

Abstract
The present study comparatively evaluated the potency of a series of new phenylethyl[1,2,4]methyltriazines which are analogues of the classical metabotropic glutamate (mGlu) receptor subtype 5 (mGluR5) anatgonist 2-methyl-6-(phenylethynyl)pyridine (MPEP) in blocking hyperalgesia induced by the group I mGlu receptor agonist (S)-3,5-DHPG as well as in reversing morphine antinociceptive tolerance in mice. Hyperalgesia was assessed in mice using the tail immersion test. Intrathecal (i.t.) pre-treatment with the test compounds 5-methyl-3-phenylethynyl-[1,2,4]triazine (RTI-4229-707), 5-methyl-3-(4-phenoxy-phenylethynyl-[1,2,4]triazine (RTI-4229-766), and 3-(3-methylphenylethynyl)-5-methyl-[1,2,4]triazine (RTI-4229-787) resulted in a dose-dependent blockade of (S)-3,5-DHPG-induced hyperalgesia. The inhibitory dose-50 (ID50) values were 0.49, 0.72 and 0.44 nmol/mouse, for RTI-4229-707, RTI-4229-766 and RTI-4229-787, respectively, compared to 18.63 nmol/mouse for MPEP. The other two compounds tested 3-(2,5-dimethylphenylethynyl)-5-methyl[1,2,4]triazine (RTI-4229-785) and 3-(2-methylphenylethynyl)-5-methyl[1,2,4]triazine (RTI-4229-828) were totally inactive. Morphine tolerance was induced in mice by implanting a 75 mg morphine pellet and assessing morphine-induced antinociception at 72-h later. Morphine-pelleted mice showed a 5.5-fold tolerance to the antinociceptive effect of acute morphine compared to placebo-pelleted mice in the tail immersion test. Intracerebroventricular (i.c.v.) administration of the three active mGluR5 antagonists dose-dependently reversed morphine antinociceptive tolerance. The ID50 values were 57.7, 25.8 and 64.3 nmol/mouse, for RTI-4229-707, RTI-4229-766 and RTI-4229-787, respectively, compared to 1050 nmol/mouse for MPEP. Similar to the hyperalgesia study, test compounds RTI-4229-785 and RTI-4229-828 were totally inactive in reversing morphine tolerance. These results are in agreement with our previous study in which we demonstrated that the same active mGluR5 antagonists blocked glutamate-mediated mobilization of internal calcium in a selective mGluR5 in vitro efficacy assay.
Keywords: mGluR5, mGluR5 antagonist, hyperalgesia, morphine tolerance, antinociception
Introduction
Glutamate, the major excitatory transmitter in the central nervous system (CNS), acts on two classes of receptors; the ionotropic glutamate receptors and the metabotropic glutamate receptors (Marino and Conn, 2006). Extensive research into the functions of glutamate and glutamate receptors in CNS has shown an essential role of metabotropic glutamate (mGlu) receptors in normal brain functions, but also in neurological and psychiatric disorders (Slassi et al., 2005).
The group I mGlu receptor 5 (mGluR5), is well positioned to regulate and fine-tune neuronal excitability and synaptic transmission through its modulation of various signal transduction pathways and interactions with other transmitter systems. Therefore, the mGlu5 receptor may be an important therapeutic target for the treatment of disorders of the central nervous system (Slassi et al., 2005). The discovery of 2-methyl-6-(phenylethynyl)pyridine (MPEP; Gasparini et al., 1999), a non-competitive mGluR5 antagonist, provided a potent, selective, systemically active compound for proof of concept studies in animal models of various disease states. These studies have led to greater understanding of possible therapeutic applications of mGlu5 receptor antagonists in recent years, suggesting their use in a number of disease states, including chronic pain, various psychiatric and neurological disorders, substance abuse and withdrawal, obesity and gastroesophageal reflux disease.
Several studies suggest that mGluR5 plays a crucial role in regulating the positive reinforcing properties of cocaine as well as other drugs of abuse. Chiamulera et al., (2001) reported that mice lacking the mGluR5 gene did not acquire cocaine self-administration behavior and did not exhibit cocaine-induced hyperlocomotor activity even though food responding was unaffected. The studies also showed that the selective noncompetitive mGluR5 antagonist MPEP decreased cocaine self-administration in wild-type mice. Moreover, MPEP decreased nicotine self-administration in rats and mice (Paterson et al., 2003) attenuated morphine and cocaine-induced condition place preference in mice (Popick and Wrobel, 2002; McGeehan and Olive, 2003) and decreased ethanol drinking and ethanol-seeking in rats (Backstrom et al., 2004).
The role of group I mGlu receptors in nociception has been well established. Activation of mGluRs has been demonstrated to produce nociception in animals. Intrathecal (i.t.) administration of the mGluR5 agonist (S)-3,5-dihydroxyphenylglycine ((S)-3,5-DHPG) was shown to induce persistent thermal hyperalgesia and mechanical allodynia (Fisher and Coderre, 1998). In addition, carrageenan-induced thermal and mechanical hyperalgesia in rats with hindpaw inflammation were blocked by i.t. administration of the group I mGlu receptors antagonist (S)-4-carboxy-3-hydroxyphenylglycine ((S)-4C3HPG) (Young et al., 1997). We recently demonstrated that (S)-3,5-DHPG-induced hyperalgesia is mediated via the coupling between group I mGlu receptors and the N-methyl-D-aspartic acid (NMDA) channels through the NR2B subunit in the spinal cord, and that this coupling involves the activation of PKC and PKA (Gabra et al., 2007).
We have also hypothesized a possible significant role for group I mGlu receptors antagonists in reversing morphine antinociceptive tolerance in mice (Smith et al., 2004). The activation of group I mGlu receptors leads to Gαq or Gα11 subunit binding to the C2 domain of the phospholipase C (PLC) β1, β3 and β4 isoforms and subsequent increases in the phosphatidylinositol 4,5-bisphosphate (PIP2) turnover (Conn and Pin, 1997). Furthermore, the βγ subunits from Gq, G11, and other G-proteins (e.g., Gi/o from μ-opioid receptor stimulation) can bind the pleckstrin homology domain of PLC to stimulate enzyme activity. Thus, chronic administration of morphine could lead to higher levels of Group I mGlu receptors, or more efficient receptor coupling to Gq/11 proteins, resulting in increases in the activity of the phosphatidylinositol (PI) cascade leading to activation of protein kinase C (PKC) and consequently μ-opioid receptor desensitization and the development of tolerance. This led to the hypothesis that group I mGlu receptor antagonists would reverse antinociceptive tolerance in mice implanted with morphine pellets, while having no effect in non-tolerant placebo-pelleted mice.
