

Abstract
D-peptides, as the enantiomers of the naturally occurring L-peptides, usually resist endogenous proteases and presumably insensitive to most enzymes. But it is unclear whether or how a phosphatase catalyzes the dephosphorylation from D-peptides. In this work, we examine the formation of the nanofibers of D-peptides via enzymatic dephosphorylation. By comparing the enzymatic hydrogelation of L-peptide and D-peptide based hydrogelators, we find that the chirality of the precursors of the hydrogelators affects little on the enzymatic hydrogelation resulted from the removal of phosphate group from a tyrosine phosphate residue. The attachment of a therapeutic agent (e.g., taxol) or a fluorophore (e.g., 4-nitro-2,1,3-benzoxadiazole (NBD)) to the D-peptide based hydrogelators afford a new type of biostable or biocompatible hydrogelators, which may find applications in intratumoral chemotherapy or intracellular imaging, respectively. This work, as the first comprehensive and systematic study of the unexpected enzymatic dephosphorylation of D-peptides, illustrates a useful approach to generate supramolecular hydrogels that have both biostability and other desired functions.
INTRODUCTION
This study investigates the use of alkaline phosphatase to generate supramolecular hydrogels of D-peptide derivatives and explores the potential applications of this apparently anti-intuitive enzyme-instructed self-assembly process. As the result of the self-assembly of certain small-molecules (i.e., hydrogelators1,2,3,4) in water, supramolecular nanofibers act as entangled matrices for holding large amounts of water and result in hydrogels that are referred as supramolecular hydrogels.2 Largely because of their inherent biocompatibility and biodegradability originated from the supramolecular (i.e., noncovalent) nature of the nanofibers formed by molecular self-assembly, supramolecular hydrogels are emerging as a relatively new class of biomaterials and are finding increased applications in biomedicine, ranging from tissue engineering,5 drug delivery, 3,6 biosensing,7,8 wound healing,9 enzyme assays,10 gel electrophoresis,11 nucleic acid sequestration,12 and protein separation.13 Among a variety of molecules that serve as hydrogelators, small peptide-based hydrogelators14 have attracted considerable attentions because of the well-established synthesis procedure (e.g., SPPS)15 and the obvious biological relevance of peptides. Most of the peptide- based hydrogelators, being made of L-amino acids (i.e., L-peptides), not only preserve the biological functions of a peptide motif, but also serve as the native substrates of enzymes.
As an alternative process of the use of enzymes to cross-link polymers to cause rapid hydrogelation,16 small peptides made of L-amino acid residues undergo a process referred as enzymatic hydrogelation that the solution of a precursor of hydrogelator, upon the addition of an enzyme, turns into the gel of the corresponding hydrogelator. 17 As a useful strategy for generating supramolecular nanofibers/hydrogels, enzymatic hydrogelation has already found a wide range of applications, such as screening the inhibitors of enzymes,18 measuring enzyme activity, 8 modulating biomineralization,19 typing bacteria,20 delivering drugs or proteins,21,22 stabilizing enzymes,23 and regulating the fate of cells.24 Despite the merits of L-peptides as the substrates for enzymatic hydrogelation, L-peptides, however, are susceptible of degradation catalyzed by a various of endogenous proteases, which limits the applications of supramolecular hydrogels when longterm biostability are required (such as controlled drug release,6,25 intracellular imaging,26 or other in vivo applications). Therefore, it is advantageous to develop a system that not only undergoes enzymatic hydrogelation, but also forms hydrogels or nanofibers that are stable for a prolong period inside cells or in vivo. Among the strategies for improving stability of peptidic materials, the use of D-amino acid to replace L-amino acid is an effective one.27 In addition to protease resistant, D-peptides also are of considerable biological relevance.28 For example, D-peptides can play a special role in defense mechanisms as “alien” agents from other organisms,29 act as potent inhibitors to inhibit HIV-1 entry,30 inhibit tumor cell migration,31 reduce adverse drug reactions (ADRs),4 control the formation and disassembly of bacteria biofilms,32 bind to DNA,33 form β-sheet,34 and dissociate Alzheimer’s amyloid to reduce the cytotoxicity induced by amyloid.35
The merits of D-peptides encourage us to explore the D-amino acid-based supramolecular hydrogels, which would provide stable scaffolds for long term drug release. Our previous works show that D-peptide based hydrogels are resistant to protease while the corresponding L-amino acid derived molecules undergo proteolytic hydrolysis. Recently, we also have found that D-peptides are able to improve the selectivity of non-steroid anti-inflammatory drugs (NSAID) and the D-peptide derivatives can serve as the substrate of an enzyme for hydrogelation catalyzed by alkaline phosphatase (ALP).4 However, it is unclear why and how the D-peptide derivatives to serve as the substrates of phosphatases for hydrogelation and what other potential applications are. To address these unanswered questions, we synthesize the precursors of hydrogelators that are made of D-amino acid residues or L-amino acid residues and evaluate the formation of the nanofibers of the hydrogelators via enzymatic dephosphorylation of this pair of enantiomeric substrates. Our results from kinetic studies performed by 31P NMR and rheology indicate that the chirality of the precursors affect little on the enzymatic hydrogelation when the dephosphorylation occurs from the L- or D-tyrosine phosphate residue of the precursors. Moreover, the attachment of therapeutic agents or fluorophores to the side chain of the phosphorylated D-peptides results in new precursors, which confer biostable or biocompatible hydrogels/nanofibers that may find applications in intratumoral chemotherapy or intracellular imaging. This work, as the first study that confirms the enzymatic hydrogelation of D-peptides and L-peptides to occur at almost the same rate, illustrates a useful approach and provides a new class of molecular platforms for generating supramolecular hydrogels that have both biostability and other desired functions for potential application inside cells or in vivo.
