

Summary
The pathology and severity of toxoplasmosis results from the rapid replication cycle of the apicomplexan parasite Toxoplasma gondii. The tachyzoites divide asexually through endodyogeny, wherein two daughter cells bud inside the mother cell. Before mitosis is completed, the daughter buds form around the duplicated centrosomes and subsequently elongate to serve as the scaffold for organellogenesis and organelle partitioning. The molecular control mechanism of this process is poorly understood. Here, we characterized a T. gondii NIMA-related kinase (Nek) ortholog that was identified in a chemical mutagenesis screen. A temperature-sensitive mutant, V-A15, possesses a Cys316Arg mutation in TgNek1 (a novel mutant allele in Neks), which is responsible for growth defects at the restrictive temperature. Phenotypic analysis of V-A15 indicated that TgNek1 is essential for centrosome splitting, proper formation of daughter cells and faithful segregation of genetic material. In vitro kinase assays showed that the mutation abolishes the kinase activity of TgNek1. TgNek1 is recruited to the centrosome prior to its duplication and localizes on the duplicated centrosomes facing the spindle poles in a cell-cycle-dependent manner. Mutational analysis of the activation loop suggests that localization and activity are spatio-temporally regulated by differential phosphorylation. Collectively, our results identified a novel temperature-sensitive allele for a Nek kinase and highlight its essential function in centrosome splitting in Toxoplasma. Moreover, these results conclusively show for the first time that Toxoplasma bud assembly is facilitated by the centrosome because defective centrosome splitting results in single daughter cell budding.
Key words: NIMA, Nek, Kinase, Toxoplasma, Centrosome, Endodyogeny
Introduction
Cell division of the apicomplexan parasite Toxoplasma gondii unfolds through an internal budding mechanism producing two new daughter parasites per division round (Striepen et al., 2007). This process is driven by assembly of the cortical cytoskeleton (Anderson-White et al., 2012), serving as a scaffold for de novo formation of secretory organelles as well as for partitioning of the nucleus, plastid, mitochondrion and the Golgi apparatus (Nishi et al., 2008). As in most eukaryotes, progression through the cell cycle is ultimately controlled by cyclin-cdk pairs in the parasite, but it is unclear how these are translated into the unique coordination of assembling cytoskeletal elements with the progression of mitosis (Gubbels et al., 2008b). It has been shown that mitosis is required for daughter budding and that daughter assembly initiates before the completion of DNA synthesis (Radke et al., 2001). A pivotal hub in coordinating these processes is the centrosome, which duplicates prior to mitosis and subsequently provides the spatial cue for daughter formation (Anderson-White et al., 2012; Gubbels et al., 2008b; Hu et al., 2002; Striepen et al., 2000). Notably, the internal assembly of the daughters starts before mitosis is complete; therefore, the checkpoints on these processes appear to be coordinated differently from the well-studied eukaryotic model systems (Gubbels et al., 2008b).
Across the eukaryotes, centrosomes play key roles in the spatio-temporal organization of mitosis and cell division. It has been suggested that the centrosome serves as a signaling platform for cell cycle regulation since many regulatory complexes, including tumor suppressors and checkpoint proteins, localize in these structures. In human cells, surgical removal of the centrosome or perturbation of centrosomal protein levels induces G1 arrest (Doxsey et al., 2005). In yeast, the analogous structure of the centrosome, the spindle pole body (SPB), sequesters regulatory molecules that are essential for mitotic exit and cytokinesis (Seshan and Amon, 2004). Like nuclear DNA, the centrosome duplicates only once per cell cycle, making these two events tightly regulated. A set of mitotic kinases and phosphatases controls the progression through mitosis and its coordination with cytokinesis. NIMA (never in mitosis-A)-related kinases (Nek or NRK) represent a conserved family of serine/threonine kinases implicated in regulation of distinct cellular events. The NIMA kinase was first identified in filamentous fungus, Aspergillus nidulans and shown to be essential for G2/M transition (Morris et al., 1989; O'Regan et al., 2007; Osmani et al., 1987). Orthologs have been identified in a wide variety of eukaryotes and in general play a crucial role in mitotic progression (O'Connell et al., 2003). Mammals have an expanded family of NIMA related kinases, named Nek1-11. Several of these Neks have important roles in maintenance of centrosome function and structure, mitotic microtubule organization, and regulation of axonemal microtubule in cilia and flagella (O'Regan et al., 2007). The functional ortholog of NIMA is HsNek2, which functions in mitotic progression. Mutations in HsNek2 are associated with chromosome instability and cancer and, although a variety of substrates have been identified, the role of HsNek2 in centrosome separation appears to be the most crucial (Hayward and Fry, 2006; Mardin et al., 2010). In fission yeast, the NIMA ortholog is known as Fin1 which acts in duplication of the SPB (Salisbury, 2007). KIN3, the NIMA ortholog in Saccharomyces cerevisiae, is not essential for mitotic progression but there is an indication it acts on kinetochore attachment to the microtubules analogous to an additional function reported for HsNek2 (Chen et al., 2002).
Although the function of NIMA-related kinases has not been studied in much detail in protists, a range of divergent roles has been reported. For instance, in the ciliate Tetrahymena thermophila an extended family of 39 Neks controls cilium length and stability (Wloga et al., 2006). Three NIMA kinases named NRK were identified in Trypanosoma brucei, the parasite responsible for sleeping sickness in human and animals. TbNRKA and TbNRKB were suggested to be involved in parasite differentiation, where as TbNRKC is involved in regulating basal body separation and cytokinesis (Pradel et al., 2006). In Giardia lamblia, two Nek kinases were identified with roles in mitosis and excystation (Smith et al., 2012). In Plasmodium falciparum, the causative agent of malaria, four Neks were identified, two of which (PfNek2 and PfNek4) are essential for sexual development in the mosquito vector, whereas PfNek1 is required for progression through the erythrocyte cycle (Reininger et al., 2005; Reininger et al., 2009; Xue et al., 2011). Seven Neks were identified in the Toxoplasma genome (Peixoto et al., 2010), but have not been experimentally addressed except one: TgNek1 has been genetically connected to lytic cycle progression as a temperature-sensitive (ts) mutant named V-A15 was identified in a chemical mutagenesis screen for lytic cycle progression (Gubbels et al., 2008a). In this preliminary analysis, mutant V-A15 displayed uncoupling of cytokinetic progression from mitotic progression.
Here, we further dissect the phenotype of ts-mutant V-A15 and TgNek1. We show that its gene product localizes to the centrosomes and regulates the splitting of duplicated centrosomes in the G1/S phase transition. This suggests TgNek1 is a functional ortholog of mitotic Neks in mammals and yeast. Interestingly, the unsplit centrosomes in the mutant result in formation of only a single daughter bud, subsequently resulting in failed cell division. Sequence analysis, modeling and in vitro studies showed that the TgNek1 structure deviates from its orthologs in other systems, since TgNek1 lacks the coiled-coil domain functioning in dimerization and TgNek1 does not autophosphorylate. Together with the observation that the typical mitotic Nek substrates are absent from the Toxoplasma genome, this suggests that TgNek1 may share its function with HsNek2 but likely has differently interaction partners and substrates.
Results
Single daughter formation in mutant V-A15 originates in a centrosome-splitting defect
We have previously reported that the temperature-sensitive growth mutant V-A15 displays an uncoupling phenotype, which is caused by a point mutation in TgNek1 (Gubbels et al., 2008a). To dissect further the origin of the uncoupling, we performed a detailed phenotypic analysis using a battery of developmental markers in immunofluorescence and live imaging assays. First, we established at which point the normal cell division cycle derails by incubating synchronized RH and V-A15 parasites at 40°C and staining with a centrosome (anti-HsCentrin) and budding marker (anti-TgIMC3) (Fig. 1A); we have used this marker combination in the past to determine the stage of arrested development (Szatanek et al., 2012). Parasites were synchronized by a 3-methyladenine (3-MA) block, which arrests parasites just before centrosome duplication (supplementary material Fig. S1) (Wang et al., 2010). Three hours after release of the 3-MA block about 60% of the wild-type RH parasites have progressed through centrosome duplication (Fig. 1A). However, hardly any of the V-A15 parasites have progressed to centrosome duplication at this point (5.7%), or even at the next time point (6.6% at 4.5 hours; Fig. 1A). These results indicate there is either a defect with centrosome duplication or a defect with entering or progression through S phase, which is consistent with the role of Neks in other systems (O'Regan et al., 2007).
Fig. 1.