In the present study we comparatively evaluated the efficacy of some selected newly synthesized phenylethyl[1,2,4]methyltriazine analogues: 5-methyl-3-phenylethynyl[1,2,4]triazine (RTI-4229-707), 5-methyl-3-(4-phenoxyphenylethynyl[1,2,4]triazine (RTI-4229-766), 3-(2,5-dimethylphenylethynyl)-5-methyl[1,2,4]triazine (RTI-4229-785), 3-(3-methylphenylethynyl)-5-methyl[1,2,4]triazine (RTI-4229-787) and 3-(2-methylphenylethynyl)-5-methyl[1,2,4]triazine (RTI-4229-828) versus MPEP in blocking (S)-3,5-DHPG-induced hyperalgesia as well as in reversing morphine antinociceptive tolerance in mice.
2. Results
2.1. Blockade of (S)-3,5-DHPG-induced hyperalgesia
As shown in Fig. 1A-D, i.t. administration of the mGluR5 agonist (S)-3,5-DHPG at 15 nmol/mouse (a dose tested to produce a maximal hyperalgesia in our previous studies; Gabra et al., 2007) induced a significant (≅ 50 %) decrease in the tail immersion reaction time (P < 0.001), 30 min following its administration. The maximum possible effect was 54.1 ± 1.7% in (S)-3,5-DHPG-treated mice compared to 99.6 ± 2.0% in vehicle-treated control mice. The mGluR5 antagonist MPEP, injected i.t. 5 min prior to (S)-3,5-DHPG, blocked in a dose-dependent manner the (S)-3,5-DHPG-mediated hyperalgesia [F(4,25)= 32.2; P < 0.001] (Fig. 1A) with and ID50 value of 18.63 (95% C.L. 14.88 to 23.81) nmol/mouse (Table 1). Post-hoc analyses indicated that the 12.5, 25 and 50 nmol/mouse of MPEP producing a 44, 60 and 95% inhibition of (S)-3,5-DHPG-induced hyperalgesia, respectively, were all significant (P < 0.001; Fig. 1A).
Figure 1. Inhibition of(S)-3,5-DHPG-mediated hyperalgesi.
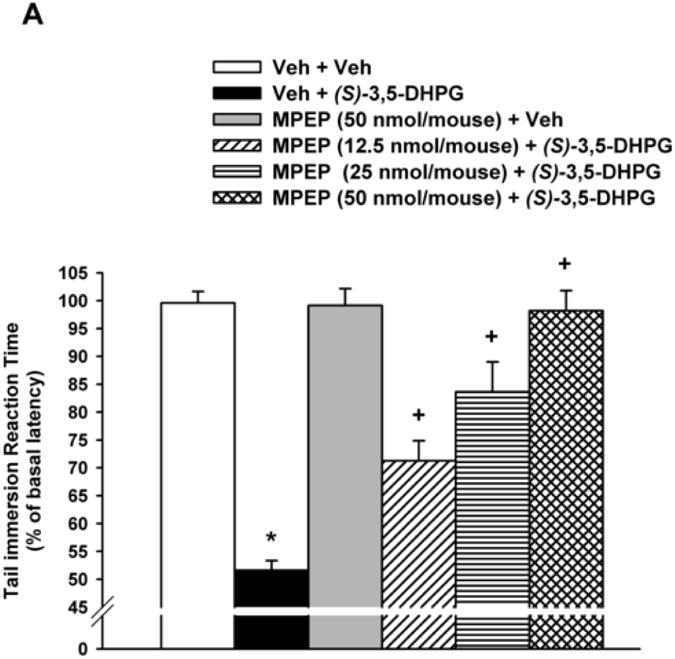
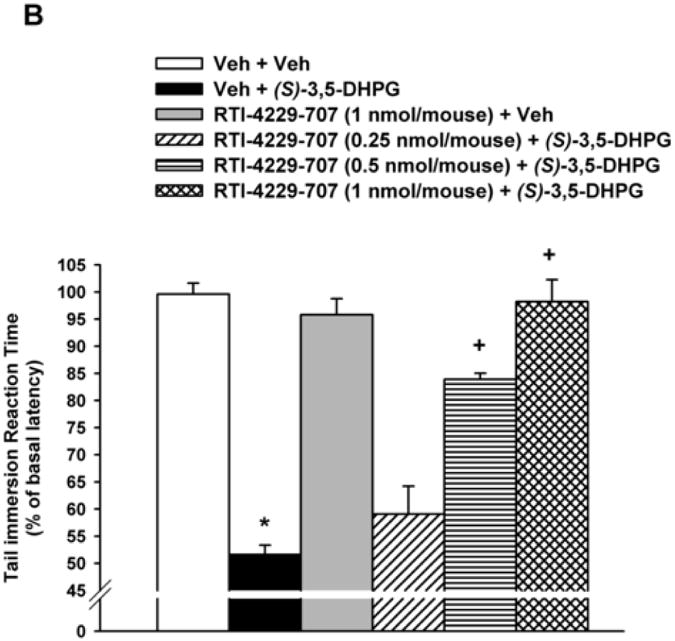
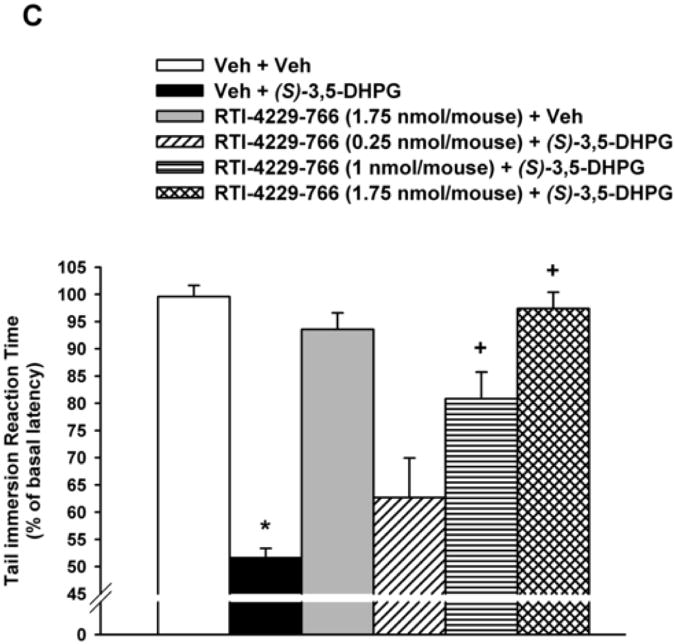
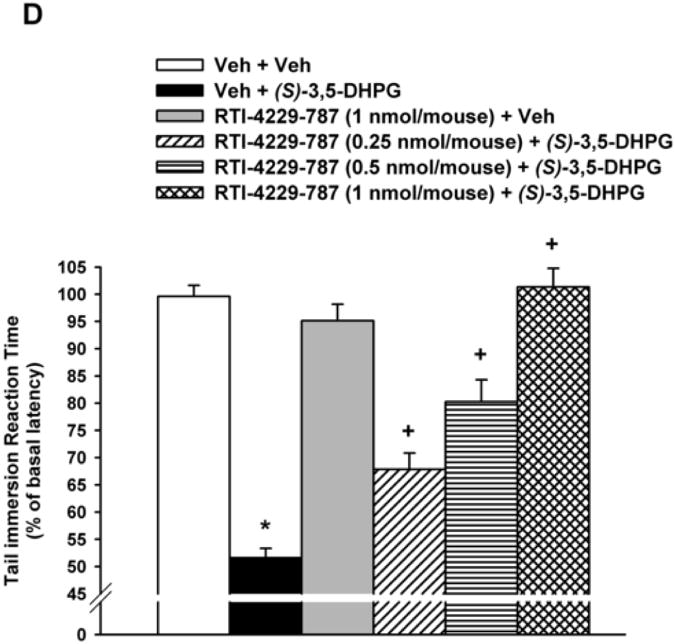
Mice (n= 6-8), with predetermined tail immersion baseline, were injected i.t. with either the vehicle (Veh) or the mGluR5 antagonists MPEP (12.5, 25 and 50 nmol/mouse), RTI-4229-707 (0.25, 0.5 and 1 nmol/mouse), RTI-4229-766 (0.25, 1 and 1.75 nmol/mouse) or RTI-4229-787 (0.25, 0.5 and 1 nmol/mouse).). The mGluR5 agonist, (S)-3,5-DHPG was administered to the mice 5 min later and at 30 min from its injection, the mice were re-assessed for their tail immersion reaction time. Data are expressed as mean % of basal latency ± S.E.M. *Value significantly different from vehicle-injected mice at P<0.05, +Value significantly different from (S)-3,5-DHPG-injected mice at P<0.05.