RESULTS AND DISCUSSION
Molecular Design
In our previous studies, we have found that small dipeptide derivative, 2-(naphthalen-2-yl)acetic-Phe-Phe (NapFF) is an excellent motif for enabling self-assembly and hydrogelation due to its strong supramolecular interactions arising from aromatic-aromatic interactions and hydrogen bonds among the molecules.36 Since lysine (K) possesses an ε-amine site for the attachment of biofunctional molecules on the side chain, and tyrosine phosphate (Y(p)) offers a handle for enzyme instructed hydrogelation, the incorporation of K and Y(p) with NapFF provides a versatile hydrogelator precursor NapFFKY(p) (1a), which undergoes enzymatic hydrogelation. Those studies suggest that one can use D-amino acids–D-Phe (f), D-Lys (k), and D-Tyr phosphate (y(p))—to replace the corresponding L-amino acids for making a more biostable precursor Napffky(p) (1b). To evaluate whether the dephosphorylation of D-tyrosine phosphate (y(p)) from the D-peptide by the phosphatase still would be possible, we first examine the binding of the tyrosine phosphate on 1a or 1b with ALP according to the crystal structure of ALP.37 With the phosphate groups (represented by the yellow and red spheres in Figure 1) being anchored to the active site of ALP (represented by the solid ribbons in Figure 1), the structures of the phosphatase that binds with L-peptide/D-peptide based precursors 1a and 1b are shown in Figure 1A and Figure 1B, respectively. Although there are stereochemical differences between 1a and 1b, the phosphate groups appear to be able to bind the same active site without any hinderance. According to the top view (Figure 1C), the opening in the structure of ALP is large enough to accommodate either 1a or 1b. Similarly, the side view (Figure 1D) clearly indicates that the phosphate groups on 1a or 1b are able to bind the active site of ALP. Thus, we choose to investigate the enzymatic hydrogelation of 1b, and to compare it with 1a by the rate of formation, morphology, and viscoelastic property of the corresponding hydrogels.
Figure 1.
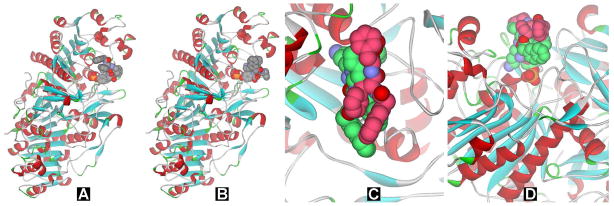
The binding of the phosphate precursors (presented as CPK model: yellow, phosphorous; red, oxygen.) to the active site of an ALP (presented as solid ribbons). (A) L-peptide based precursor (1a) and (B) D-peptide based precursor (1b) binding to the phosphatase. (C) Top view and (D) side view of 1a (green) and 1b (dark pink) in the active site. Purple atoms represent the parts of 1a and 1b that would occupy same space in the active site.
To explore the biological and biomedical applications of 1b, we attach small functional molecules, such as 4-nitro-2,1,3-benzoxadiazole (NBD), a fluorophore used in cell imaging, and taxol, a clinically-used anti-cancer drug, to 1b. Our previous works have established the synthetic route for the incorporation of the fluorophore or taxol to the hydrogelator precursor 1a. For example, we have developed a fluorescent hydrogelator precursor that undergoes intracellular enzymatic hydrogelation and forms fluorescent molecular aggregates inside cells.38 We have also attached taxol to 1a to afford the precursor of an anticancer hydrogelator that increases the solubility of taxol and achieves controlled drug delivery.22 These studies offer the necessary synthetic routes that allow us to conjugate precursor 1b with NBD group or taxol, which affords Napffk(NBD)y(p) (4b) or Napffk(taxol)y(p) (9b). These molecules represent a new type of precursors to result in the hydrogelators that are both biostable and multifunctional.
Synthesis
Scheme 1 shows the chemical structures of precursors 1a and 1b. Utilizing Fmoc-protected D-amino acids, we prepare 1b by standard solid phase synthesis with 2-chlorotrityl chloride resin (100~200 mesh and 0.3~0.8 mmol/g), followed by HPLC purification. We conjugate NBD group at the side chain of lysine to afford the precursor Napffk(NBD)y(p) (4b). As shown in Scheme 1, we dissolve 7-chloro-4-nitro-2,1,3-benzoxadiazole (NBD-Cl) (3) in methanol, followed by adding the basic aqueous solution of 1b (pH 9). The reaction of the mixed solution at 50 °C for 2 hours yields 4b as red precipitates after work-up and purification by reverse phase HPLC.
Scheme 1.

The synthetic route of the precursor of the NBD or taxol-containing hydrogelator based on a D-peptide.
Using a similar approach, we obtain the conjugate of taxol and 1b. As shown in Scheme 1, we add succinic an- hydride and 4-dimethylamino-pyridine (DMAP) into the clear solution of taxol (10) in pyridine. After stirring the mixture at room temperature overnight, we extract the solution with dichloromethane (DCM) and obtain taxol-succinic acid (7). The conjugation of 7 and N-hydroxysuccinimide (NHS) with the aid of N,N′-dicyclohexylcarbodiimide (DCC) affords taxol-succinic-NHS ester (8). Purified with column chromatography, we collect pure 8 and re-dissolve it with acetone. Then we add the acetone solution into a basic aqueous solution (pH 8.5) of 5b, which reacts for 24 hours. After working up the reaction and using reverse phase HPLC for the purification, we obtain compound 9b as the conjugate of taxol and 1b. These results indicate that it is convenient to apply the synthesis of L-peptidic precursors for producing the corresponding D-peptidic precursors.