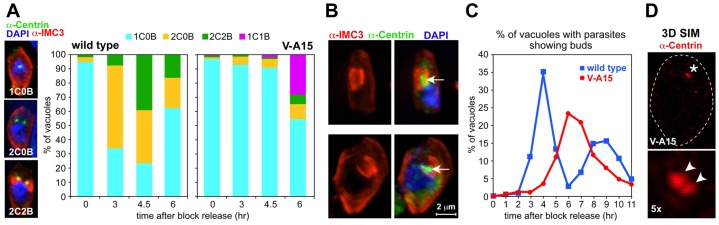
Mutant V-A15 forms single daughter cells. (A) V-A15 exhibits a delay in cell cycle progression following release from a 3-MA synchronization block at 40°C. Images show the cell division stages differentiated using antibody markers for the centrosome (anti-HsCentrin) and daughter buds (anti-TgIMC3). B, bud; C, centrosome. Numbers reflect counts of these structures. Graphs show quantification data of cell cycle stages in RH parasites and the V-A15 mutant after 3-MA block and release at 40°C. (B) Two examples of single daughter buds observed in V-A15 6 hours after release of the 3-MA block. Side by side panels show the same parasite. Arrows mark the centrosome in the single daughter parasite. (C) Time-resolved quantification of bud development following release of the 3-MA block. (D) Super resolution microscopy (3D-SIM) of V-A15 parasites stained with anti-HsCentrin shows that the single centrosome observed by widefield microscopy is actually a centrosome-splitting defect. Lower panel shows 5× magnification of the pair of duplicated centrosomes (marked with arrowheads; marked with an asterisk in the top panel). Dashed line indicates the outline of the parasite.
At later time points of synchronized V-A15 parasites (>4.5 hours post 3-MA block release), we still observed a single centrosome but these were associated with a single daughter bud (1C1B) (Fig. 1A,B). Six hours after release from the block, 23.4% of the mutant parasites contain single daughter buds (Fig. 1C). We noted that the morphology of the centrosomes in the mutant is different compared with the control parasites (marked by arrows in Fig. 1B); the mutant centrosomes are bulkier and irregularly shaped instead of globular. To study this in more detail, we performed 3D-SIM super-resolution microscopy on the mutant parasites (Fig. 1D; supplementary material Movie 1). The irregular centrosome shape can now be resolved as composed of two connected globular structures (Fig. 1D, arrowhead), resulting in a slightly dumbbell-shaped morphology at lower resolution. As such, these results show that the centrosome does duplicate but it fails to split and segregate. These duplicated yet unsplit centrosomes do promote, albeit with a little delay, the formation of the single daughter bud. Therefore, these results suggest that in normal progression of cytokinesis the physically separated centrosomes serve as platforms for the daughter buds assembly and their physical separation is a key requirement to initiate bud formation. Conversely, if the centrosomes are physically too closely apposed as in mutant V-A15, only a single daughter bud can be assembled.
Mutant V-A15 displays a cell division defect but not a DNA replication defect
To analyze whether the defects observed in V-A15 correspond with a delay or defect in S-phase progression, we analyzed the development of DNA contents upon induction of the phenotype. Following cell cycle synchronization by a limited 2 hours invasion period, we analyzed the DNA content after 6, 12 and 18 hours, representing the first three rounds of DNA replication cycle (Radke et al., 2001). As shown in Fig. 2A, we observe a normal distribution after 6 hours suggesting there is no delay in S phase compared with wild-type parasites. Therefore, the delay in centrosome splitting and the defect in daughter formation are not accompanied by a defect in DNA synthesis in the early development of the phenotype. However, after 12 hours a population with >2N DNA content appears, indicating the start of a new round of DNA replication without having undergone cytokinesis. This corresponds with the previously observed uncoupling phenotype (Gubbels et al., 2008a) and likely originates in failed cytokinesis due to the formation of a single daughter bud.
Fig. 2.

Mutant V-A15 displays a cell division defect. (A) DNA content analysis by flow cytometry of synchronized parasites (2 hours limited invasion) shows that DNA replication of V-A15 is not arrested by the temperature-sensitive defects compared with the control RH parasites. DNA contents were analyzed at 6, 12 and 18 hours post invasion. (B) RH and V-A15 mutant parasites expressing CherryRFP-MORN1 induced at 40°C were examined by live cell imaging. The arrowheads mark the ring representing the forming daughter basal complexes: only one daughter complex forms in V-A15. At the bottom of the images MORN1 resides in the basal complex of the mother. Dashed lines indicate the outline of the parasites. (C) V-A15 displays mis-segregation of the nucleus upon long induction (16 hours at 40°C). V-A15 is stained with anti-TgIMC3, anti-HsCentrin and DAPI. Arrowheads indicate fragmented DNA.
Although the DNA content analysis suggests that the nuclear DNA undergoes a normal replication cycle in V-A15 mutants (Fig. 2A), it is insufficient to demonstrate whether mitosis occurs or whether nuclear division takes place. Since Nek1 orthologs have been associated with defects in mitotic progression (e.g. Chen et al., 2002; O'Regan et al., 2007; Smith et al., 2012; Woodbury and Morgan, 2007), we used MORN1 as a marker for the spindle, spindle poles and basal complex (the growing end of the daughter bud's cytoskeleton) to monitor the progression of mitosis and cytokinesis (Gubbels et al., 2006; Hu et al., 2006). As shown in the pre-mitotic panel of Fig. 2B, the mutant forms a single daughter bud as indicated by the single arrowhead marking the formation of a single circular basal complex in the daughter. However, the bar-shaped MORN1 signal to the right indicates the parasite is progressing through mitosis, since here MORN1 marks the spindle. Furthermore, in the post-mitotic panel two globularly shaped MORN1 signals are present, which represent the separated spindle poles. These static observations were confirmed by the dynamics of a time-lapse movie that showed the development of Cherry-MORN1 in mutant V-A15 undergoing mitosis and cytokinesis; mitosis, or at least spindle pole segregation, does progress normally in the ts-mutant while only a single daughter basal complex is formed (supplementary material Movie 2). When we studied the fidelity of nuclear integrity at later stages we observed a high incidence of nuclear fragmentation suggesting that nuclear division is not accurately completed (Fig. 2C). However, it is more likely that this reflects a defect in karyokinesis, correlating with the fact that Toxoplasma cytokinesis starts before mitosis is complete and both progress in parallel. Hence, it is conceivable that the single daughter buds as observed in Fig. 1B result in a secondary defect in karyokinesis.
Two TgNek1 splice variants
We previously showed that TgNek1 contains the etiological SNP causing a temperature-sensitive growth defect in mutant V-A15 (Gubbels et al., 2008a). To analyze further the gene and the role of its product, we cloned the TgNek1 cDNA. Because the agreement among available gene models on http://toxodb.org/toxo was poor, we first performed 5′- and 3′-RACE PCR reactions using primers originating in the conserved kinase domain. These confirmed the predicted 5′-end of the TGME49_092140 gene model; however, two different splice variants were identified at the 3′-end of the gene, both of which disagreed with any of the available gene models (Fig. 3A). In particular, the large predicted last exon, which is outside the N-terminally located active site, was significantly shorter. Subsequently, we PCR amplified the full length coding sequence of the two different splice variants and aligned them to the ToxoDB consensus gene model. Splice variants 1 and 2 encode 448 and 499 amino acids, respectively, compared to the 1595 amino acids of the predicted ORF. The RACE-validated CDS sequences have been deposited as user comments on the TGME49_092140 gene page in ToxoDB.
Fig. 3.
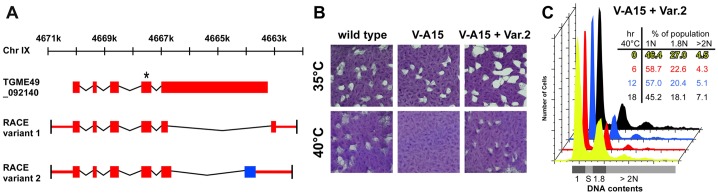
Characterization of TgNek1. (A) Identification of two TgNek1 splice variants by RACE PCR. Coding sequence is indicated by colored blocks, and UTRs by colored lines. The blue block in variant 2 indicates a different reading frame compared with the other two models. The asterisk marks the location of the V-A15 point mutation. (B) Plaque assays of wild-type RH, V-A15 mutant and V-A15 complemented by the pnek-TgNek1-2/sagCAT construct (V-A15 + Var.2) at indicated temperatures. (C) DNA content analysis of the complemented V-A15 clonal line (V-A15 + Var.2) at 40°C displays wild-type phenotype restoration.
TgNek1 splice variant 2 complements ts-mutant V-A15
To examine the functionality of the identified TgNek1 splice variants we performed genetic complementation experiments of the V-A15 mutant. Hereto we cloned the coding sequence of the two variants into vectors wherein the open reading frames were driven by the α-tubulin promoter (ptub) or the endogenous promoter (pnek1), either with a C-terminal YFP tag, an N-terminal DD-YFP tag, or without any tag. These constructs were transfected into mutant V-A15 and complementation was screened by growth restoration at 40°C. The only construct resulting in complementation was the untagged splice variant 2 driven by the pnek1 promoter (Fig. 3B). To further validate phenotype complementation of V-A15 we performed DNA content analysis. The defect displayed in V-A15 was completely restored (Fig. 3C). Based on this functional information we focused on splice variant 2 (TgNek1-2) in the rest of this study.