Table 1. ID50 values for inhibition of (S) 3,5 DHPG-mediated hyperalgesisa.
| Tail immersion test | ID50 value (nmol/mouse (95% C.L.)) |
|---|---|
| MPEP | 18.63 (95% C.L. 14.88 to 23.81) |
| RTI-4229-707 | 0.49 (95% C.L. 0.41 to 0.58) |
| RTI-4229-766 | 0.72 (95% C.L. 0.49 to 1.03) |
| RTI-4229-787 | 0.44 (95% C.L. 0.36 to 0.55) |
| RTI-4229-785 | Inactive |
| RTI-4229-828 | Inactive |
Mice (n= 6-8), with pre-determined tail immersion baseline, were injected i.t. with either the vehicle or the mGluR5 antagonists MPEP (12.5, 25 and 50 nmol/mouse), RTI-4229-707 (0.25, 0.5 and 1 nmol/mouse), RTI-4229-766 (0.25, 1 and 1.75 nmol/mouse) or RTI-4229-787 (0.25, 0.5 and 1 nmol/mouse). The mGluR5 agonist, (S)-3,5-DHPG was administered to the mice 5 min later and at 30 min from its injection, the mice were re-assessed for their tail immersion reaction time. The inhibitory dose-50 (ID50) values with 95% confidence limits were calculated for the antagonists co-administered with (S)-3,5-DHPG.
In a similar but more efficacious fashion, the three test selective mGluR5 antagonists RTI-4229-707, RTI-4229-766 and RTI-4229-787 were significantly more potent than MPEP in blocking (S)-3,5-DHPG-induced hyperalgesia. The calculated ID50 values for the three compounds were 0.49 (95% C.L. 0.41 to 0.58), 0.72 (95% C.L. 0.49 to 1.03) and 0.44 (95% C.L. 0.36 to 0.55) nmol/mouse, respectively (Table 1). Compound RTI-707, administered i.t. 5 min prior to (S)-3,5-DHPG, resulted in a dose-dependent inhibition of the (S)-3,5-DHPG-induced hyperalgesia [F(4,25)= 48.2; P < 0.001] (Fig. 1B). Post-hoc analyses indicated that the 0.5 and 1 nmol/mouse of RTI-4229-707 producing a 63 and 95% inhibition of (S)-3,5-DHPG-induced hyperalgesia, respectively, were significant (P < 0.001), whereas the dose of 0.25 nmol/mouse had no significant effect on the (S)-3,5-DHPG-induced hyperalgesia (Fig. 1B). Compound RTI-4229- 766 also resulted in a dose-dependent inhibition of the (S)-3,5-DHPG-induced hyperalgesia [F(4,25)= 24.1; P < 0.001] (Fig 1C). Post-hoc analyses indicated that the 1 and 1.75 nmol/mouse of RTI-4229-766 producing a 61 and 95% inhibition of (S)-3,5-DHPG-induced hyperalgesia, respectively, were significant (P < 0.001), whereas the dose of 0.25 nmol/mouse had no significant effect on the (S)-3,5-DHPG-induced hyperalgesia (Fig. 1C). Finally, compound RTI-4229-787 blocked in a dose-dependent inhibition of the (S)-3,5-DHPG-induced hyperalgesia [F(4,25)= 36.3; P < 0.001] (Fig 1D). Post-hoc analyses indicated that the 0.25, 0.5 and 1 nmol/mouse of RTI-4229-787 producing a 32, 56 and 97% inhibition of (S)-3,5-DHPG-induced hyperalgesia, respectively, were all significant (P < 0.01) (Fig. 1D). All the three antagonists tested at their highest doses were inactive when administered alone to mice (Fig 1B-D). It is noteworthy that compounds RTI-4229-785 and RTI-4229-828 tested at doses up to 5 nmol/mouse were totally inactive in blocking (S)-3,5-DHPG-induced hyperalgesia.
2.2. Reversal of morphine antinociceptive tolerance
Morphine administered s.c. elicited dose-dependent antinociception in the tail immersion test in both placebo and morphine pellet-implanted mice at 72-h following implantation (Fig. 2A-D). Morphine-pelleted mice showed a 5.5-fold tolerance to the antinociceptive effect of acute morphine compared to placebo-pelleted mice in the tail immersion test. I.c.v. administration of the selective mGluR5 antagonist MPEP to morphine-tolerant mice dose-dependently reversed morphine antinociceptive tolerance with and ED50 value of 1050 (95% C.L. 869.4 to 1213.6) nmol/mouse (Table 2). Complete reversal of morphine antinociceptive tolerance in the tail immersion test was achieved at a dose of 2000 nmol/mouse. Acutely administered morphine s.c. was equally potent in both placebo- and morphine-pelleted mice injected with bisindolylmaleimide I (Fig. 2A; Table 3).
Figure 2. Reversal of morphine antinociceptive tolerance.