Hydrogelation of the D-peptidic hydrogelator (2b)
To investigate the enzymatic hydrogelation of the D-peptidic precursor 1b, we prepare a series of hydrogels formed by using ALP to treat 1b at different concentrations. After dissolving 1.0, 2.0, 3.0, 4.0, and 5.0 mg of 1b in 0.5 mL of water (pH 7.6), respectively, we obtain clear solutions of 1b with different concentrations. The treatment of the solutions of 1b with ALP (1.0 U/mL) affords the molecules of hydrogelator 2b, which are less soluble than 1b and thus self-assemble in water to form hydrogels when the concentrations of 2b are sufficient. As shown in Figure 2, except the solution of 0.2 wt% of 1b, solutions of 1b with the concentration of 0.4, 0.6, 0.8, or 1.0 wt% form a stable transparent hydrogel within 24 h after the addition of 1.0 U/mL ALP into the solutions. Furthermore, as shown of the optical images in Figure 2, the higher concentration of the solutions of 1b gives the less transparent hydrogels of 2b, which also exhibits little birefringence (Figure S6), indicating that excess overlapping of the nanofibers to form large domains in the hydrogels of 2b cause the scattering of the light.
Figure 2.

The TEM images of the hydrogels formed by using ALP (1.0 U/mL) to treat 1b at pH 7.6 and concentrations of (A) 0.4 wt%, (B) 0.6 wt% (C) 0.8%wt, and (D) 1.0 wt%. Inset: optical images. Scale bar is 100 nm.
Being complementary to the optical images that serve as a simple way for proving the macroscopic phase transition (i.e., hydrogelation) triggered by the addition of ALP, transmission electron microscopy (TEM) images reveal the ordered nanostructures (e.g., nanofibers), formed by the self-assembly of the hydrogelators, that lead to hydrogelation. As shown in Figure 2, the TEM images of all the hydrogels, which consist of different concentrations of 2b, exhibit long, flexible, and uniform nanofibers that entangle to form stable networks. With the increase of the concentrations of hydrogelator 2b (0.4, 0.6, 0.8, and 1.0 wt%), the densities of the nanofibers in the hydrogels increase, but the widths of the nanofibers in the hydrogels remain similar (around 9±2 nm). These results indicate that concentration of the hydrogelator 2b hardly affect the self-assembling process controlled by the enzymatic hydrogelation so that the nanofibers exhibit similar morphology regardless the concentrations of the precursor solutions. The concentrations of the hydrogelators correlate well with the densities of nanofibers, which should match with the viscoelastic behaviors of the hydrogels.
The oscillatory rheological measurement of the hydrogels of 2b agrees with the density of the nanofibers in the hydrogels. The dynamic strain sweep, under constant oscillation frequencies and various oscillation strains, indicates that the storage moduli (G′s) of all these hydrogels are independent to strain until their critical strains reach, and G′s start to decrease drastically due to the breakdown of the networks of the hydrogels. After obtaining the maximum G′s of the hydrogels in dynamic strain sweep, we measure the frequency dependence of their storage moduli (G′s) and loss moduli (G″s) using dynamic frequency sweep at constant oscillation stain (the strain for maximum G′s) and temperature (25 °C) but varying oscillation frequency (0.1~200 rad/s). All the hydrogels of 2b exhibit viscoelastic properties of solid-like materials, evidenced by that the values of their G′s are significant higher (more than five times) than those of their G″s and are independent of the frequency during dynamic frequency sweep (Figure S8). As listed in Table 1, the hydrogels of 2b at the concentrations of 0.4, 0.6, 0.8, and 1.0 wt% exhibit strains of 4.7%, 5.0%, 14%, and 16%, respectively. In addition, their values of G′ (at the frequency of 6.28 rad/s) in dynamic frequency sweep are 6.5 × 102 Pa, 1.8 × 103 Pa, 2.7 × 103 Pa, and 3.8 × 103 Pa. While the critical strains of the resulting hydrogels of 2b show little correlation with the concentrations of the hydrogelators (Figure S8C and S8D), the storage moduli of hydrogels of 2b increase with the concentrations of 2b. This result agrees with that more physical cross-linking of the nanofibers at high concentrations of the hydrogelators.
Table 1.
The rheological properties and TEM characteristics of the hydrogels of 2a, 2b, 5b, and 10b.
| Compound | Conc. (wt%) | Dynamic Strain Sweep | Dynamic Frequency Sweep | ||
|---|---|---|---|---|---|
|
| |||||
| Maximum G′ (Pa) | Critical strain (%) | G′a (Pa) | Width of fiber (nm) | ||
| 2a | 0.4 | 7.2 × 102 | 3.7 | 8.6 × 102 | 8 ± 2 |
| 2b | 0.4 | 6.4 × 102 | 4.7 | 6.5 × 102 | 8 ± 2 |
| 0.6 | 1.7 × 103 | 5.0 | 1.8 × 103 | 9 ± 2 | |
| 0.8 | 2.7 × 103 | 14 | 2.7 × 103 | 9 ± 2 | |
| 1.0 | 3.9 × 103 | 16 | 3.8 × 103 | 9 ± 2 | |
| 5bb | 0.4 | 46 | 6.9 | 62 | 8 ± 2 |
| 10bb | 1.8 | 43 | 0.59 | 16 | 9 ± 2 |
The value is taken at frequency equals 6.28 rad/s.
The hydrogel is formed at pH7.4, while others are formed at pH 7.6.