TgNek1-2 localizes to the duplicating centrosomes
To understand the role of TgNek1-2 in the centrosome cycle and its coordination with the cytokinetic cycle, we reasoned that the sub-cellular localization pattern throughout the cell cycle would provide hints to the nature of the processes in which TgNek1-2 exerts its function. We raised a specific antiserum against the unique 396–499 C-terminal amino acid portion of TgNek1-2 expressed in E. coli. The polyclonal anti-TgNek1-2 antibody recognized a single band of ∼55 kDa on western blot (supplementary material Fig. S2), close to the 54.9 kDa predicted for TgNek1-2. Interestingly, the TgNek1-2 protein could only be detected in intracellular parasites, suggesting that the expression and/or stability of TgNek1-2 is tightly regulated and restricted to dividing parasites. Immunofluorescence analysis showed that anti-TgNek1-2 highlights the centrosomes in a narrow window around their duplication, and did not highlight any structures in interphase or dividing parasites (Fig. 4A, arrowheads; supplementary material Fig. S3).
Fig. 4.

Spatio-temporal localization dynamics of TgNek1-2 throughout the tachyzoite division cycle. (A) Endogenous TgNek1-2 localization in RH parasites was analyzed by immunofluorescence assay (IFA) using anti-TgNek1-2 and anti-HsCentrin. Arrowheads mark individual parasites without anti-TgNek1-2 signal: the two parasites on the bottom left are in G1 whereas the parasite on the bottom right is in cell division but has completed centrosome splitting and mitosis but not nuclear division. (B) RH parasites expressing Cherry-MORN1 were analyzed by IFA using anti-TgNek1-2 and anti-HsCentrin antibodies. Progressive stages of the early mitotic apparatus are shown (mitosis progresses from top to bottom). Arrows in the third panel mark the localization of the fluorescence density profile shown in the fourth panel. Full size vacuoles are shown in supplementary material Fig. S3. (C) Mutant V-A15 induced at 40°C for 16 hours was analyzed by anti-TgNek1-2 and anti-HsCentrin. Insets display magnification of the mitotic apparatus (in the top panel of the one marked by the arrowhead). Top panel is pre-centrosome division; bottom panel shows arrested centrosome separation. (D). Schematic summarizing TgNek1-2 localization during early mitotic division in wild-type and V-A15 parasites with unsplit centrosome and single daughter bud.
TgNek1-2 localizes between the spindle pole and the centrosome
Although anti-TgNek1-2 colocalized with the anti-HsCentrin marker, the colocalization is imperfect. Careful analysis of the centrosome localization revealed that TgNek1-2 localizes only to a small portion of the centrosome, on the inner side of duplicated centrosomes (Fig. 4A,B). This indicates that TgNek1-2 localizes to the proximal ends of duplicated centrosomes, implying that the protein may functionally resemble its mammalian counterpart HsNek2 (Fry et al., 1998b). To fully orient the localization of TgNek1-2 on the centrosome, we reasoned that the mitotic spindle apparatus could provide further spatio-temporal detail. Hereto we co-stained CherryRFP-MORN1 expressing parasites with anti-TgNek1-2 and anti-HsCentrin. As shown in Fig. 4B (left panels), TgNek1-2 localizes to the space between centrosome and spindle pole throughout the transition from S phase to mitosis. The sequential peaks observed in the fluorescence intensity profiles for the three markers confirm this assignment and the localization of TgNek1-2 to the proximal end of the centrosome (Fig. 4B, right panels).
To establish whether the point mutation affects its localization in mutant V-A15 we applied the TgNek1-2 antiserum to the mutant. As shown in Fig. 4C, the mutant allele of TgNek1-2 still associates with the unsplit centrosomes; however, it is now located on the outside of the unsplit centrosome. Hence, the ts mutation does not affect localization to the centrosome but interferes with the splitting of the centrosomes. We cannot differentiate whether this originates in a defect in the recruitment of other proteins, e.g. its substrates, or could affect the kinase activity. It has been reported that centrosomal recruitment of HsNek2A depends on two Hippo pathway components, the mammalian sterile 20-like kinase 2 (Mst2) and the scaffold protein Salvador (hSav1) (Mardin et al., 2010). This finding suggests that an upstream regulatory mechanism of TgNek1-2 localization remains to be identified. The schematics in Fig. 4D summarize the TgNek1-2 localization of the early division apparatus in the wild-type and V-A15 mutant phenotype. This schematic incorporates the observation made in Fig. 2B showing that spindle formation and spindle pole separation as highlighted by MORN1 still occurs in V-A15 at the restrictive temperature. Based on this we conclude that the primary function of TgNek1-2 lies in controlling centrosome splitting.
We also generated an YFP fusion reporter of TgNek1-2 to enable live cell imaging. The TgNek1-2-YFP encoding plasmid was transiently transfected into a line stably expressing CherryRFP-MORN1 (Fig. 5). These experiments identified a dynamic localization pattern for TgNek1-2-YFP throughout the division cycle (Fig. 5A). TgNek1-2 partially colocalizes with the spindle pole as marked by MORN1 in G1 (Fig. 5Aa), where it relocates to the duplicated centrosomes in S phase (Fig. 5Ab). In later stages, the signal translocates to the initial assembly site of daughter buds (Fig. 5Ac,d) and then expands to the cortical daughter cells (Fig. 5Ae). During bud formation and elongation TgNek1-2-YFP is also observed at the centrosomes, the spindle poles and the basal complexes (Fig. 5Ad,e). The dynamic localization pattern of TgNek1-2-YFP is summarized in the schematic provided in Fig. 5B; however, the result does not match with the anti-TgNek1-2 staining (Fig. 4A). We reasoned that the YFP fusion constructs driven by the ptub promoter results in artificial localization to the assembly site of daughter buds and bud cortex. However, it is also possible that the endogenous level of TgNek1-2 on the budding sites is too low for antibody detection.
Fig. 5.

Catalytic activity is required for cell-cycle-dependent localization of TgNek1-2. (A) Live imaging of transient expression of TgNek1-2-YFP driven by the α-tubulin promoter in a clonal RH line expressing Cherry-MORN1 shows a dynamic localization throughout late S phase (a), mitosis (b–d) and cytokinesis (b–e). (B) Schematic summary of TgNek1-2 localization throughout the cell cycle representing the stages shown in A. (C) Transient expression of TgNek1-2K135M-YFP driven by α-tubulin promoter in a clonal line expressing Cherry-MORN1 shows a cell-cycle-dependent localization (progressing from left to right).
It is of note that we were unable to establish stably transfected parasite lines expressing either the TgNek1-1 or the TgNek1-2 variants, either driven by the ptub or pnek1 promoter, or with an N- or C-terminal tag. This suggests that the tag interferes with the function of TgNek1 and is deleterious to tachyzoite survival. Lastly, we were unable to directly tag the genomic locus of TgNek1 at neither the stop-codon predicted by the ToxoDB predicted gene model, nor the stop-codons in the two splice variants identified by RACE PCR. As such, the YFP tag appears to confer an increased stability and/or mislocalization to TgNek1, which is ultimately lethal.
To test whether the kinase activity is required for TgNek1-2 localization, we generated a kinase dead allele of TgNek1-2 (TgNek1-2K135M) tagged at the C-terminus with YFP, and analyzed its localization by live cell imaging. Unlike wild-type TgNek1-2-YFP, TgNek1-2K135M-YFP disperses throughout the cytoplasm and associates with the initial budding site and the daughter cortex (Fig. 5C). Moreover, it is neither observed at the centrosomes nor at the spindle poles throughout the cell cycle. Surprisingly, we easily obtained parasites stably expressing this construct driven by the strong ptub promoter. This result indicates that the kinase activity is required for localization to the centrosome and spindle pole, but not for the potentially artificial localization to the cortex of the daughter buds. Moreover, the data show that overexpression of the kinase dead variant does not convey a dominant negative phenotype as is the case for the orthologous kinase in Giardia lamblia, where overexpression of GlNek1 results in a delay in disassembly of parental attachment disc and cytokinesis, as well as results in inhibition of excystation (Smith et al., 2012).