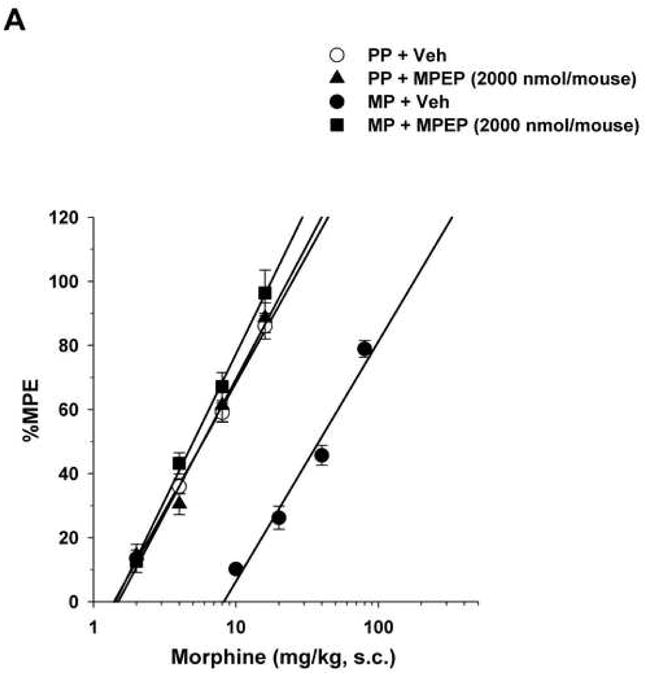
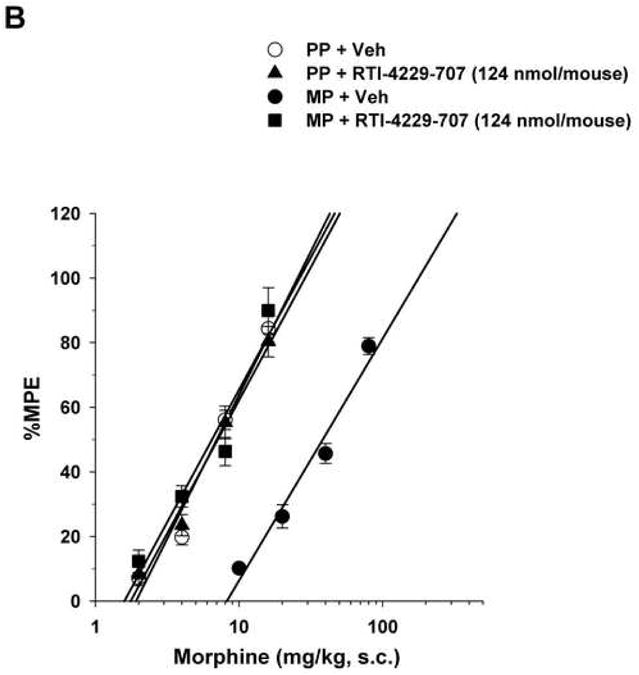
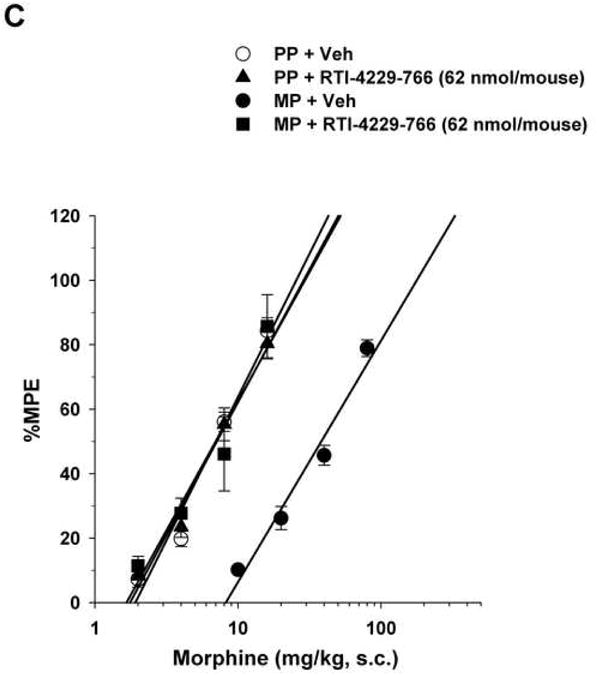
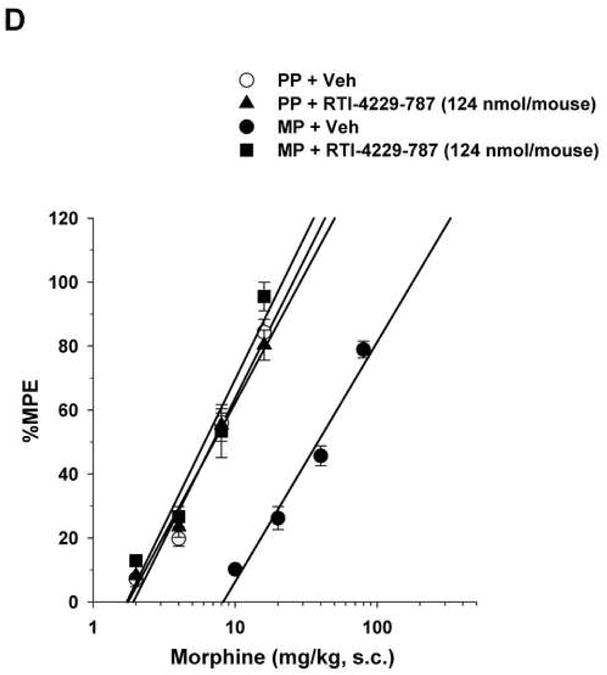
Mice were surgically implanted with placebo pellets (PP), or 75 mg morphine pellets (MP). Seventy-two hours later, baseline measures were obtained before the i.c.v. administration of vehicle (Veh) or the mGluR5 antagonists MPEP (2000 nmol/mouse), RTI-4229-707 (124 nmol/mouse), RTI-4229-766 (62 nmol/mouse) or RTI-4229-787 (124 nmol/mouse). Immediately afterwards, the mice were challenged with various doses of morphine s.c. for construction of dose-response curves. The ED50 values with 95% confidence limits were calculated for the antagonists to reverse morphine antinociceptive tolerance.
Table 2. ID50 values for reversal of morphine antinociceptive tolerance.
| Tail immersion test | ID50 value (nmol/mouse (95% C.L.)) |
|---|---|
| MPEP | 1050 (95% C.L. 869.4 to 1213.6) |
| RTI-4229-707 | 57.7 (95% C.L. 50.5 to 65.8) |
| RTI-4229-766 | 25.8 (95% C.L. 22.8 to 29.3) |
| RTI-4229-787 | 64.3 (95% C.L. 42.0 to 98.7) |
| RTI-4229-785 | Inactive |
| RTI-4229-828 | Inactive |
Mice (n= 6-8) were surgically implanted with either placebo pellets (PP) or morphine pellets (MP). At 72-h baseline measures were obtained before i.c.v. administration of vehicle or the mGluR5 antagonists MPEP (500, 1000 and 2000 nmol/mouse), RTI-4229-707 (31, 62 and 124 nmol/mouse), RTI-4229-766 (15.5, 31 and 62 nmol/mouse) or RTI-4229-787 (31, 62 and 124 nmol/mouse). Immediately afterwards, the mice were challenged with various doses of morphine s.c. for construction of dose-response curves. The inhibitory dose-50 (ID50) values with 95% confidence limits were calculated for the antagonists to reverse morphine antinociceptive tolerance.
