). Pharmacological chaperones rescue cell-surface expression and function of misfolded V2 vasopressin receptor mutants. J. Clin. Invest. 105, 887–895 10.1172/JCI8688 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newton C. L., Whay A. M., McArdle C. A., Zhang M., van Koppen C. J., van de Lagemaat R., Segaloff D. L., Millar R. P. (2011). Rescue of expression and signaling of human luteinizing hormone G protein-coupled receptor mutants with an allosterically binding small-molecule agonist. Proc. Natl. Acad. Sci. USA 108, 7172–7176 10.1073/pnas.1015723108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicola J. P., Basquin C., Portulano C., Reyna-Neyra A., Paroder M., Carrasco N. (2009). The Na+/I− symporter mediates active iodide uptake in the intestine. Am. J. Physiol. 296, C654–C662 10.1152/ajpcell.00509.2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicola J. P., Nazar M., Mascanfroni I. D., Pellizas C. G., Masini-Repiso A. M. (2010). NF-kappaB p65 subunit mediates lipopolysaccharide-induced Na(+)/I(-) symporter gene expression by involving functional interaction with the paired domain transcription factor Pax8. Mol. Endocrinol. 24, 1846–1862 10.1210/me.2010-0102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicola J. P., Nazar M., Serrano-Nascimento C., Goulart-Silva F., Sobrero G., Testa G., Nunes M. T., Muñoz L., Miras M., Masini-Repiso A. M. (2011). Iodide transport defect: functional characterization of a novel mutation in the Na+/I- symporter 5′-untranslated region in a patient with congenital hypothyroidism. J. Clin. Endocrinol. Metab. 96, E1100–E1107 10.1210/jc.2011-0349 [DOI] [PubMed] [Google Scholar]
- Nicola J. P., Reyna-Neyra A., Carrasco N., Masini-Repiso A. M. (2012). Dietary iodide controls its own absorption through post-transcriptional regulation of the intestinal Na+/I- symporter. J. Physiol. 590, 6013–6026 10.1113/jphysiol.2012.241307 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paroder V., Spencer S. R., Paroder M., Arango D., Schwartz S., Jr, Mariadason J. M., Augenlicht L. H., Eskandari S., Carrasco N. (2006). Na(+)/monocarboxylate transport (SMCT) protein expression correlates with survival in colon cancer: molecular characterization of SMCT. Proc. Natl. Acad. Sci. USA 103, 7270–7275 10.1073/pnas.0602365103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paroder-Belenitsky M., Maestas M. J., Dohán O., Nicola J. P., Reyna-Neyra A., Follenzi A., Dadachova E., Eskandari S., Amzel L. M., Carrasco N. (2011). Mechanism of anion selectivity and stoichiometry of the Na+/I- symporter (NIS). Proc. Natl. Acad. Sci. USA 108, 17933–17938 10.1073/pnas.1108278108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Partridge C. J., Beech D. J., Sivaprasadarao A. (2001). Identification and pharmacological correction of a membrane trafficking defect associated with a mutation in the sulfonylurea receptor causing familial hyperinsulinism. J. Biol. Chem. 276, 35947–35952 10.1074/jbc.M104762200 [DOI] [PubMed] [Google Scholar]
- Pohlenz J., Duprez L., Weiss R. E., Vassart G., Refetoff S., Costagliola S. (2000). Failure of membrane targeting causes the functional defect of two mutant sodium iodide symporters. J. Clin. Endocrinol. Metab. 85, 2366–2369 10.1210/jc.85.7.2366 [DOI] [PubMed] [Google Scholar]
- Purtell K., Paroder-Belenitsky M., Reyna-Neyra A., Nicola J. P., Koba W., Fine E., Carrasco N., Abbott G. W. (2012). The KCNQ1-KCNE2 K+ channel is required for adequate thyroid I− uptake. FASEB J. 26, 3252–3259 10.1096/fj.12-206110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reed-Tsur M. D., De la Vieja A., Ginter C. S., Carrasco N. (2008). Molecular characterization of V59E NIS, a Na+/I- symporter mutant that causes congenital I- transport defect. Endocrinology 149, 3077–3084 10.1210/en.2008-0027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riedel C., Levy O., Carrasco N. (2001). Post-transcriptional regulation of the sodium/iodide symporter by thyrotropin. J. Biol. Chem. 276, 21458–21463 10.1074/jbc.M100561200 [DOI] [PubMed] [Google Scholar]