The comparisons of L and D enantiomers of the precursors and hydrogelators
The comparison of the enzymatic hydrogelation process of 1a and 1b under the same conditions reveals that the chirality of 1a and 1b affects little on their dephosphorylation and the subsequent hydrogelation. To evaluate the rate of enzymatic hydrogelation process, we use 31P NMR and rheology to study the transformation of the precursors 1a and 1b upon the treatment of ALP (Figure 3). We first dissolve 10 mg of 1a and 1b into 1.0 mL of water at pH 7.6, respectively, to afford clear solutions with concentrations of 1.0 wt%. Once adding 0.02 U/mL of alkaline phosphates, we immediately monitor the solutions of 1a and 1b by 31P NMR and oscillatory rheology at 25 °C. The 31P NMR spectra at 3 min, 1 h, 2 h, 4h, 6h, 8h, 10h, 12h, 14h, 18h, 24h, and 48h indicate that the phosphate groups on the L-tyrosine of 1a and D-tyrosine 1b (δ = −2.7) become free phosphates (δ = 0.0) at almost same rate, and dephosphorylation finishes after 48 hours. This result suggests that the precursors 1a and 1b undergo dephosphorylation with similar rates upon being treated with ALP. Figures 3C and 3D display the time dependent rheology studies of 1a and 1b. At the beginning, values of G″ are higher than the values of G′ for the solutions of 1a and 1b, indicating both of them are fluids. However, as 1a and 1b slowly turn into hydrogelators 2a and 2b by enzymatic dephosphorylation, the solutions start to form solid-like hydrogels with values of G′ become higher than those of G″. The gelation points for 2a and 2b, at where G′s intersect with G″s, are both achieved around 5 hours after the addition of enzyme. This result, together with the 31P NMR experiment, suggests that the chirality of 1a and 1b exhibits almost the same influence on the enzymatic hydrogelation catalyzed by ALP. The oscillatory shear during rheological measurement may accelerates enzymatic dephosphorylation so that the gelation points reach at the time (5 hours) much shorter than the time for completely dephosphorylation during the NMR experiment (48 hours).
Figure 3.

The 31P NMR shows the conversion of 1.0 wt% of (A) 1a and (B) 1b catalyzed by the phosphatase (0.02 U/mL) at pH 7.6 at 3 minutes and 4, 12, 24, and 48 h; The time dependent rheology study of 1.0 wt% of (C) 1a and (D) 1b catalyzed by the phosphatase (0.02 U/mL) at pH 7.6.
After comparing the rate of the dephosphorylation of the L- and D-enantiomeric precursors (1a and 1b), we examine the morphology of the microstructures and viscoelastic properties of the corresponding hydrogels (2a and 2b). By sonication, we dissolve 2.0 mg of 1a or 1b into 0.5 mL of water at pH 7.6 to afford a clear solution. The addition of 1.0 U/mL of ALP into the solution of 1a or 1b turns the hydrogelator precursor to its corresponding hydrogelator, 2a or 2b, which results in a transparent hydrogel (0.4 wt%) within 24 hours. As shown in Figure 4A and 4B, both hydrogelators 2a and 2b self-assemble to form long, flexible, and uniform nanofibers with average width around 8±2 nm, which entangle to develop physically cross-linked networks and to afford stable hydrogels. The similarity of the nanofibers in these two hydrogels indicates that chirality of 2a and 2b has little influence on the morphology of their nanofibers. Oscillatory rheology of the hydrogels of 2a and 2b indicates that both hydrogels behave as solid-like materials that have storage moduli (G′) to be significantly higher than loss moduli (G″) and exhibit weak frequency dependence in dynamic frequency sweep (Figure 4C and 4D). As shown in Table 1, hydrogels of 2a and 2b have critical strains of 3.7% and 4.7% during the dynamic strain sweep, and their values of G′ (at the frequency of 6.28 rad/s) in dynamic frequency sweep are 8.6 × 102 Pa and 6.5 × 102 Pa, respectively. These results suggest that the chirality of these two hydrogelators causes negligible differences on the viscoelastic properties of the corresponding hydrogels.
Figure 4.

The optical images and TEM images of the hydrogels formed by using ALP (1.0 U/mL) to treat 0.4 wt% of (A) 1a and (B) 1b at pH 7.6. (C) The strain sweep and (D) the frequency sweep of the hydrogels 2a and 2b.
The application of the D-enantiomer hydrogelator (1b) for potential intracellular imaging
According to the molecular design, the attachment of functional molecules to 1b broadens the scope of the applications of supramolecular hydrogelators in cells or in vivo. We first examine the feasibility and characteristic of the use 4b for imaging intracellular self-assembly of D-peptidic hydrogelators. After dissolving 2.0 mg of 4b into 0.5 mL of water at pH 7.4, we treat the clear orange solution with 20.0 U/mL of ALP, which turns 4b into the fluorescent hydrogelator 5b. The self-assembly of 5b affords a transparent orange hydrogel (Figure 5A, inset) that is stable over weeks. The TEM image of hydrogel of 5b exhibits long and uniform nanofibers with average width of 8±2 nm that entangle to afford stable network (Figure 5A). According to our previous study, the unassociated molecules of NBD containing hydrogelators in aqueous solutions exhibit little fluorescence unless they aggregate to form nanofibers.38 This important feature makes NBD containing hydrogelator be a useful candidate for imaging molecular self-assembly inside cells.
Figure 5.
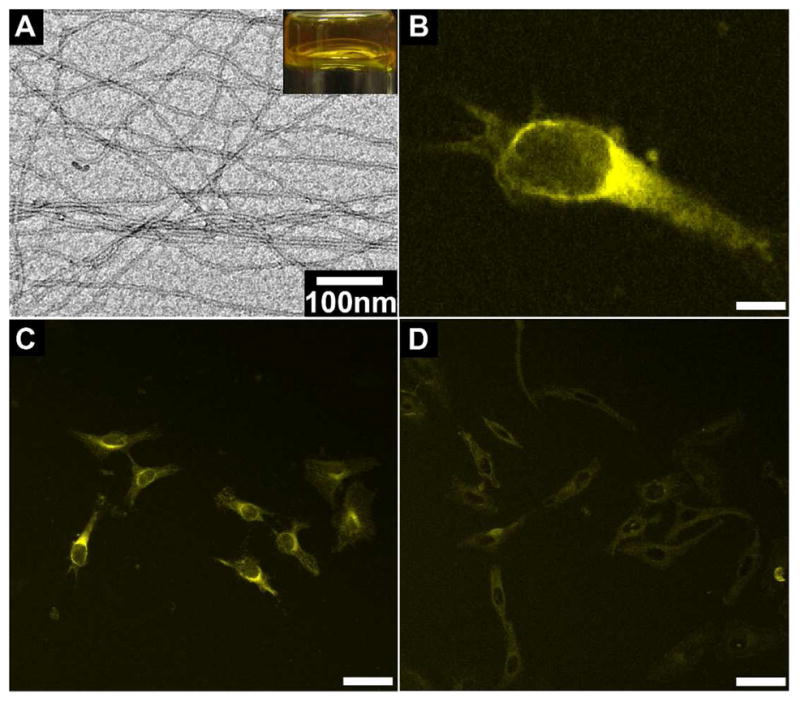
(A) The optical image and TEM image of hydrogel formed by 0.4 wt% of 4b at pH 7.4 upon the catalysis of ALP (20.0 U/ml). (B) The fluorescent confocal microscope image of a HeLa cell incubated with 500 μM of 4b in PBS buffer (scale bar is 10 μm). The fluorescent confocal microscope images of HeLa cells incubated with 500 μM of 4b without (C) or with (D) the PTP1B inhibitor (25 μM) (scale bar is 50 μm).