Table 3.
Morphine antinociceptive tolerance.
| Treatment | Morphine ED50 value (mg/kg (95% C.L.)) | Potency Ratio (95% C.L.) | |
|---|---|---|---|
| PP + Veh | 6.9 (6.3 to 7.6) | ||
| MP + Veh | 38.2 (34.4 to 42.4)* | vs. PP + Veh | 5.2 (4.7 to 5.9)‡ |
| PP + MPEP | 6.2 (5.7 to 6.7) | ||
| MP + MPEP | 5.8 (5.2 to 6.3) | vs. PP + MPEP | 0.9 (0.8 to 0.9) |
| PP + RTI-4229-707 | 6.4 (4.8 to 6.9) | ||
| MP + RTI-4229-707 | 6.3 (5.4 to 7.2) | vs. PP + RTI-4229-707 | 1.0 (0.9 to 01.0) |
| PP + RTI-4229-766 | 6.6 (5.2 to 7.1) | ||
| MP + RTI-4229-766 | 6.0 (5.3 to 6.8) | vs. PP + RTI-4229-766 | 0.9 (0.8 to 0.9) |
| PP + RTI-4229-787 | 6.5 (5.1 to 7.3) | ||
| MP + RTI-4229-787 | 6.0 (5.3 to 6.8) | vs. PP + RTI-4229-787 | 0.9 (0.8 to 0.9) |
Mice (n= 6-8) were surgically implanted with either placebo pellets (PP) or morphine pellets (MP). At 72-h baseline measures were obtained before i.c.v. administration of vehicle (Veh) or the mGluR5 antagonists MPEP (2000 nmol/mouse), RTI-4229-707 (124 nmol/mouse), RTI-4229-766 (62 nmol/mouse) or RTI-4229-787 (124 nmol/mouse). Immediately afterwards, the mice were challenged with various doses of morphine s.c. for construction of dose-response curves for calculation of the effective dose-50 (ED50) values and potency ratios.
Significantly different than PP + Veh group based on non-overlapping 95% C.L.s;
Significantly different potency ratio since lower 95% C.L. is greater than 1.
Similarly, but more potently, i.c.v administration of the three test selective test mGluR5 antagonists RTI-4229-707, RTI-4229-766 and RTI-4229-787 to morphine-tolerant mice resulted in a dose dependent reversal of morphine antinociceptive tolerance. The calculated ID50 values for the three compounds were 57.7 (95% C.L. 50.5 to 65.8), 25.8 (95% C.L. 22.8 to 29.3) and 64.3 (95% C.L. 42.0 to 98.7) nmol/mouse, respectively (Table 2). Fig. 2B-D shows complete reversal of morphine antinociceptive tolerance at a dose of 124, 62 and 124 nmol/mouse for compound RTI-4229-707, RTI-4229-766 and RTI-4229-787, respectively. It is noteworthy that all three antagonists at the tested doses had no effect on morphine-induced antinociception in placebo-pelleted mice. Compounds RTI-4229-785 and RTI-4229-828 at doses up to 2000 nmol/mouse were totally inactive in reversing morphine antinociceptive tolerance.
3. Discussion
Group I mGlu receptors have been recognized to play an important role in nociceptive processing (Neugebauer and Carlton, 2002) including the modulation of the ligand gated ionotropic channels activity and/or neurotransmitter release (Schkeryantz et al., 2007). In addition, a possible role for the receptors has been hypothesized in reversing morphine antinociceptive tolerance in mice (Smith et al., 2004).
We recently reported on the synthesis and in vitro effects of a series of phenylethyl[1,2,4]methyltriazines which are analogues of the classical mGluR5 anatgonist MPEP (Carroll et al., 2007). Some but not all of the compounds tested selectively antagonized glutamate-mediated mobilization of internal calcium in the mGluR5 assay without possessing any efficacy at the mGlu receptor subtype 1 (mGluR1). In the present study, we characterized some of the in vivo pharmacological properties of five compounds from this series by comparing their efficacy to that of the well-known mGluR5 antagonist MPEP in blocking the hyperalgesia mediated by the group I mGlu receptors agonist (S)-3,5-DHPG as well as in reversing morphine antinociceptive tolerance in mice.
In agreement with the previously mentioned study, we found that three of the compounds that are active mGluR5 antagonists in vitro are also effective in blocking (S)-3,5-DHPG-induced hyperalgesia in vivo. On the other hand, the other two compounds which were inactive in vitro, were also totally ineffective in vivo in this test. We recently reported that the group I mGluR agonist (S)-3,5-DHPG produces a maximal hyperalgesia effect when administered i.t. at 15 nmol/mouse (Gabra et al., 2007). In the same study, we showed that the (S)-3,5-DHPG-induced hyperalgesia was blocked by pre-treatment with mGluR1, mGluR5 or NMDA receptor antagonists, PKC or protein kinase A (PKA) inhibitors as well as by knocking-down of the NR2B subunit of the NMDA receptor. These results supported our hypothesis of a coupling between the group I mGlu receptors and the N-methyl-D-aspartic acid (NMDA) receptor in mediated hyperalgesia in the spinal cord. It is known that phosphorylation of the NMDA receptor results in an increase in NMDA receptor channel current and Ca2+ influx through the channel (Wang and Salter, 1994; Ali and Salter, 2001). Thus, one functional consequence of mGluR activation under painful stimuli is to prime NMDA receptor for further enhanced hyperexcitability. This mechanism may be a critical initiator for central nociceptive sensitization. The three active compounds tested were found to be 26-42 times more potent than MPEP in blocking (S)-3,5-DHPG-induced hyperalgesia, thus providing new potent and selective mGluR5 antagonists as a tool in managing acute and chronic pain.