- Sato S., Ward C. L., Krouse M. E., Wine J. J., Kopito R. R. (1996). Glycerol reverses the misfolding phenotype of the most common cystic fibrosis mutation. J. Biol. Chem. 271, 635–638 10.1074/jbc.271.2.635 [DOI] [PubMed] [Google Scholar]
- Shepshelovich J., Goldstein-Magal L., Globerson A., Yen P. M., Rotman-Pikielny P., Hirschberg K. (2005). Protein synthesis inhibitors and the chemical chaperone TMAO reverse endoplasmic reticulum perturbation induced by overexpression of the iodide transporter pendrin. J. Cell Sci. 118, 1577–1586 10.1242/jcs.02294 [DOI] [PubMed] [Google Scholar]
- Spitzweg C., Morris J. C. (2010). Genetics and phenomics of hypothyroidism and goiter due to NIS mutations. Mol. Cell. Endocrinol. 322, 56–63 10.1016/j.mce.2010.02.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szinnai G., Kosugi S., Derrien C., Lucidarme N., David V., Czernichow P., Polak M. (2006). Extending the clinical heterogeneity of iodide transport defect (ITD): a novel mutation R124H of the sodium/iodide symporter gene and review of genotype-phenotype correlations in ITD. J. Clin. Endocrinol. Metab. 91, 1199–1204 10.1210/jc.2005-1832 [DOI] [PubMed] [Google Scholar]
- Tamarappoo B. K., Verkman A. S. (1998). Defective aquaporin-2 trafficking in nephrogenic diabetes insipidus and correction by chemical chaperones. J. Clin. Invest. 101, 2257–2267 10.1172/JCI2303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tazebay U. H., Wapnir I. L., Levy O., Dohan O., Zuckier L. S., Zhao Q. H., Deng H. F., Amenta P. S., Fineberg S., Pestell R. G. et al. (2000). The mammary gland iodide transporter is expressed during lactation and in breast cancer. Nat. Med. 6, 871–878 10.1038/78630 [DOI] [PubMed] [Google Scholar]
- van den Heuvel L. P., Assink K., Willemsen M., Monnens L. (2002). Autosomal recessive renal glucosuria attributable to a mutation in the sodium glucose cotransporter (SGLT2). Hum. Genet. 111, 544–547 10.1007/s00439-002-0820-5 [DOI] [PubMed] [Google Scholar]
- Wapnir I. L., van de Rijn M., Nowels K., Amenta P. S., Walton K., Montgomery K., Greco R. S., Dohán O., Carrasco N. (2003). Immunohistochemical profile of the sodium/iodide symporter in thyroid, breast, and other carcinomas using high density tissue microarrays and conventional sections. J. Clin. Endocrinol. Metab. 88, 1880–1888 10.1210/jc.2002-021544 [DOI] [PubMed] [Google Scholar]
- Wolff S. C., Qi A. D., Harden T. K., Nicholas R. A. (2010). Charged residues in the C-terminus of the P2Y1 receptor constitute a basolateral-sorting signal. J. Cell Sci. 123, 2512–2520 10.1242/jcs.060723 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wright E. M., Turk E., Martin M. G. (2002). Molecular basis for glucose-galactose malabsorption. Cell Biochem. Biophys. 36, 115–121 10.1385/CBB:36:2-3:115 [DOI] [PubMed] [Google Scholar]
- Wright E. M., Loo D. D., Hirayama B. A. (2011). Biology of human sodium glucose transporters. Physiol. Rev. 91, 733–794 10.1152/physrev.00055.2009 [DOI] [PubMed] [Google Scholar]
- Zeitlin P. L. (1999). Novel pharmacologic therapies for cystic fibrosis. J. Clin. Invest. 103, 447–452 10.1172/JCI6346 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Z., Gong Q., January C. T. (1999). Correction of defective protein trafficking of a mutant HERG potassium channel in human long QT syndrome. Pharmacological and temperature effects. J. Biol. Chem. 274, 31123–31126 10.1074/jbc.274.44.31123 [DOI] [PubMed] [Google Scholar]
- Zimmermann M. B. (2009). Iodine deficiency. Endocr. Rev. 30, 376–408 10.1210/er.2009-0011 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.