After treating HeLa cells with 500 μM of hydrogelator precursor 4b for two minutes, we observe strong fluorescence emerging from the region near the nuclei of cells (Figure 5B, C), suggesting that the self-assembly of 5b results in formation of the nanofibers of 5b around the endoplasmic reticulum (ER). There is little fluorescence outside the cells, suggesting the lack of dephosphorylation and/or self-assembly of 5b. To confirm that the dephosphorylation of 4b and self-assembly of 5b take place in ER, we use 25 μM of CinnGEL 2Me to inhibit protein tyrosine phosphatase-1B (PTP1B),39 a highly efficient phosphatase located at the outer membrane of ER, when the HeLa cells are incubated with 4b (500 μM). As shown in Figure 5D, the addition of the inhibitor of PTP1B significantly decreases and delays the fluorescence inside cells, confirming that the dephosphorylation of 4b and the self-assembly of 5b occur at ER. As shown by the time sequence fluorescent images of the HeLa cells incubated with 4b in the absence of the PTP1B inhibitor (Figure S11), most of the cells exhibit strong fluorescence after treated with 4b for only 2 minutes. Even being incubated with the presence of PTP1B inhibitor, the cells still show partial fluorescence after 5 minutes of the incubation. Apparently, the fluorescence of the nanofibers in the HeLa cells treated by the D-peptide precursor (4b) emerges much faster than that of L-peptide precursor (4a) (which takes about 15 min38 in the presence of CinnGEL 2Me). This result agrees with that the resulted D-peptide hydrogelator (5b) is more resistant to proteolytic degradation than the L-peptide hydrogelator (5a) does.
The application of D-enantiomer hydrogelator (1b) for potential intratumoral chemotherapy
Typically, after dissolving 9.0 mg of 9b in 0.5 mL of water at pH 7.4 by sonication, we add ALP (1.0 U/mL) into the solution of 9b to obtain hydrogelator 10b, which forms a stable and semitransparent hydrogel (Figure 6A). This result differs slightly from the behavior of precursor 9a that undergoes enzymatic hydrogelation at the concentration of 1.0 wt%,22. suggesting that precursor 9b (having a concentration up to 1.8 wt% for enzymatic hydrogelation) and hydrogelator 10b exhibit relatively good solubility. This subtle increase of the solubility should increase the amount of taxol in the hydrogel. The TEM image of the hydrogel 10b shows the uniform nanofibers with the average width of 9±2 nm. To determine the efficacies of taxol after conjugating it into the hydrogelator, we use MTT assays to examine the viability of HeLa cells incubated with taxol (6), 9b, and 10b for 72 hours at 37°C. Figure 6B shows the IC50 values of 6, 9b, and 10b, which are 45.8 nM, 61.9 nM, and 105.9 nM, respectively. This result suggests that the conjugation of taxol to the D-peptide essentially preserve the anti-tumor activity of taxol, thus encouraging us to carry out in vivo test of 10b on a mouse model.40
Figure 6.

(A) The optical and TEM images of hydrogel formed by 1.8 wt% of 10b at pH 7.4 with the catalysis of ALP (1 U/mL) with scale of 100 nm; (B) The IC50 values of 6, 9b, and 10b incubated with HeLa cells after 72 h; (C) The relative tumor sizes and (D) relative weights of mice treated with 6, 10a, and 10b for in vivo tests.
As expected, both L- and D-peptide based hydrogels of 10a and 10b exhibit similar anti-tumor activities up to 12 days of intratumoral injection of the hydrogels. After inoculating female Balb/c mice with 2 × 105 of 4T1-luciferase cells in the mammary fat pad, we allow tumors grow until their sizes reach about 500 mm3, and randomly divide them into different treatment groups: (1) intravenous injections of PBS vehicle control; (2) intravenous injection of 4 × 10 mg/kg Taxol® every other day from day 0 for indicated times; (3) a single intratumoral injection of 10 mg/kg taxol containing hydrogels in 40 μL volume. With the treatments of 6 (taxol), 10a, 10b, or PBS buffer (control) for 14 days, we monitor the relative tumor sizes (calculated by the formula: tumor volume = length × width × (Length + Width) / 2) and relative weights of mice every two days. Due to the toxicity of clinical taxol (formulated with Cremophor EL),41 the single injection of 40 mg/kg of Taxol® may cause the death of mouse immediately. Therefore, we have to divide 40 mg/kg of 6 into four injections with each injection of 10 mg/kg. As shown in Figure 6C, the intravenous injections of 40 mg/kg of 6 every other day from day 0 results in the relative tumor sizes to be smaller than those of the control group after day 8. In contrast, the intratumoral injections of the hydrogel 10a or 10b at only one dose of 10 mg/kg in the mice at day 0, which may sustain for one month, reduce the relative tumor sizes on the mice more significantly than those of the controls after day 2. At day 14, although the relative tumor sizes in the groups injected with 6 and the hydrogel of 10a are similar with the PBS buffer control group, the relative tumor size in the group injected with the hydrogel of 10b is statistically smaller than the control. This result suggests that the hydrogel of 10b exhibits higher anti-tumor efficacy than 10a or 6 does. Figure 6D shows the relative weights of mice during these 14 days treatment, suggesting that the intratumoral injection of hydrogels of 10a and 10b, only once, certainly limit the side effect of taxol to the mice. These results support that the local injection of the hydrogels appears to achieve long term drug release with higher efficacy and better biocompatibility than the intravenous injection of taxol.40 This promising result warrants further investigation of the D-peptidic hydrogels of taxol on animal models.