The neurobiological mechanisms of the development of opioid tolerance are multifaceted and only partially understood. The second messenger pathways activating protein PKC and PKA have been shown to play an integral role in the development and expression of tolerance to the inhibitory effects of opioids (Narita et al., 1996; Bernstein and Welch 1997; Smith et al., 1999). In our laboratory, we have shown that i.c.v. administration of PKC or PKA inhibitors reversed morphine antinociceptive tolerance (Javed et al., 2004; Smith et al., 2007) but also reinstated the morphine-induced behaviors of antinociception, hypothermia and Straub tail, 72-h after implantation of a 75 mg morphine pellet, to the same extent observed following acute administration of morphine (Smith et al., 2006). Chronic morphine administration is associated with increases in the activity of the PI cascade. Higher levels of group I mGlu receptors, or more efficient receptor coupling to Gq/11 proteins, could contribute to the increases in the activity of the PI cascade observed in animals chronically administered morphine. The subsequent activation of PLCβ results in higher PKC activity leading to μ-opioid receptor desensitization and the development of opioid tolerance (Conn and Pin, 1997; Smith et al., 2004). This led to the hypothesis that group I mGlu receptor antagonists would reverse antinociceptive tolerance. Sharif et al. (2002) demonstrated that i.t. infusion of antisense oligonucleotide to the mGluR1 during 6 days of s.c. morphine administration prevented the development of morphine tolerance. Morphine antinociceptive tolerance was also prevented in mice by the daily s.c. administration of the mGluR5 antagonist MPEP (Kozela et al., 2003). We have previously demonstrated that the mixed mGluR1/5 antagonist AIDA, ((RS)-1-Aminoindan-1,5-dicarboxylic acid), completely reverses morphine antinociceptive tolerance, whereas the selective mGluR5 antagonist MPEP, used at doses up to 1 μmol/mouse, resulted only in a partial tolerance reversal (Smith et al., 2004). However, in the present study we were able to achieve complete tolerance reversal with MPEP at 2 μmol/mouse. The present study showed that the three active mGluR5 antagonists resulted in a complete reversal of morphine antinociceptive tolerance but at much lower nanomol doses compared to MPEP, which reflects a higher potency and efficacy. Conversely, the inactive compounds in the in vitro assay and in blocking (S)-3,5-DHPG-induced hyperalgesia, were totally inactive in reversing morphine tolerance.
It is worth mentioning that higher doses of each of the compounds were required to reverse morphine antinociceptive tolerance than to block (S)-3,5-DHPG-induced hyperalgesia. There is a major difference in the procedure that could contribute to this difference in potency. The test compounds were given prior to the agonist (S)-3,5-DHPG that induced the hyperalgesia, thus they blocked the acute effect of the agonist. In the tolerance study, the test compounds were given after the tolerance has developed, thus in this procedure we were quantifying the potency of the compounds to reverse an established phenomenon. It is expected that compounds would be less potent in reversing than in blocking an effect. Further, the difference might just be due to the difference in the mechanisms being studied.
Taken together, the ability of the new selective mGluR5 antagonists to block the acute (S)-3,5-DHPG-induced hyperalgesia and to reverse the chronic morphine antinociceptive tolerance reflects the difference in the pathways involved in both effects. We hypothesize that the antagonism of (S)-3,5-DHPG-induced hyperalgesia by mGluR5 antagonists is a receptor mediated mechanism, whereas the reversal of morphine tolerance involves multiple downstream mechanisms including the PI cascade. These findings helped to characterize and understand some of the pharmacological properties of three novel non-competitive mGluR5 antagonists towards potential therapeutic applications of these drugs.
4. Experimental Procedure
4.1. Animals
Male Swiss Webster mice (Harlan Laboratories, Indianapolis, IN) weighing 25-30 g were housed 6 to a cage in animal care quarters and maintained at 22 ± 2 °C on a 12-hr light-dark cycle. Food and water were available ad libitum. The mice were brought to a test room (22 ± 2 °C, 12-hr light-dark cycle), marked for identification and allowed 18-hr to recover from transport and handling. Protocols and procedures were approved by the Institutional Animal Care and Use Committee (IACUC) at Virginia Commonwealth University Medical Center and comply with the recommendations of the IASP (International Association for the Study of Pain).
4.2. Surgical implantation of pellets
Mice were anesthetized with 2.5% isoflurane before shaving the hair around the base of the neck. Adequate anesthesia was noted by the absence of the righting-reflex and lack of response to toe-pinch, according to IACUC guidelines. The skin was cleansed with 10% povidone iodine (General Medical Corp., Prichard, WV) and rinsed with alcohol before making a 1 cm horizontal incision at the base of the neck. The underlying subcutaneous space toward the dorsal flanks was opened using a sterile glass rod. Maintenance of a stringent aseptic surgical field minimized any potential contamination of the pellet, incision and subcutaneous space. A placebo pellet or 75 mg morphine pellet was inserted in the space before closing the site with Clay Adams Brand, MikRon® AutoClip® 9mm Wound Clips (Becton Dickinson and Co., Sparks, MD) and again applying iodine to the surface. The animals were allowed to recover in their home cages where they remained throughout the experiment.
4.3. Intrathecal injections
Intrathecal (i.t.) injections were made through the intervertebral space in unanaesthetized mice between the L5-L6 of the spinal cord, as described by Hylden and Wilcox (1980). Briefly, a volume of 5 μl was administered i.t. with a 28 gauge, 1/2-inch stainless steel needle connected to a 50 μl Hamilton microsyringe, the animal being lightly restrained to maintain the position of the needle. Puncture of the dura was indicated behaviorally by a slight flick of the tail.
4.4. Intracerebroventricular injections
Intracerebroventricular (i.c.v.) injections were performed as described by Pedigo et al. (1975). Mice were anesthetized with isoflurane and a horizontal incision was made in the scalp. A free-hand 5 μl injection of drug or vehicle was made in the lateral ventrical (2 mm rostral and 2 mm lateral at a 45° angle from the bregma). The extensive experience of this laboratory has made it possible to inject drugs with greater than 95% accuracy. Immediately after testing, the animals were euthanized to minimize any type of distress, according to IACUC guidelines.
4.5. Tail immersion test
The warm-water tail immersion test was performed according to Coderre and Rollman (1983) using a water bath with the temperature maintained at 56 ± 0.1 °C. Before injecting the mice, a base-line (control) latency was determined. Only mice with a control reaction time from 2- to 4-sec were used. The average baseline latency for these experiments was 3.0 ± 0.1 sec. The test latency after drug treatment was assessed at the appropriate time, and a 10-sec maximum cut-off time was imposed to prevent tissue damage. Antinociception was quantified according to the method of Harris and Pierson (1964) as the percentage of maximum possible effect (% MPE) which was calculated as: %MPE = [(test latency – control latency) / (10 – control latency)] × 100. Percent MPE was calculated for each mouse using at least 6 mice per drug.
4.6. Drugs and chemicals
The placebo and 75mg morphine pellets were obtained from the National Institute on Drug Abuse (NIDA), Bethesda, MD. Morphine sulfate (Mallinckrodt, St. Louis, MO) was dissolved in pyrogen-free isotonic saline (Baxter Healthcare, Deerfield, IL). The selective mGluR5 antagonist 2-methyl-6-(phenylethynyl)pyridine hydrochloride (MPEP) was purchased from Tocris Cookson Bioscience (Ellisville, MO, USA).
The five phenylethyl[1,2,4]methyltriazine analogues: 5-methyl-3-phenylethynyl[1,2,4]triazine (RTI-4229-707), 5-methyl-3-(4-phenoxyphenylethynyl[1,2,4]triazine (RTI-4229-766), 3-(2,5-dimethylphenylethynyl)-5-methyl[1,2,4]triazine (RTI-4229-785), 3-(3-methylphenylethynyl)-5-methyl[1,2,4]triazine (RTI-4229-787) and 3-(2-methylphenylethynyl)-5-methyl[1,2,4]triazine (RTI-4229-828) were synthesized in the Center for Organic and Medicinal Chemistry, Research Triangle Institute (Research Triangle Park, NC, USA; Carroll et al., 2007). All test drugs as well as MPEP were dissolved in a mixture of 10% dimethyl sulfoxide, 20% cremophor, 70% distilled water. The control vehicle-injected mice were injected with the same mixture.