TgNek1-2 kinase activity and structure
The unexpected lack of a dominant negative phenotype upon overexpression of the active site K135M mutant lead us to question whether TgNek1-2 has kinase activity. Hereto we performed in vitro kinase assays. Bacterially expressed GST-TgNek1-2 phosphorylates the artificial kinase substrate β-casein (Fig. 6A, lane 2) but not BSA (data not shown). The kinase dead allele (GST-TgNek1-2K135M) was tested as well and its activity was completely abolished (Fig. 6A, lane 3). This suggests that the phosphorylation event is TgNek1-2 specific and does not originate from a bacterial kinase contamination, and it furthermore shows that K135 is required for catalytic activity.
Fig. 6.

Examination of TgNek1-2 kinase activity. (A) In vitro kinase assay using GST-TgNek1-2. Recombinant GST (lane 1), GST-TgNek1-2 (lane 2), GST-TgNek1-2K135M (kinase dead; lane 3) and GST-TgNek1-2C316R (ts-mutant; lane 4) were expressed and purified from bacteria and incubated with β-casein. The results were analyzed by SDS-PAGE electrophoresis and subjected to Coomassie staining and autoradiography. (B) Domain analysis of TgNek1-2 and HsNek2. Leucine zipper (LZ), coiled-coil (CC) and PEST domains are marked. The asterisk on TgNek1-2 indicates the Cys316Arg mutation. (C) Protein modeling of TgNek1-2 onto human Nek2 (Westwood et al., 2009). (D) Sequence alignment of T-loop in TgNek1-2 and HsNek2. Conserved and phosphorylated serine and threonines are marked in red. Glutamic acid and alanine substitutions in the activation loop mutants are shown in green. (E,F) Live cell images of transiently expressed TgNek1-23A-YFP (E) and TgNek1-2K135M+3E-YFP (F) driven by α-tubulin promoter in a line expressing Cherry-MORN1 show distinct localization patterns. (G) Alignment of the conserved region of TgNek1-2 containing the temperature-sensitive mutation in V-A15 with selected orthologs. Asterisks indicate conserved amino acids; dots indicate identical residues; colons indicate similar residues.
It has been reported that HsNek2 (Fry et al., 1999) and Aspergilus NIMA (Lu et al., 1993; O'Regan et al., 2007) can autophosphorylate. As shown in Fig. 6A, we did not detect autophosphorylation of TgNek1-2. For HsNek2 it has been shown that a cryptic Leu-zipper and coiled-coil domains functions in dimerization and is required for autophosphorylation (Fry et al., 1999). As such, we analyzed the domain structure of TgNek1-2 and compared it to the related kinases in human (HsNek2), Schizosaccharomyces pombe (Fin1) and Aspergillus (NIMA) (Fig. 6B). For TgNek1-2, the only conserved region is the catalytic domain and, surprisingly, no other functional or protein interacting domains were detected. For example, the coiled-coil domain and PEST sequence required for activity and regulation in fungal and several mammalian NIMA kinases are missing in TgNek1-2 (Fig. 6B) (O'Regan et al., 2007). It is of note that Aspergilus NIMA does not dimerize but still is able to autophosphorylate (Lu et al., 1993; O'Regan et al., 2007), suggesting that other, unknown regulatory mechanisms do exist. Either way, TgNek1-2 lacks the typical domains found in its relatives and therefore its activity and mechanism of action are likely different as well.
To test directly the potential role of the regions outside the kinase domain we generated N- and C-terminal deletions of TgNek1-2. As shown in Fig. 6B, the N-terminal extension sets the Toxoplasma Nek apart from its relatives. We generated an N-terminal deletion mutant (amino acids 1–89) of TgNek1-2 and analyzed its localization by a C-terminal YFP fusion protein (supplementary material Fig. S4A). Interestingly, ΔN-TgNek1-2-YFP showed a localization pattern similar to the wild-type construct. However, deletion of the N-terminus in combination with the kinase dead allele (ΔN-TgNek1-2K135M-YFP) completely abolished specific TgNek1-2 localization in the parasite and resulted in a weak cytoplasmic YFP signal (supplementary material Fig. S4B). Interestingly, deletion of the C-terminus (amino acids 104–499) had no effect on sub-cellular localization (data not shown), but it did relieve the disruption of cell division as observed for the wild-type control (supplementary material Fig. S5). This suggests that the C-terminus is a platform for binding of proteins crucial to progression of the division cycle or plays a direct rule in regulating TgNek1-2 function. Collectively, these results suggest that the kinase activity and the unique N-terminal domain are indispensable for proper function and localization of TgNek1-2.
It has been shown that the phosphorylation of T170, S171, T175 and T179 residues in the activation T-loop of HsNek2 is essential for centrosome splitting (Rellos et al., 2007). Therefore, we modeled the TgNek1-2 sequence on the crystal structures available for HsNek2 (Rellos et al., 2007; Westwood et al., 2009). As shown in Fig. 6C,D, the primary and secondary structure of the T-loop is well conserved, although the ortholog of T170 is missing in TgNek1-2. It is therefore reasonable to assume that phosphorylation of the corresponding Ser and Thr residues in TgNek1-2 would be required for activation of its activity. The current Toxoplasma phosphoproteome data does not contain any hints toward this (Treeck et al., 2011), but this is not very surprising as these data were collected from non-dividing, extracellular parasites, during which TgNek1-2 is undetectable (supplementary material Fig. S2). To determine whether the conserved residues in the activation loop (S278, S282 and T286) play a role in regulating TgNek1-2 kinase activity, we generated TgNek1-2 constructs carrying these three residues replaced by glutamic acid (TgNek1-23E) and alanine (TgNek1-23A), representing the phosphomimetic (activated) and the nonphosphorylatable (inactive) form of the regulating residues, respectively. Since we observed a distinct localization for the kinase dead allele upon expression of the YFP fusion in the parasite (Fig. 5A,C), we reasoned that we could differentiate the activation status by changes in YFP localization pattern for the activation loop mutants as well. Hence we transiently expressed these constructs in the CherryMORN1 background to track their localization pattern during cytokinesis. TgNek1-23A-YFP showed a localization pattern similar to the wild-type construct (Fig. 6E). Interestingly, we easily obtained stable lines expressing TgNek1-23A-YFP, which suggests that this construct, like the K135M mutant, is not toxic to the parasite. Moreover, these results indicate that the phosphorylatable activation loop residues are required for activating the kinase activity. Extending on this observation, we reasoned that testing the 3E mutant in the K135M background would result in unviable parasites. Indeed we were unable to establish stable parasites transfected with the TgNek1-2K135M+3E-YFP construct. Surprisingly, when this construct was transiently expressed in the CherryMORN1 background we observed cytoplasmic YFP localization at all times (Fig. 6F). Hence, these data show that both the actual active catalytic site in combination with the phosphorylated activation loop are required for localization to the centrosome and that loss of either feature can overcome the cytotoxic effect. Finally, we tested the in vitro kinase activity of both constructs. As anticipated, for neither TgNek1-2K135M+3E nor TgNek1-23A could we detect in vitro kinase activity (supplementary material Fig. S6). Collectively, the phosphorylation status of the T-loop regulates kinase activity of TgNek1-2, which is intrinsically linked to its sub-cellular localization.
Finally, we tested the kinase activity of the V-A15 ts-mutant allele (GST-TgNek1-2C316R). We could not detect kinase activity of the ts variant (Fig. 6A, lane 4). By protein alignment, the Cys316 is conserved among the NIMA family of kinases and is located at the edge of the active site at the end of a conserved α-helix (Fig. 6C,G), suggesting a conserved and crucial role in the function of the protein (to our knowledge, a mutant allele in this conserved residue has not been reported before). It is possible that the C316R variant completely misfolds explaining its lack of activity (it is of note that we did test a comparable amount of protein in the kinase assay: Fig. 6A, left panel). Therefore we performed in silico mutagenesis to test a potential effect of the point mutation on protein structure (data not shown). We did not observe a major effect on the conformation of the protein besides that the change into arginine has the ability to form hydrogen bonds with the adjacent helixes, which could make the structure of the protein more rigid resulting in loss of activity. Alternatively, it is possible that the C-terminal GST tag interferes with dimerization of the kinase; however, we cannot differentiate these possibilities based on the current data.
Discussion
We have shown that Toxoplasma TgNek1-2 functions in centrosome splitting (Figs 1, 4) and therefore is the functional ortholog of related kinases found in Aspergillus (NIMA), S. pombe (Fin1) and humans (HsNek2). Moreover, a mutant allele of TgNek1 shows that a defect in centrosome splitting results in only a single daughter bud (Figs 1, 2). Therefore, our data provide the formal proof for the long held model that the centrosome is the spatial scaffold for daughter cell formation in apicomplexan parasites (Anderson-White et al., 2012; Gubbels et al., 2008b; Hu et al., 2002; Striepen et al., 2000). Moreover, our data show the tight coordination of mitotic events with cytokinetic events mediated by the centrosome. How daughters are initiated on the centrosome is not well understood. Very recently, it was shown that a striated fiber assembly (SFA) protein structure emanates from the centrosome and is required for deposition of the daughter microtubular cytoskeleton (Francia et al., 2012). Vesicles are targeted to he microtubules and fuse into the alveolar vesicles mediated by Rab11B (Agop-Nersesian et al., 2010). The occurrence of Rab11B in the daughter bud coincides with the transition of intermediate filament-like protein IMC15 from the centrosome to bud, followed by the assembly of numerous other cytoskeleton scaffold proteins (Anderson-White et al., 2012). However, how these events are coordinated remains a mystery, and our observations on TgNek1 provide a first glimpse at this mechanism.