CONCLUSION
In conclusion, taking the advantages of D-amino acids, we have developed biostable and biocompatible supramolecular hydrogels made of D-amino acid residues. Similar to other D-amino acid containing peptides, 1b resists to protease while the corresponding L-amino acid derived molecule (1a) undergo proteolytic hydrolysis (Figure S12). Although these D-peptidic derivatives are intrinsically resistant to proteolytic hydrolysis, which make the hydrogels as stable platform materials for long term biomedical applications, D-peptide based hydrogelator precursor still acts as a substrate of phosphatase for enzyme-instructed self-assembly. While it appears unexpected that the rates of dephosphorylation of L-peptide and D-peptide precursors are comparable, the crystal structure of the phosphatase confirms it is feasible for such an observation. This work, thus, illustrates a protein structure-based approach for designing the substrates of enzyme-instructed self-assembly and hydrogelation. While the conjugation of 1b with taxol affords a biostable hydrogel that exhibits improved drug efficacy in anti-cancer activity, the fast accumulation of molecular nanofibers of D-peptide (in the case of 4b) is particularly intriguing because it indicates the introduction of D-peptide may result in certain biological effects much faster than intuitively thought, which is a subject worthwhile for the further exploration.
Supplementary Material
Acknowledgments
This work was partially supported by HFSP (RGP0056 /2008), a start-up fund from Brandeis University, and NIH (R01CA142746) The TEM images were taken at the Brandeis EM and Optical Imaging facilities.
Footnotes
Supporting Information. The details of the synthesis, NMR Spectra and LC-MS data for all compounds, rheological data, cytotoxicity data, and in vivo tests. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.(a) Terech P, Weiss RG. Chem Rev. 1997;97:3133–3159. doi: 10.1021/cr9700282. [DOI] [PubMed] [Google Scholar]; (b) Heeres A, van der Pol C, Stuart MCA, Friggeri A, Feringa BL, van Esch J. J Am Chem Soc. 2003;125:14252–14253. doi: 10.1021/ja036954h. [DOI] [PubMed] [Google Scholar]; (c) Mukhopadhyay S, Maitra U, Ira, Krishnamoorthy G, Schmidt J, Talmon Y. J Am Chem Soc. 2004;126:15905–15914. doi: 10.1021/ja046788t. [DOI] [PubMed] [Google Scholar]; (d) Zhang Y, Yang ZM, Yuan F, Gu HW, Gao P, Xu B. J Am Chem Soc. 2004;126:15028–15029. doi: 10.1021/ja044401g. [DOI] [PubMed] [Google Scholar]; (e) Vemula PK, Li J, John G. J Am Chem Soc. 2006;128:8932–8938. doi: 10.1021/ja062650u. [DOI] [PubMed] [Google Scholar]; (f) Yang ZM, Liang GL, Wang L, Xu B. J Am Chem Soc. 2006;128:3038–3043. doi: 10.1021/ja057412y. [DOI] [PubMed] [Google Scholar]; (g) Cai W, Wang GT, Du P, Wang RX, Jiang XK, Li ZT. J Am Chem Soc. 2008;130:13450–13459. doi: 10.1021/ja8043322. [DOI] [PubMed] [Google Scholar]; (h) Ma ML, Kuang Y, Gao Y, Zhang Y, Gao P, Xu B. J Am Chem Soc. 2010;132:2719–2728. doi: 10.1021/ja9088764. [DOI] [PubMed] [Google Scholar]; (i) Li XM, Kuang Y, Shi JF, Gao Y, Lin HC, Xu B. J Am Chem Soc. 2011;133:17513–17518. doi: 10.1021/ja208456k. [DOI] [PMC free article] [PubMed] [Google Scholar]; (j) Zheng WT, Gao J, Song LJ, Chen CY, Guan D, Wang ZH, Li ZB, Kong DL, Yang ZM. J Am Chem Soc. 2013;135:266–271. doi: 10.1021/ja308690y. [DOI] [PubMed] [Google Scholar]; (k) Xing BG, Yu CW, Chow KH, Ho PL, Fu DG, Xu B. J Am Chem Soc. 2002;124:14846–14847. doi: 10.1021/ja028539f. [DOI] [PubMed] [Google Scholar]
- 2.Estroff LA, Hamilton AD. Chem Rev. 2004;104:1201–1217. doi: 10.1021/cr0302049. [DOI] [PubMed] [Google Scholar]
- 3.(a) Komatsu H, Matsumoto S, Tamaru S, Kaneko K, Ikeda M, Hamachi I. J Am Chem Soc. 2009;131:5580–5585. doi: 10.1021/ja8098239. [DOI] [PubMed] [Google Scholar]; (b) Boekhoven J, Koot M, Wezendonk TA, Eelkema R, van Esch JH. J Am Chem Soc. 2012;134:12908–12911. doi: 10.1021/ja3051876. [DOI] [PubMed] [Google Scholar]