4.7. Experimental design and statistical analysis
In the first series of experiments, the effect of MPEP as well as the test drugs on (S)-3,5-DHPG-induced hyperalgesia was evaluated. Mice, with pre-determined tail immersion baseline, were injected i.t. with either MPEP or test drug at various doses and 5 min later they were administered (S)-3,5-DHPG at a dose of 15 nmol/mouse, i.t. This dose was previously shown to produce maximal hyperaslgesia in mice (Gabra et al., 2007). Mice were re-assessed for their tail immersion reaction time 20 min later. Effective dose-50 (ED50) values were calculated using least-squares linear regression analysis followed by calculation of 95% confidence limits (95% C.L.) by the method of Bliss (1967).
In the second series of experiments, morphine dose-response curves were generated in both placebo-pelleted (PP) and morphine-pelleted (MP) mice 72-h following pellets implantation, to test for the development of tolerance. Baseline measures of tail immersion were obtained prior to subcutaneous (s.c.) morphine administration.
The following experiments then examined the effects of i.c.v. administration of different doses of MPEP as well as the test drugs to reverse morphine tolerance. Mice were surgically implanted with placebo pellets (PP) or morphine pellets (MP). At 72-h baseline measures were obtained before i.c.v. administration of vehicle, MPEP or test drugs at various doses. Immediately afterwards, the mice were challenged with various doses of morphine s.c. for construction of dose-response curves. Effective dose-50 (ED50) and inhibitory dose-50 (ID50) values were calculated as previously shown. Tests for parallelism were conducted before calculating the potency-ratio values with 95% C.L. by the method of Colquhoun (1971). Colquhoun notes that a potency-ratio value of greater than one, with the lower 95% C.L. greater than one, is considered a significant difference in potency between groups.
Data are expressed as mean values ± S.E.M. Analysis of variance (ANOVA) followed by the post hoc “Student-Newman-Keuls” test were performed to assess significance using the Instat 3.0 software (GraphPad Software, San Diego, CA, U.S.A.). P < 0.05 was considered significant.
Acknowledgments
We thank Joshua A. Seager and David L. Stevens for valuable technical assistance during these studies. This work was funded by the National Institute on Drug Abuse grants: DA-01647, K05-DA00480, DA-020836, DA05477, DA016472 and K05-DA00480
Abbreviations
- CNS
Central nervous system
- mGlu
metabotropic glutamate
- mGluR1
mGlu receptor subtype 1
- mGluR5
mGlu receptor subtype 5
- (S)-35-DHPG
(S)-3,5-dihydroxyphenylglycine
- NMDA
N-methyl-D-aspartic acid
- PKC
protein kinase C
- PKA
protein kinase A
- ED50
effective dose-50
- ID50
inhibitory dose-50
- %MPE
percent maximum possible effect
- i.t
intrathecal
- i.c.v
intracerebroventricular
- s.c
subcutaneous
- MPEP
2-methyl-6-(phenylethynyl)pyridine
- RTI-4229-707
5-methyl-3-phenylethynyl[1,2,4]triazine
- RTI-4229-766
5-methyl-3-(4-phenoxyphenylethynyl[1,2,4]triazine
- RTI-4229-785
3-(2,5-dimethylphenylethynyl)-5-methyl[1,2,4]triazine
- RTI-4229-787
3-(3-methylphenylethynyl)-5-methyl[1,2,4]triazine
- RTI-4229-828
3-(2-methylphenylethynyl)-5-methyl[1,2,4]triazine
- AIDA
(RS)-1-Aminoindan-1,5 dicarboxylic acid
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Ali DW, Salter MW. NMDA receptor regulation by Src kinase signaling in excitatory synaptic transmission and plasticity. Curr Opin Neurobiol. 2001;11:336–342. doi: 10.1016/s0959-4388(00)00216-6. [DOI] [PubMed] [Google Scholar]
- Backstrom P, Bachteler D, Koch S, Hyytia P, Spanagel R. mGluR5 antagonist MPEP reduces ethanol-seeking and relapse behavior. Neuropsychopharmacology. 2004;29:921–928. doi: 10.1038/sj.npp.1300381. [DOI] [PubMed] [Google Scholar]
- Bernstein MA, Welch SP. Effects of spinal versus supraspinal administration of cyclic nucleotide-dependent protein kinase inhibitors on morphine tolerance in mice. Drug and Alcohol Depend. 1997;44:41–46. doi: 10.1016/s0376-8716(96)01320-8. [DOI] [PubMed] [Google Scholar]
- Bliss CI. Statistics in Biology. McGraw-Hill; New York, NY: 1967. p. 439. [Google Scholar]
- Carroll FI, Kotturi SV, Navarro HA, Mascarella SW, Gilmour BP, Smith FL, Gabra BH, Dewey WL. Synthesis and pharmacological evaluation of phenylethynyl[1,2,4]methyltriazines as analogues of 3-methyl-6-(phenylethynyl)pyridine. J Med Chem. 2007;50:3388–3391. doi: 10.1021/jm070078r. [DOI] [PubMed] [Google Scholar]
- Chiamulera C, Epping-Jordan MP, Zocchi A, Marcon C, Cottiny C, Tacconi S, Corsi M, Orzi F, Conquet F. Reinforcing and locomotor stimulant effects of cocaine are absent in mGluR5 null mutant mice. Nat Neurosci. 2001;4:873–874. doi: 10.1038/nn0901-873. [DOI] [PubMed] [Google Scholar]
- Coderre TJ, Rollman GB. Naloxone hyperalgesia and stress-induced analgesia in rats. Life Sci. 1983;32:2139–2146. doi: 10.1016/0024-3205(83)90103-0. [DOI] [PubMed] [Google Scholar]
- Colquhoun D. Lectures on biostatistics: An introduction to statistics with applications in biology and medicine. Oxford: Clarendon Press; 1971. pp. 327–333. [Google Scholar]
- Conn PJ, Pin JP. Pharmacology and functions of metabotropic glutamate receptors. Annu Rev Pharmacol Toxicol. 1997;37:205–237. doi: 10.1146/annurev.pharmtox.37.1.205. [DOI] [PubMed] [Google Scholar]
- Fisher K, Coderre TJ. Hyperalgesia and allodynia induced by intrathecal (RS)-dihydroxyphenylglycine in rats. Neuroreport. 1998;9:1169–1172. doi: 10.1097/00001756-199804200-00038. [DOI] [PubMed] [Google Scholar]