In many other eukaryotic systems, centrosomes also play a key role in chromosome segregation by mediating formation of the mitotic spindle. Toxoplasma mitosis progresses by closed mitosis accompanied by very limited chromosome condensation wherein the spindle poles are closely apposed rather than on opposite sides of the nucleus (Brooks et al., 2011; Gubbels et al., 2008b; Striepen et al., 2007). The centrosome residing in the cytoplasm serve as microtubule organizing center for the microtubules making up the spindle poles embedded in the nuclear lamina (Gubbels et al., 2006). Note that this is dramatically different from yeast with the merged spindle pole and centrosome structure in the SPB, even though mitosis also progresses in a closed fashion. Deletion and overexpression of S. pombe Fin1 results in spindle formation defects, and fin1 genetically interacts with the spindle checkpoint proteins Mad2 and Bub1 (Grallert and Hagan, 2002). In humans, Nek2 also regulates chromosome alignment and signaling of the spindle assembly checkpoint by phosphorylating the kinetochore protein Ndc80/Hec1 (Wei et al., 2011). Our observations using MORN1 as a marker for the spindle indicate that in mutant V-A15 the spindle pole divides and the structure we observe in Fig. 2B likely reflects the spindle. As such, it appears that mitosis progresses normally in mutant V-A15. The nuclear fragmentation observed in later stages indicates that karyokinesis rather than mitosis is compromised in the mutant. Since karyokinesis is tightly linked to cytokinesis, which with a single bud does not progress normally, it is difficult to assess whether the karyokinesis defect is a direct or indirect effect of the mutation in TgNek1.
Our data also show that with a single amino acid change in TgNek1 the centrosome duplicates normally but fails to split properly (Figs 1, 2, 4). Furthermore, the spatio-temporal localization pattern of TgNek1-2 fits a role in the centrosome duplication process (Fig. 4). This observation is consistent with the function of HsNek2, which phosphorylates several components required for centrosome splitting: the centriole-associated filament proteins C-Nap1 and rootletin (Bahe et al., 2005; Fry et al., 1998a). In Toxoplasma we could not identify clear C-Nap1 or rootletin by reciprocal BLAST searches. In yeasts, spindle pole body separation is mediated by phosphorylation of Sfi1p, which is a protein coating the centrin fiber connecting duplicated, unsplit SPBs (Salisbury, 2007). A convincing ortholog of Sfi1p is absent from the Toxoplasma genome but the different centrin proteins are encoded. It has not been studied whether any of these centrins has a role in centrosome splitting. Furthermore, in other systems a highly conserved mitotic kinesin assists in pushing the duplicated centrosomes apart: Eg5 in humans, BimC in A. nidulans, Kip1 in S. cerevisiae and Cut7 in S. pombe (Mardin et al., 2010). In Toxoplasma we could not identify a convincing Eg5 ortholog by reciprocal BLAST searches. Lastly, it has been shown that HsNek2 overexpression does promote premature separation of centrosomes (Fry et al., 1998b). We performed a comparable experiment in Toxoplasma using a destabilization domain (DD) TgNek1-2 allele whose stability can be controlled by addition of the small molecule Shield-1 (Herm-Götz et al., 2007). As shown in supplementary material Fig. S5A, we did not observe an early defect associated with misregulated centrosome duplication but rather observed a late defect in cytokinesis. Taken together, this observation and the comparison with other well-studied systems suggest that TgNek1-2 likely regulates centrosome splitting through a mechanism different from mammalian and yeast centrosome/SPB separation. Our future work will therefore focus on identification of the substrates of TgNek1-2 to uncover its exact mechanism of action.
The interpretation of sub-cellular localization studies of various TgNek1-2 mutation and deletion constructs is slightly complicated by the differences between the exclusive centrosome localization of the specific antiserum (Fig. 4A) and the additional localization of the wild-type TgNek1-2-YFP fusion to the daughter buds (Fig. 5A). Moreover, the ectopic expression of TgNek1-2-YFP induces lethality, suggesting the YFP tag interferes with correct function. However, careful direct comparison of the wild-type constructs with the mutant constructs permits us to draw some conclusion on structure function relationships. In the expression of the YFP-tagged versions we showed that kinase activity is responsible for the lethality to parasite, since a stable transgenic line expressing the kinase dead K135M allele driven by the tubulin promoter is completely viable (Fig. 5C). Note that we experimentally validated that this mutation results in abolition of kinase activity (Fig. 6A). Removal of the unique N-terminal extension does not dramatically change its localization pattern (supplementary material Fig. S4). However, specific localization was abolished in combination with the kinase dead (K135M) mutation, suggesting the N-terminus has a function in localization to the centrosome, dependent on kinase activity. Interestingly deletion of the C-terminal domain relieves the dominant lethal effect of the full-length control construct (supplementary material Fig. S5). This suggests that the C-terminus likely serves as a platform for binding of proteins crucial to progression of the division cycle (likely checkpoint proteins) that are not timely removed in overexpression of the full-length control.
In general, Nek kinases are activated during the G2/M transition by autophosphorylation and phosphorylation by mitotic kinases, i.e. cyclin-dependent kinase (cdk) and nuclear Dbf2-related (NDR) kinase (Grallert et al., 2012; O'Regan et al., 2007; Ye et al., 1995). The crosstalk between other mitotic kinases (notably Polo kinase) and phosphatases (Cdc25 and PP1) and the positive feedback loop of mitosis promoting factors may also contribute to early activation of Nek kinases. Three of the four phosphorylated residues responsible for HsNek2 activation are conserved in TgNek1-2, and our results indicate that TgNek1-2 activity and localization are similarly controlled by phosphorylation status of the activation loop (Fig. 6; supplementary material Fig. S6). Moreover, amino acid replacements mimicking the phosphorylated or unphosphorylated stated for the activation loop resembled the difference in lethality and sub-cellular localizations observed with the K135M mutation in the active site. Mammalian Polo-kinase Plk1 has been shown to control centrosome disjunction by regulating the HsNek2–PP1γ-Mst1 complex. Recruitment of a phosphatase PP1γ counteracts phosphorylation events of HsNek2 on C-Nap1 (Mardin et al., 2011). How these pathways operate in Toxoplasma has not been explored, although orthologs of a Polo and NDR kinases are not part of the annotated kinome (Peixoto et al., 2010), making a cdk the most likely candidate to activate TgNek1 (e.g. TPK2; Khan et al., 2002). Once Neks have done their duty, their degradation is mediated by PEST domains. We could not identify a PEST domain in TgNek1-2 (Fig. 6), so the mechanism of its inactivation or degradation is unclear. However, TgNek1-2's transcript level cycles strongly throughout the division cycle (Behnke et al., 2010) and could contribute to timing its presence.
In conclusion, TgNek1-2 clearly plays a role in centrosome splitting, but how it is activated and how it exerts its function remains largely unknown. It is tempting to hypothesize that TgNek1-2 functions as a checkpoint protein to monitor completion of centrosome segregation. If not, it remains associated with the unsplit centrosome until the parasite progresses through later events of mitosis (Fig. 4D). Given the clear differences in TgNek1-2 compared with the well-studied orthologs in mammals and yeasts this provides an opportunity to explore its mechanism as a potential specific drug target. This promise is supported by the very existence of lethal temperature-sensitive mutant of TgNek1, suggesting its activity is a strict requirement for progression through the replication cycle. Further characterization of TgNek1 and its roles in mitosis and cell division requires the expansion of the Toxoplasma toolbox with various reagents, which will be an exciting direction of our future work.
Material and Methods
Parasites
RH Toxoplasma gondii tachyzoites and its transgenic derivatives were grown in human foreskin fibroblast (HFF) as described in (Roos et al., 1995). Stable transgenics were established by chloramphenicol, pyrimethamine or phleomycin selection and cloned by limiting dilution. V-A15 ts-mutant was obtained by chemical mutagenesis described in (Gubbels et al., 2008a). The mutant was grown at 35°C (permissive), and the temperature-sensitive phenotypes were analyzed under 40°C (restrictive) incubation.