- 4.Li JY, Kuang Y, Gao Y, Du XW, Shi JF, Xu B. J Am Chem Soc. 2013;135:542–545. doi: 10.1021/ja310019x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.(a) Hartgerink JD, Beniash E, Stupp SI. Science. 2001;294:1684–1688. doi: 10.1126/science.1063187. [DOI] [PubMed] [Google Scholar]; (b) Holmes TC, de Lacalle S, Su X, Liu GS, Rich A, Zhang SG. Proc Natl Acad Sci U S A. 2000;97:6728–6733. doi: 10.1073/pnas.97.12.6728. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Haines LA, Rajagopal K, Ozbas B, Salick DA, Pochan DJ, Schneider JP. J Am Chem Soc. 2005;127:17025–17029. doi: 10.1021/ja054719o. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Galler KM, Aulisa L, Regan KR, D’Souza RN, Hartgerink JD. J Am Chem Soc. 2010;132:3217–3223. doi: 10.1021/ja910481t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zhao F, Ma ML, Xu B. Chem Soc Rev. 2009;38:883–891. doi: 10.1039/b806410p. [DOI] [PubMed] [Google Scholar]
- 7.(a) Wada A, Tamaru S, Ikeda M, Hamachi I. J Am Chem Soc. 2009;131:5321–5330. doi: 10.1021/ja900500j. [DOI] [PubMed] [Google Scholar]; (b) Rajagopalan A, Kroutil W. Mater Today. 2011;14:144–152. [Google Scholar]
- 8.Bremmer SC, Chen J, McNeil AJ, Soellner MB. Chem Commun. 2012;48:5482–5484. doi: 10.1039/c2cc31537h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yang ZM, Liang GL, Ma ML, Abbah AS, Lu WW, Xu B. Chem Commun. 2007:843–845. doi: 10.1039/b616563j. [DOI] [PubMed] [Google Scholar]
- 10.Kiyonaka S, Sada K, Yoshimura I, Shinkai S, Kato N, Hamachi I. Nat Mater. 2004;3:58–64. doi: 10.1038/nmat1034. [DOI] [PubMed] [Google Scholar]
- 11.Yamamichi S, Jinno Y, Haraya N, Oyoshi T, Tomitori H, Kashiwagi K, Yamanaka M. Chem Commun. 2011;47:10344–10346. doi: 10.1039/c1cc13826j. [DOI] [PubMed] [Google Scholar]
- 12.Yang ZM, Kuang Y, Li XM, Zhou N, Zhang Y, Xu B. Chem Commun. 2012;48:9257–9259. doi: 10.1039/c2cc34935c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gao Y, Long MJC, Shi JF, Hedstrom L, Xu B. Chem Commun. 2012;48:8404–8406. doi: 10.1039/c2cc33631f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.(a) Cui HG, Webber MJ, Stupp SI. Biopolymers. 2010;94:1–18. doi: 10.1002/bip.21328. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Williams RJ, Mart RJ, Ulijn RV. Biopolymers. 2010;94:107–117. doi: 10.1002/bip.21346. [DOI] [PubMed] [Google Scholar]; (c) Rajagopal K, Ozbas B, Pochan DJ, Schneider JP. Biopolymers. 2005;80:487–487. [Google Scholar]; (d) Gao Y, Yang ZM, Kuang Y, Ma ML, Li JY, Zhao F, Xu B. Biopolymers. 2010;94:19–31. doi: 10.1002/bip.21321. [DOI] [PubMed] [Google Scholar]
- 15.Chan WC, White P. Fmoc Solid Phase Peptide Synthesis: A Practical Approach. OUP Oxford; 2000. [Google Scholar]
- 16.Hu BH, Messersmith PB. J Am Chem Soc. 2003;125:14298–14299. doi: 10.1021/ja038593b. [DOI] [PubMed] [Google Scholar]
- 17.(a) Yang ZM, Gu HW, Fu DG, Gao P, Lam JK, Xu B. Adv Mater. 2004;16:1440. [Google Scholar]; (b) Toledano S, Williams RJ, Jayawarna V, Ulijn RV. J Am Chem Soc. 2006;128:1070–1071. doi: 10.1021/ja056549l. [DOI] [PubMed] [Google Scholar]; (c) Yang Z, Liang G, Xu B. Acc Chem Res. 2008;41:315–326. doi: 10.1021/ar7001914. [DOI] [PubMed] [Google Scholar]; (d) Hirst AR, Roy S, Arora M, Das AK, Hodson N, Murray P, Marshall S, Javid N, Sefcik J, Boekhoven J, van Esch JH, Santabarbara S, Hunt NT, Ulijn RV. Nat Chem. 2010;2:1089–1094. doi: 10.1038/nchem.861. [DOI] [PubMed] [Google Scholar]; (e) Williams RJ, Smith AM, Collins R, Hodson N, Das AK, Ulijn RV. Nat Nanotechnol. 2009;4:19–24. doi: 10.1038/nnano.2008.378. [DOI] [PubMed] [Google Scholar]
- 18.Yang ZM, Xu B. Chem Commun. 2004:2424–2425. doi: 10.1039/b408897b. [DOI] [PubMed] [Google Scholar]
- 19.Schnepp ZAC, Gonzalez-McQuire R, Mann S. Adv Mater. 2006;18:1869. [Google Scholar]
- 20.Yang ZM, Ho PL, Liang GL, Chow KH, Wang QG, Cao Y, Guo ZH, Xu B. J Am Chem Soc. 2007;129:266–267. doi: 10.1021/ja0675604. [DOI] [PubMed] [Google Scholar]
- 21.(a) Vemula PK, Cruikshank GA, Karp JM, John G. Biomaterials. 2009;30:383–393. doi: 10.1016/j.biomaterials.2008.09.045. [DOI] [PubMed] [Google Scholar]; (b) Williams RJ, Hall TE, Glattauer V, White J, Pasic PJ, Sorensen AB, Waddington L, McLean KM, Currie PD, Hartley PG. Biomaterials. 2011;32:5304–5310. doi: 10.1016/j.biomaterials.2011.03.078. [DOI] [PubMed] [Google Scholar]
- 22.Gao Y, Kuang Y, Guo ZF, Guo ZH, Krauss IJ, Xu B. J Am Chem Soc. 2009;131:13576. doi: 10.1021/ja904411z. [DOI] [PubMed] [Google Scholar]