- Gabra BH, Kessler FK, Ritter JK, Dewey WL, Smith FL. Decrease in NMDAR-NR2B subunit levels by intrathecal shRNA blocks group I mGluR-mediated hyperalgesia. J Pharmacol Exp Ther. 2007;322:186–194. doi: 10.1124/jpet.107.120071. [DOI] [PubMed] [Google Scholar]
- Gasparini F, Lingenhohl K, Stoehr N, Flor PJ, Heinrich M, Vranesic I, Biollaz M, Allgeier H, Heckendorn R, Urwyler S, Varney MA, Johnson EC, Hess SD, Rao SP, Sacaan AI, Santori EM, Velicelebi G, Kuhn R. 2-Methyl-6-(phenylethynyl)-pyridine (MPEP), a potent, selective and systemically active mGlu5 receptor antagonist. Neuropharmacology. 1999;38:1493–1503. doi: 10.1016/s0028-3908(99)00082-9. [DOI] [PubMed] [Google Scholar]
- Hylden JL, Wilcox GL. Intrathecal morphine in mice: a new technique. Eur J Pharmacol. 1980;67:313–316. doi: 10.1016/0014-2999(80)90515-4. [DOI] [PubMed] [Google Scholar]
- Iso Y, Grajkowska E, Wroblewski JT, Davis J, Goeders NE, Johnson KM, Sanker S, Roth BL, Tueckmantel W, Kozikowski AP. Synthesis and structure-activity relationships of 3-[(2-methyl-1,3-thiazol-4-yl)ethynyl]pyridine analogues as potent, noncompetitive metabotropic glutamate receptor subtype 5 antagonists; search for cocaine medications. J Med Chem. 2006;49:1080–1100. doi: 10.1021/jm050570f. [DOI] [PubMed] [Google Scholar]
- Javed RR, Dewey WL, Smith PA, Smith FL. PKC and PKA inhibitors reverse tolerance to morphine-induced hypothermia and supraspinal analgesia in mice. Eur J Pharmacol. 2004;492:149–157. doi: 10.1016/j.ejphar.2004.03.061. [DOI] [PubMed] [Google Scholar]
- Kozela E, Pilc A, Popik P. Inhibitory effects of MPEP, an mGluR5 antagonist, and memantine, an N-methyl-D-aspartate receptor antagonist, on morphine antinociceptive tolerance in mice. Psychopharmacology. 2003;165:245–251. doi: 10.1007/s00213-002-1287-8. [DOI] [PubMed] [Google Scholar]
- Marino MJ, Conn PJ. Glutamate-based therapeutic approaches: allosteric modulators of metabotropic glutamate receptors. Curr Opin Pharmacol. 2006;6:98–102. doi: 10.1016/j.coph.2005.09.006. [DOI] [PubMed] [Google Scholar]
- McGeehan AJ, Olive MF. The mGluR5 antagonist MPEP reduces the conditioned rewarding effects of cocaine but not other drugs of abuse. Synapse. 2003;47:240–242. doi: 10.1002/syn.10166. [DOI] [PubMed] [Google Scholar]
- Narita M, Mizoguchi H, Kampine JP, Tseng LF. Role of protein kinase C in desensitization of spinal delta-opioid-mediated antinociception in the mouse. Br J Pharmacol. 1996;118:1829–1835. doi: 10.1111/j.1476-5381.1996.tb15610.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neugebauer V, Carlton SM. Peripheral metabotropic glutamate receptors as drug targets for pain relief. Expert Opin Ther Targets. 2002;6:349–361. doi: 10.1517/14728222.6.3.349. [DOI] [PubMed] [Google Scholar]
- Paterson NE, Semenova S, Gasparini F, Markou A. The mGluR5 antagonist MPEP decreased nicotine self-administration in rats and mice. Psychopharmacology. 2003;167:257–264. doi: 10.1007/s00213-003-1432-z. [DOI] [PubMed] [Google Scholar]
- Pedigo NW, Dewey WL, Harris LS. Determination and characterization of the antinociceptive activity of intraventricularly administered acetylcholine in mice. J Pharmacol Exp Ther. 1975;193:845–852. [PubMed] [Google Scholar]
- Popik P, Wrobel M. Morphine conditioned reward is inhibited by MPEP, the mGluR5 antagonist. Neuropharmacology. 2002;43:1210–1217. doi: 10.1016/s0028-3908(02)00309-x. [DOI] [PubMed] [Google Scholar]
- Schkeryantz JM, Kingston AE, Johnson MP. Prospects for metabotropic glutamate 1 receptor antagonists in the treatment of neuropathic pain. J Med Chem. 2007;50:2563–2568. doi: 10.1021/jm060950g. [DOI] [PubMed] [Google Scholar]
- Sharif RN, Osborne M, Coderre TJ, Fundytus ME. Attenuation of morphine tolerance after antisense oligonucleotide knock-down of spinal mGluR1. Br J Pharmacol. 2002;136:865–872. doi: 10.1038/sj.bjp.0704792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slassi A, Isaac M, Edwards L, Minidis A, Wensbo D, Mattsson J, Nilsson K, Raboisson P, McLeod D, Stormann TM, Hammerland LG, Johnson E. Recent advances in non-competitive mGlu5 receptor antagonists and their potential therapeutic applications. Curr Top Med Chem. 2005;5:897–911. doi: 10.2174/1568026054750236. [DOI] [PubMed] [Google Scholar]
- Smith FL, Gabra BH, Smith PA, Redwood MC, Dewey WL. Determination of the role of conventional, novel and atypical PKC isoforms in the expression of morphine tolerance in mice. Pain. 2007;127:129–139. doi: 10.1016/j.pain.2006.08.009. [DOI] [PubMed] [Google Scholar]
- Smith FL, Javed RR, Smith PA, Dewey WL, Gabra BH. PKC and PKA inhibitors reinstate morphine-induced behaviors in morphine tolerant mice. Pharmacol Res. 2006;54:474–480. doi: 10.1016/j.phrs.2006.09.007. [DOI] [PubMed] [Google Scholar]
- Smith FL, Smith PA, Dewey WL, Javed RR. Effects of mGlu1 and mGlu5 metabotropic glutamate antagonists to reverse morphine tolerance in mice. Eur J Pharmacol. 2004;492:137–142. doi: 10.1016/j.ejphar.2004.03.055. [DOI] [PubMed] [Google Scholar]
- Young MR, Fleetwood-Walker SM, Dickinson T, Blackburn-Munro G, Sparrow H, Birch PJ, Bountra C. Behavioural and electrophysiological evidence supporting a role for group I metabotropic glutamate receptors in the mediation of nociceptive inputs to the rat spinal cord. Brain Res. 1997;777:161–169. [PubMed] [Google Scholar]
- Wang YT, Salter MW. Regulation of NMDA receptors by tyrosine kinases and phosphatases. Nature. 1994;369:233–235. doi: 10.1038/369233a0. [DOI] [PubMed] [Google Scholar]