RACE PCR
We determined the 5′- and 3′-ends of TgNek1 by Rapid Amplification of cDNA Ends (RACE) using the GeneRacer kit (Invitrogen) according to the manufacturer's instruction. Amplified fragments were TOPO cloned (Stratagene) and sequenced. Based on these results full-length cDNA for the two different splice variants was amplified from RH strain cDNA by nested PCR and verified by sequencing. All primer sequences are provided in supplementary material Table S1.
Plasmids
YFP tagging plasmids are based on the ptub-YFP2(MCS)/sagCAT plasmid (Gubbels et al., 2003; Anderson-White et al., 2011) and were generated by replacing modular segment, either the α-tubulin promoter (PmeI/BglII), the first YFP (BglII/AvrII), the second YFP (AvrII/EcoRV) or the entire tandem YFP cassette (BglII/EcoRV). The sagCATsag cassette was replaced by the sagBLEsag cassette (van Dooren et al., 2008) by XhoI digest swapping. ptub-TgNek1-2-YFP(MCS)/sagCAT was cloned by BamHI/AvrII. Approximately 1500 kb endogenous promoter was PCR amplified from genomic DNA and cloned by PmeI/BglII sites into ptub-TgNek1-2-YFP(MCS)/sagCAT. ptub-DDYFP(MCS)/sagCAT plasmid was generated by cloning PCR amplified DDYFP with BglII/AvrII from ptub-DDYFPIMC5(MCS)/sagCAT (Anderson-White et al., 2011) into ptub-YFP2(MCS)/sagCAT for N-terminal fusion. C-terminal TgNek1-2 (396-499 aa) was amplified and cloned into AVA0421 (Alexandrov et al., 2004) to create N-terminal His6 fusion. Full length TgNEK1-2 were PCR amplified and cloned by EcoRI/NotI sites into pGEX4T-1 plasmid to generate N-terminal GST fusions. The TgNek1-2K135M, TgNek1-2K135M+3E and TgNek1-23A mutant alleles were generated by megaprimer as described in (Lai et al., 2003) using GST-TgNek1-2 as template. The mutant was also cloned into ptub-YFP2(MCS)/sagCAT plasmid by BamHI/AvrII. Endogenous tagging of the TgNek1 locus was performed as described (Huynh and Carruthers, 2009) using pYFP-LIC-DHFR vector.
Immunofluorescence assay and light microscopy
Immunofluorescence assays were performed as described (Gubbels et al., 2006). The following primary antibodies were used: rabbit anti-HsCentrin (1∶1000) (kindly shared by Dr Iain Cheeseman, Whitehead Institute); rabbit anti-TgMORN1 (1∶2000) (Gubbels et al., 2006), rat anti-TgIMC3 (1∶2000) (Anderson-White et al., 2011), mouse anti-c-Myc (1∶50; Santa Cruz Biotech), rabbit anti-GFP (1∶200; Torrey Pines Biolabs), mouse anti-GFP (1∶500; Abgent), guinea pig anti-TgNek1-2 (1∶1000; this study). Alexa Fluor 350, 488, 568, 594 and 633 goat anti-mouse, -rabbit, -rat and -guinea pig secondary antibodies were used (1∶200; Invitrogen). 4′,6′-diamidino-2-phenylindole (DAPI) was used to stain nuclear material. Images were collected using a Zeiss Axiovert 200M wide-field fluorescent microscope equipped with an a-Plan-Fluor 100×/1.45 NA oil objective and a Hamamatsu C4742-95 CCD camera. Images were collected, deconvolved and adjusted for phase-contrast using Volocity software (Improvision). Super-resolution images were obtained by 3D structured illumination (DeltaVision Core; Applied Precision). 3D datasets were deconvolved using constrained iterative restoration as implemented in SoftWoRx software (Applied Precision). Live cell time-lapse movies were acquired using a Leica TCS SP5 inverted confocal microscope with a 63×/1.4 oil objective.
3-MA synchronization
3-methyladenine (Sigma) synchronization was performed essentially as described by (Wang et al., 2010). In short, parasites were seeded on coverslips in 24-well plates confluent with HFF cells and grown at the desired temperature. After two hours, extracellular parasites were removed by a 1×PBS wash and remaining parasites were treated with 10 mM 3-MA for 6 hours in ED1 medium (Roos et al., 1995); the block was released by replacement with fresh ED1 medium. At least 200 vacuoles per time point were analyzed by immunofluorescence and experiments were three times independently replicated.
Generation of antisera
Plasmid AVA0421-TgNek1-2 (396–499 amino acid) was expressed in BL21 Escherichia coli (Invitrogen) and purified over TALON Resin (Clontech). Polyclonal guinea pig antisera were generated by Covance (Denver, PA).
DNA content analysis
Protocol essentially as previously described (Jammallo et al., 2011). In short, parasites were seeded and grown at the desired temperature and after 2 hours extracellular parasites were removed to synchronize the culture. Parasites were harvested after 6, 12 and 18 hours, and filtered through a 12 µm pore polycarbonate filter (Millipore) to allow detection of large, mutant parasites. DNA was stained with Sytox Green and samples were analyzed on FACSCanto flow cytometer (Becton Dickinson) with FITC filter set. Data were analyzed using FloJo software (Treestar).
In vitro kinase assay
GST-TgNek1-2 variants were expressed in BL21 E. coli bacteria and purified using glutathione sepharose resin (GE). Recombinant kinase was incubated with 5 µCi of gamma-32P (Perkin Elmer), 10 µM cold ATP, 1×kinase buffer (10 mM Tris/HCl pH 7.4 and 10 mM MgCl2) and 5 µg of β-casein (Sigma). The reaction was incubated at 30°C for 30 minutes and terminated by addition of 10 µl 4×SDS sample buffer and boiling for 3 minutes. The samples were examined by SDS-PAGE electrophoresis and radiation imaged by PhosphorImager (Molecular Dynamics).
Western blot analysis
Purified parasites collected after needle passage, 3 µm filtering and centrifugation were washed twice in 1×PBS and lysed in RIPA buffer. Cleared parasite lysates representing 20 million parasites separated by SDS-PAGE gels and transferred to nitrocellulose membrane. The blots were probed by anti-TgNek1-2 (1∶1000) and α-tubulin 12G10 (1∶500) [Hybridoma Bank, University of Iowa (Jerka-Dziadosz et al., 1995)], goat anti-guinea pig IgG-HRP sc-2438 (1∶3000; Santa Cruz) and goat anti-mouse HRP (1∶10,000; Invitrogen).
Sequence analysis and modeling
The identified protein sequences were subject to searches through PFam, SMART and ProSite databases. Coiled-coil regions were predicted using the on line Coils-server (Lupas et al., 1991) and PEST sequences using http://emboss.bioinformatics.nl/cgi-bin/emboss/epestfind using default settings. Sequences were aligned using default settings in ClustalW (Larkin et al., 2007). TgNek1-2 was searched against available 3D structures on Swiss-model (http://swissmodel.expasy.org) using automated mode (Arnold et al., 2006). The obtained TgNek1-2 model coordinates were visualized using PyMOL (http://www.pymol.org). Successful modeling information was obtained for TgNek1-2 on the following sections: Residues 102 to 385 (out of a total length of 499 residues) to the structure of human NEK2 ATPγS-bound (Swissmodel template 2w5bA) (Westwood et al., 2009). The C316R point mutation was modeled using DeepView (http://spdbv.vital-it.ch) (Guex and Peitsch, 1997).
Supplementary Material
Acknowledgments
We thank Brain Benenati for assistance with protein expression; Drs Fay Dufort and Thomas Chiles for assistance with flow cytometry; Bret Judson for the time-lapse imaging; and the 2011 ‘Yale Microscopy Workshop and Symposium’ for the 3D SIM images.
Footnotes
Author contributions
C.-T.C. and M.-J.G. designed the study, interpreted the data and co-wrote the paper; C.-T.C. performed the experiments.
Funding
This work was funded by a National Institutes of Health grant [grant number AI081924 to M.J.G.]; a March of Dimes Basil O'Connor Starter Scholar Research Award [5-FY09-08 to M.-J.G.]; and a Knights Templar Eye Foundation Inc. Research Award [to C.-T.C.]. Deposited in PMC for release after 12 months.