- 23.Wang QG, Yang ZM, Gao Y, Ge WW, Wang L, Xu B. Soft Matter. 2008;4:550–553. doi: 10.1039/b715439a. [DOI] [PubMed] [Google Scholar]
- 24.(a) Yang ZM, Xu KM, Guo ZF, Guo ZH, Xu B. Adv Mater. 2007;19:3152. [Google Scholar]; (b) Yang Z, Liang G, Guo Z, Xu B. Angew Chem Intl Ed. 2007;46:8216–8219. doi: 10.1002/anie.200701697. [DOI] [PubMed] [Google Scholar]
- 25.Cheetham AG, Zhang PC, Lin YA, Lock LL, Cui HG. J Am Chem Soc. 2013;135:2907–2910. doi: 10.1021/ja3115983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gao Y, Shi JF, Yuan D, Xu B. Nat Commun. 2012;3 doi: 10.1038/ncomms2040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.(a) Li XM, Du XW, Li JY, Gao Y, Pan Y, Shi JF, Zhou N, Xu B. Langmuir. 2012;28:13512–13517. doi: 10.1021/la302583a. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Liang GL, Yang ZM, Zhang RJ, Li LH, Fan YJ, Kuang Y, Gao Y, Wang T, Lu WW, Xu B. Langmuir. 2009;25:8419–8422. doi: 10.1021/la804271d. [DOI] [PubMed] [Google Scholar]
- 28.(a) Van Regenmortel MHV, Muller S. Curr Opin Biotechnol. 1998;9:377–382. doi: 10.1016/s0958-1669(98)80011-6. [DOI] [PubMed] [Google Scholar]; (b) Nair DT, Kaur KJ, Singh K, Mukherjee P, Rajagopal D, George A, Bal V, Rath S, Rao KVS, Salunke DM. J Immunol. 2003;170:1362–1373. doi: 10.4049/jimmunol.170.3.1362. [DOI] [PubMed] [Google Scholar]
- 29.Konno R. D-amino acids: a new frontier in amino acids and protein research : practical methods and protocols. Vol. 35 Nova Science Publishers; 2007. [Google Scholar]
- 30.Welch BD, VanDemark AP, Heroux A, Hill CP, Kay MS. Proc Natl Acad Sci U S A. 2007;104:16828–16833. doi: 10.1073/pnas.0708109104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.(a) Sroka TC, Pennington ME, Cress AE. Carcinogenesis. 2006;27:1748–1757. doi: 10.1093/carcin/bgl005. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Liu M, Li C, Pazgier M, Li CQ, Mao YB, Lv YF, Gu B, Wei G, Yuan WR, Zhan CY, Lu WY. Proc Natl Acad Sci U S A. 2010;107:14321–14326. doi: 10.1073/pnas.1008930107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kolodkin-Gal I, Romero D, Cao SG, Clardy J, Kolter R, Losick R. Science. 2010;328:627–629. doi: 10.1126/science.1188628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.(a) Reich Z, Schramm O, Brumfeld V, Minsky A. J Am Chem Soc. 1996;118:6345–6349. [Google Scholar]; (b) Morii T, Tanaka T, Sato S, Hagihara M, Aizawa Y, Makino K. J Am Chem Soc. 2002;124:180–181. doi: 10.1021/ja017078f. [DOI] [PubMed] [Google Scholar]; (c) Michaud M, Jourdan E, Villet A, Ravel A, Grosset C, Peyrin E. J Am Chem Soc. 2003;125:8672–8679. doi: 10.1021/ja034483t. [DOI] [PubMed] [Google Scholar]
- 34.Swanekamp RJ, DiMaio JTM, Bowerman CJ, Nilsson BL. J Am Chem Soc. 2012;134:5556–5559. doi: 10.1021/ja301642c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.(a) Esteras-Chopo A, Pastor MT, Serrano L, de la Paz ML. J Mol Biol. 2008;377:1372–1381. doi: 10.1016/j.jmb.2008.01.028. [DOI] [PubMed] [Google Scholar]; (b) Chalifour RJ, McLaughlin RW, Lavoie L, Morissette C, Tremblay N, Boule M, Sarazin P, Stea D, Lacombe D, Tremblay P, Gervais F. J Biol Chem. 2003;278:34874–34881. doi: 10.1074/jbc.M212694200. [DOI] [PubMed] [Google Scholar]
- 36.(a) Lehn JM. Angew Chem Int Ed Engl. 1988;27:89–112. [Google Scholar]; (b) Whitesides GM, Grzybowski B. Science. 2002;295:2418–2421. doi: 10.1126/science.1070821. [DOI] [PubMed] [Google Scholar]
- 37.Coleman JE. Annu Rev Biophys Biomol Struct. 1992;21:441–483. doi: 10.1146/annurev.bb.21.060192.002301. [DOI] [PubMed] [Google Scholar]
- 38.Gao Y, Shi JF, Yuan D, Xu B. Nat Commun. 2012;3:1033. doi: 10.1038/ncomms2040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Frangioni JV, Beahm PH, Shifrin V, Jost CA, Neel BG. Cell. 1992;68:545–560. doi: 10.1016/0092-8674(92)90190-n. [DOI] [PubMed] [Google Scholar]
- 40.Wang HM, Wei J, Yang CB, Zhao HY, Li DX, Yin ZN, Yang ZM. Biomaterials. 2012;33:5848–5853. doi: 10.1016/j.biomaterials.2012.04.047. [DOI] [PubMed] [Google Scholar]
- 41.Rowinsky EK, Eisenhauer EA, Chaudhry V, Arbuck SG, Donehower RC. Semin Oncol. 1993;20:1–15. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.