Supplementary material available online at http://jcs.biologists.org/lookup/suppl/doi:10.1242/jcs.123364/-/DC1
References
- Agop-Nersesian C., Egarter S., Langsley G., Foth B. J., Ferguson D. J., Meissner M. (2010). Biogenesis of the inner membrane complex is dependent on vesicular transport by the alveolate specific GTPase Rab11B. PLoS Pathog. 6, e1001029 10.1371/journal.ppat.1001029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexandrov A., Vignali M., LaCount D. J., Quartley E., de Vries C., De Rosa D., Babulski J., Mitchell S. F., Schoenfeld L. W., Fields S. et al. (2004). A facile method for high-throughput co-expression of protein pairs. Mol. Cell. Proteomics 3, 934–938 10.1074/mcp.T400008-MCP200 [DOI] [PubMed] [Google Scholar]
- Anderson-White B. R., Ivey F. D., Cheng K., Szatanek T., Lorestani A., Beckers C. J., Ferguson D. J., Sahoo N., Gubbels M. J. (2011). A family of intermediate filament-like proteins is sequentially assembled into the cytoskeleton of Toxoplasma gondii. Cell. Microbiol. 13, 18–31 10.1111/j.1462-5822.2010.01514.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson-White B. R., Beck J. R., Chen C. T., Meissner M., Bradley P. J., Gubbels M. J. (2012). Cytoskeleton assembly in Toxoplasma gondii cell division. Int. Rev. Cell Mol. Biol. 298, 1–31 10.1016/B978-0-12-394309-5.00001-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arnold K., Bordoli L., Kopp J., Schwede T. (2006). The SWISS-MODEL workspace: a web-based environment for protein structure homology modelling. Bioinformatics 22, 195–201 10.1093/bioinformatics/bti770 [DOI] [PubMed] [Google Scholar]
- Bahe S., Stierhof Y. D., Wilkinson C. J., Leiss F., Nigg E. A. (2005). Rootletin forms centriole-associated filaments and functions in centrosome cohesion. J. Cell Biol. 171, 27–33 10.1083/jcb.200504107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Behnke M. S., Wootton J. C., Lehmann M. M., Radke J. B., Lucas O., Nawas J., Sibley L. D., White M. W. (2010). Coordinated progression through two subtranscriptomes underlies the tachyzoite cycle of Toxoplasma gondii. PLoS ONE 5, e12354 10.1371/journal.pone.0012354 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brooks C. F., Francia M. E., Gissot M., Croken M. M., Kim K., Striepen B. (2011). Toxoplasma gondii sequesters centromeres to a specific nuclear region throughout the cell cycle. Proc. Natl. Acad. Sci. USA 108, 3767–3772 10.1073/pnas.1006741108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y., Riley D. J., Zheng L., Chen P. L., Lee W. H. (2002). Phosphorylation of the mitotic regulator protein Hec1 by Nek2 kinase is essential for faithful chromosome segregation. J. Biol. Chem. 277, 49408–49416 10.1074/jbc.M207069200 [DOI] [PubMed] [Google Scholar]
- Doxsey S., Zimmerman W., Mikule K. (2005). Centrosome control of the cell cycle. Trends Cell Biol. 15, 303–311 10.1016/j.tcb.2005.04.008 [DOI] [PubMed] [Google Scholar]
- Francia M. E., Jordan C. N., Patel J. D., Sheiner L., Demerly J. L., Fellows J. D., de Leon J. C., Morrissette N. S., Dubremetz J. F., Striepen B. (2012). Cell division in Apicomplexan parasites is organized by a homolog of the striated rootlet fiber of algal flagella. PLoS Biol. 10, e1001444 10.1371/journal.pbio.1001444 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fry A. M., Mayor T., Meraldi P., Stierhof Y. D., Tanaka K., Nigg E. A. (1998a). C-Nap1, a novel centrosomal coiled-coil protein and candidate substrate of the cell cycle-regulated protein kinase Nek2. J. Cell Biol. 141, 1563–1574 10.1083/jcb.141.7.1563 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fry A. M., Meraldi P., Nigg E. A. (1998b). A centrosomal function for the human Nek2 protein kinase, a member of the NIMA family of cell cycle regulators. EMBO J. 17, 470–481 10.1093/emboj/17.2.470 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fry A. M., Arnaud L., Nigg E. A. (1999). Activity of the human centrosomal kinase, Nek2, depends on an unusual leucine zipper dimerization motif. J. Biol. Chem. 274, 16304–16310 10.1074/jbc.274.23.16304 [DOI] [PubMed] [Google Scholar]
- Grallert A., Hagan I. M. (2002). Schizosaccharomyces pombe NIMA-related kinase, Fin1, regulates spindle formation and an affinity of Polo for the SPB. EMBO J. 21, 3096–3107 10.1093/emboj/cdf294 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grallert A., Connolly Y., Smith D. L., Simanis V., Hagan I. M. (2012). The S. pombe cytokinesis NDR kinase Sid2 activates Fin1 NIMA kinase to control mitotic commitment through Pom1/Wee1. Nat. Cell Biol. 14, 738–745 10.1038/ncb2514 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gubbels M. J., Li C., Striepen B. (2003). High-throughput growth assay for Toxoplasma gondii using yellow fluorescent protein. Antimicrob. Agents Chemother. 47, 309–316 10.1128/AAC.47.1.309-316.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gubbels M. J., Vaishnava S., Boot N., Dubremetz J. F., Striepen B. (2006). A MORN-repeat protein is a dynamic component of the Toxoplasma gondii cell division apparatus. J. Cell Sci. 119, 2236–2245 10.1242/jcs.02949 [DOI] [PubMed] [Google Scholar]
- Gubbels M. J., Lehmann M., Muthalagi M., Jerome M. E., Brooks C. F., Szatanek T., Flynn J., Parrot B., Radke J., Striepen B. et al. (2008a). Forward genetic analysis of the apicomplexan cell division cycle in Toxoplasma gondii. PLoS Pathog. 4, e36 10.1371/journal.ppat.0040036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gubbels M. J., White M., Szatanek T. (2008b). The cell cycle and Toxoplasma gondii cell division: tightly knit or loosely stitched? Int. J. Parasitol. 38, 1343–1358 10.1016/j.ijpara.2008.06.004 [DOI] [PubMed] [Google Scholar]
- Guex N., Peitsch M. C. (1997). SWISS-MODEL and the Swiss-PdbViewer: an environment for comparative protein modeling. Electrophoresis 18, 2714–2723 10.1002/elps.1150181505 [DOI] [PubMed] [Google Scholar]
- Hayward D. G., Fry A. M. (2006). Nek2 kinase in chromosome instability and cancer. Cancer Lett. 237, 155–166 10.1016/j.canlet.2005.06.017 [DOI] [PubMed] [Google Scholar]
- Herm-Götz A., Agop-Nersesian C., Münter S., Grimley J. S., Wandless T. J., Frischknecht F., Meissner M. (2007). Rapid control of protein level in the apicomplexan Toxoplasma gondii. Nat. Methods 4, 1003–1005 10.1038/nmeth1134 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu K., Mann T., Striepen B., Beckers C. J., Roos D. S., Murray J. M. (2002). Daughter cell assembly in the protozoan parasite Toxoplasma gondii. Mol. Biol. Cell 13, 593–606 10.1091/mbc.01-06-0309 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu K., Johnson J., Florens L., Fraunholz M., Suravajjala S., DiLullo C., Yates J., Roos D. S., Murray J. M. (2006). Cytoskeletal components of an invasion machine – the apical complex of Toxoplasma gondii. PLoS Pathog. 2, e13 10.1371/journal.ppat.0020013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huynh M. H., Carruthers V. B. (2009). Tagging of endogenous genes in a Toxoplasma gondii strain lacking Ku80. Eukaryot. Cell 8, 530–539 10.1128/EC.00358-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jammallo L., Eidell K., Davis P. H., Dufort F. J., Cronin C., Thirugnanam S., Chiles T. C., Roos D. S., Gubbels M. J. (2011). An insertional trap for conditional gene expression in Toxoplasma gondii: identification of TAF250 as an essential gene. Mol. Biochem. Parasitol. 175, 133–143 10.1016/j.molbiopara.2010.10.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jerka-Dziadosz M., Jenkins L. M., Nelsen E. M., Williams N. E., Jaeckel-Williams R., Frankel J. (1995). Cellular polarity in ciliates: persistence of global polarity in a disorganized mutant of Tetrahymena thermophila that disrupts cytoskeletal organization. Dev. Biol. 169, 644–661 10.1006/dbio.1995.1176 [DOI] [PubMed] [Google Scholar]
- Khan F., Tang J., Qin C. L., Kim K. (2002). Cyclin-dependent kinase TPK2 is a critical cell cycle regulator in Toxoplasma gondii. Mol. Microbiol. 45, 321–332 10.1046/j.1365-2958.2002.03026.x [DOI] [PubMed] [Google Scholar]
- Lai R., Bekessy A., Chen C. C., Walsh T., Barnard R. (2003). Megaprimer mutagenesis using very long primers. Biotechniques 34, 52–54, 56 [DOI] [PubMed] [Google Scholar]
- Larkin M. A., Blackshields G., Brown N. P., Chenna R., McGettigan P. A., McWilliam H., Valentin F., Wallace I. M., Wilm A., Lopez R. et al. (2007). Clustal W and Clustal X version 2.0. Bioinformatics 23, 2947–2948 10.1093/bioinformatics/btm404 [DOI] [PubMed] [Google Scholar]
- Lu K. P., Osmani S. A., Means A. R. (1993). Properties and regulation of the cell cycle-specific NIMA protein kinase of Aspergillus nidulans. J. Biol. Chem. 268, 8769–8776 [PubMed] [Google Scholar]
- Lupas A., Van Dyke M., Stock J. (1991). Predicting coiled coils from protein sequences. Science 252, 1162–1164 10.1126/science.252.5009.1162 [DOI] [PubMed] [Google Scholar]
- Mardin B. R., Lange C., Baxter J. E., Hardy T., Scholz S. R., Fry A. M., Schiebel E. (2010). Components of the Hippo pathway cooperate with Nek2 kinase to regulate centrosome disjunction. Nat. Cell Biol. 12, 1166–1176 10.1038/ncb2120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mardin B. R., Agircan F. G., Lange C., Schiebel E. (2011). Plk1 controls the Nek2A-PP1gamma antagonism in centrosome disjunction. Curr. Biol. 21, 1145–1151 [DOI] [PubMed] [Google Scholar]
- Morris N. R., Osmani S. A., Engle D. B., Doonan J. H. (1989). The genetic analysis of mitosis in Aspergillus nidulans. Bioessays 10, 196–201 10.1002/bies.950100605 [DOI] [PubMed] [Google Scholar]
- Nishi M., Hu K., Murray J. M., Roos D. S. (2008). Organellar dynamics during the cell cycle of Toxoplasma gondii. J. Cell Sci. 121, 1559–1568 10.1242/jcs.021089 [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Connell M. J., Krien M. J., Hunter T. (2003). Never say never. The NIMA-related protein kinases in mitotic control. Trends Cell Biol. 13, 221–228 10.1016/S0962-8924(03)00056-4 [DOI] [PubMed] [Google Scholar]
- O'Regan L., Blot J., Fry A. M. (2007). Mitotic regulation by NIMA-related kinases. Cell Div. 2, 25 10.1186/1747-1028-2-25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osmani S. A., May G. S., Morris N. R. (1987). Regulation of the mRNA levels of nimA, a gene required for the G2-M transition in Aspergillus nidulans. J. Cell Biol. 104, 1495–1504 10.1083/jcb.104.6.1495 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peixoto L., Chen F., Harb O. S., Davis P. H., Beiting D. P., Brownback C. S., Ouloguem D., Roos D. S. (2010). Integrative genomic approaches highlight a family of parasite-specific kinases that regulate host responses. Cell Host Microbe 8, 208–218 10.1016/j.chom.2010.07.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pradel L. C., Bonhivers M., Landrein N., Robinson D. R. (2006). NIMA-related kinase TbNRKC is involved in basal body separation in Trypanosoma brucei. J. Cell Sci. 119, 1852–1863 10.1242/jcs.02900 [DOI] [PubMed] [Google Scholar]
- Radke J. R., Striepen B., Guerini M. N., Jerome M. E., Roos D. S., White M. W. (2001). Defining the cell cycle for the tachyzoite stage of Toxoplasma gondii. Mol. Biochem. Parasitol. 115, 165–175 10.1016/S0166-6851(01)00284-5 [DOI] [PubMed] [Google Scholar]
- Reininger L., Billker O., Tewari R., Mukhopadhyay A., Fennell C., Dorin-Semblat D., Doerig C., Goldring D., Harmse L., Ranford-Cartwright L. et al. (2005). A NIMA-related protein kinase is essential for completion of the sexual cycle of malaria parasites. J. Biol. Chem. 280, 31957–31964 10.1074/jbc.M504523200 [DOI] [PubMed] [Google Scholar]
- Reininger L., Tewari R., Fennell C., Holland Z., Goldring D., Ranford-Cartwright L., Billker O., Doerig C. (2009). An essential role for the Plasmodium Nek-2 Nima-related protein kinase in the sexual development of malaria parasites. J. Biol. Chem. 284, 20858–20868 10.1074/jbc.M109.017988 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rellos P., Ivins F. J., Baxter J. E., Pike A., Nott T. J., Parkinson D. M., Das S., Howell S., Fedorov O., Shen Q. Y. et al. (2007). Structure and regulation of the human Nek2 centrosomal kinase. J. Biol. Chem. 282, 6833–6842 10.1074/jbc.M609721200 [DOI] [PubMed] [Google Scholar]
- Roos D. S., Donald R. G., Morrissette N. S., Moulton A. L. (1995). Molecular tools for genetic dissection of the protozoan parasite Toxoplasma gondii. Methods Cell Biol. 45, 27–63 10.1016/S0091-679X(08)61845-2 [DOI] [PubMed] [Google Scholar]
- Salisbury J. L. (2007). A mechanistic view on the evolutionary origin for centrin-based control of centriole duplication. J. Cell. Physiol. 213, 420–428 10.1002/jcp.21226 [DOI] [PubMed] [Google Scholar]
- Seshan A. A., Amon A. (2004). Linked for life: temporal and spatial coordination of late mitotic events. Curr. Opin. Cell Biol. 16, 41–48 10.1016/j.ceb.2003.11.003 [DOI] [PubMed] [Google Scholar]
- Smith A. J., Lauwaet T., Davids B. J., Gillin F. D. (2012). Giardia lamblia Nek1 and Nek2 kinases affect mitosis and excystation. Int. J. Parasitol. 42, 411–419 10.1016/j.ijpara.2012.03.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Striepen B., Crawford M. J., Shaw M. K., Tilney L. G., Seeber F., Roos D. S. (2000). The plastid of Toxoplasma gondii is divided by association with the centrosomes. J. Cell Biol. 151, 1423–1434 10.1083/jcb.151.7.1423 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Striepen B., Jordan C. N., Reiff S., van Dooren G. G. (2007). Building the perfect parasite: cell division in apicomplexa. PLoS Pathog. 3, e78 10.1371/journal.ppat.0030078 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szatanek T., Anderson-White B. R., Faugno-Fusci D. M., White M., Saeij J. P., Gubbels M. J. (2012). Cactin is essential for G1 progression in Toxoplasma gondii. Mol. Microbiol. 84, 566–577 10.1111/j.1365-2958.2012.08044.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Treeck M., Sanders J. L., Elias J. E., Boothroyd J. C. (2011). The phosphoproteomes of Plasmodium falciparum and Toxoplasma gondii reveal unusual adaptations within and beyond the parasites' boundaries. Cell Host Microbe 10, 410–419 10.1016/j.chom.2011.09.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Dooren G. G., Tomova C., Agrawal S., Humbel B. M., Striepen B. (2008). Toxoplasma gondii Tic20 is essential for apicoplast protein import. Proc. Natl. Acad. Sci. USA 105, 13574–13579 10.1073/pnas.0803862105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y., Karnataki A., Parsons M., Weiss L. M., Orlofsky A. (2010). 3-Methyladenine blocks Toxoplasma gondii division prior to centrosome replication. Mol. Biochem. Parasitol. 173, 142–153 10.1016/j.molbiopara.2010.05.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei R., Ngo B., Wu G., Lee W. H. (2011). Phosphorylation of the Ndc80 complex protein, HEC1, by Nek2 kinase modulates chromosome alignment and signaling of the spindle assembly checkpoint. Mol. Biol. Cell 22, 3584–3594 10.1091/mbc.E11-01-0012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westwood I., Cheary D. M., Baxter J. E., Richards M. W., van Montfort R. L., Fry A. M., Bayliss R. (2009). Insights into the conformational variability and regulation of human Nek2 kinase. J. Mol. Biol. 386, 476–485 10.1016/j.jmb.2008.12.033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wloga D., Camba A., Rogowski K., Manning G., Jerka-Dziadosz M., Gaertig J. (2006). Members of the NIMA-related kinase family promote disassembly of cilia by multiple mechanisms. Mol. Biol. Cell 17, 2799–2810 10.1091/mbc.E05-05-0450 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woodbury E. L., Morgan D. O. (2007). The role of self-association in Fin1 function on the mitotic spindle. J. Biol. Chem. 282, 32138–32143 10.1074/jbc.M705344200 [DOI] [PubMed] [Google Scholar]
- Xue M., Giagtzoglou N., Bellen H. J. (2011). Dueling Ca2+ sensors in neurotransmitter release. Cell 147, 491–493 10.1016/j.cell.2011.10.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye X. S., Xu G., Pu R. T., Fincher R. R., McGuire S. L., Osmani A. H., Osmani S. A. (1995). The NIMA protein kinase is hyperphosphorylated and activated downstream of p34cdc2/cyclin B: coordination of two mitosis promoting kinases. EMBO J. 14, 986–994 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.