

Abstract
Gene context determines gene expression, with local chromosomal environment most influential. Comparative genomic analysis is often limited in scope to conserved or divergent gene and protein families, and fungi are well suited to this approach with low functional redundancy and relatively streamlined genomes. We show here that one aspect of gene context, the amount of potential upstream regulatory sequence maintained through evolution, is highly predictive of both molecular function and biological process in diverse fungi. Orthologs with large upstream intergenic regions (UIRs) are strongly enriched in information processing functions, such as signal transduction and sequence-specific DNA binding, and, in the genus Aspergillus, include the majority of experimentally studied, high-level developmental and metabolic transcriptional regulators. Many uncharacterized genes are also present in this class and, by implication, may be of similar importance. Large intergenic regions also share two novel sequence characteristics, currently of unknown significance: they are enriched for plus-strand polypyrimidine tracts and an information-rich, putative regulatory motif that was present in the last common ancestor of the Pezizomycotina. Systematic consideration of gene UIR in comparative genomics, particularly for poorly characterized species, could help reveal organisms’ regulatory priorities.
Keywords: genome architecture, gene regulation, comparative genomics, regulatory complexity, fungi, polypyrimidine
Introduction
DNA is transcribed into RNA by proteins that interpret and remodel aspects of gene context, the influence of which decays with distance. Typically, information encoded in DNA and chromatin structure immediately flanking a gene is most influential, although in principle regulatory regions can be located anywhere in a genome read in the four-dimensional space of the active nucleus.
Established modes of gene expression regulation encompass alternative transcription initiation, alternative mRNA splicing, posttranscriptional gene silencing and RNA editing. Such complexity, and the interactive capacity of information processing signal transduction and transcription factors, is broadly correlated with organismal complexity (in the sense of enabling rather than causing, because evolutionary and life history relating to population size may be at least as important) (Lynch and Conery 2003; Tordai et al. 2005; Basu et al. 2008; Nilsen and Graveley 2010; Raffaele and Kamoun 2012).
Fungi are a diverse group of osmotrophs, ranging from obligate, single-celled pathogens to large, multicellular heterotrophs, with intimate associations across the spectrum of life. Most fungal genomes sequenced to date are streamlined by eukaryotic standards (20–60 Mb, 30–70% coding sequence, low-to-moderate levels of transposable elements, 2–3 introns per gene, and low levels of alternative transcription), though current extremes range from just 2.3 Mb for the microsporidian Encephalitozoon intestinalis to 160 Mb for the Ascomycete Golovinomyces orontii, both obligate pathogens (Kelkar and Ochman 2012). The most familiar and first sequenced fungi, the model “yeasts” Saccharomyces cerevisiae and Schizosaccharomyces pombe, have proven to be atypical relative to the majority of Ascomycetes with a reduced genome size and gene count, few introns and a greater proportion of coding sequence. As predominantly unicellular yeasts that are niche adapted, they also show reduced developmental and metabolic diversity (though both display cellular morphological flexibility under certain conditions).
The model yeasts are also clearly atypical in that their genomes are exceptionally well studied. Approximately 85% of S. cerevisiae genes are experimentally verified at present, whereas for S. pombe the figure is 38%. Study of other fungi, including those of applied interest, is often hampered by difficulty in genetic manipulation, lack of or impractical modes of sexual reproduction, and a high frequency of uncharacterized or entirely novel genes, typified by rapidly evolving families of transporters and transcription factors that have undergone expansion in a number of fungal lineages (Lespinet et al. 2002; Cornell et al. 2007).
In addressing this through comparative genomic analysis, most studies of fungi to date have focused on conserved or lineage-specific protein-coding genes (Galagan, Calvo, et al. 2005; Galagan, Henn, et al. 2005; Cornell et al. 2007). A complementary approach is to examine the evolution and function of noncoding sequence, such as gene upstream intergenic regions (UIRs) and untranslated regions (UTRs), as gene expression is equally relevant to phenotype as gene product function. This study has taken a pan-fungal approach examining noncoding sequence with the aim of identifying conserved attributes and elements that may control gene expression, with focus on the genus Aspergillus where multiple experimentally annotated genomes are available. We find that local genomic architecture is highly predictive of gene function: large UIR genes show strong enrichment in regulatory functions. Although UIR correlates inversely with evolutionary stability of gene order at the genome level, this is not seen for genes with large UIR maintained across species, suggesting functional constraint. In the Aspergilli, large UIR genes are involved predominantly in development and high-level metabolic regulation, reflected in a marked experimental characterization bias, and show unique sequence characteristics including a novel, evolutionarily mobile putative cis-regulatory motif.
Materials and Methods
Genome Data Sets and Intergenic Sequence Analysis
Genome sequence, annotations, and Pfam predictions were obtained from AspGD (A. nidulans FGSC A4, A. fumigatus Af293, A. oryzae RIB40, and A. niger CBS 513.88) (Arnaud et al. 2010), CGD (Candida albicans SC5314), SGD (S. cerevisiae S288C), and the Broad Institute (A. clavatus NRRL 1, A. terreus NIH2624, C. immitis rs3, Fusarium oxysporum 4287, Histoplasma capsulatum g186ar, Magnaporthe oryzae 70–15, Paracoccidioides brasiliensis pb01, Rhizopus oryzae RA 99–880, and Uncinocarpus reesii, downloaded May 2012. Penicillium marneffei ATCC 18224 and Talaromyces stipitatus ATCC10500 data were obtained from JCVI.
Where available, pairwise orthology predictions generated by a reciprocal best hit approach using either InParanoid (BLOSUM80, InParanoid bootstrap score of 100%) or Jaccard clustering (80% BLASTP percent identity threshold, e value threshold of 1e−5, minimum Jaccard similarity coefficient of 0.6) were downloaded from AspGD (http://www.aspergillusgenome.org/download/homology/orthologs/), covering A. fumigatus, A. nidulans, A. oryzae, A. niger, and S. cerevisiae. For other species, predictions were generated by InParanoid bidirectional best hit algorithm using (default settings, bootstrap score of 100%) (Ostlund et al. 2010). As we are primarily interested in broadly surveying orthologs across progressively narrowing taxonomic ranks relative to the Aspergilli, we did not attempt complete reconstruction of all orthologous clusters across all lineages. Instead, we calculated pairwise predictions relative to A. nidulans, and so the caveat that prediction sensitivity varies with genetic distance applies here. With this method, orthology across all 17 study species was limited to 1,062 genes. For analysis limited to the Aspergilli, we included gene clusters where one of the six orthology predictions failed, after manual follow-up of cases suggested this was most often due to variation or complete failure of automated gene prediction models, usually for A. niger. The full orthology matrix generated is provided as supplementary data, Supplementary Material online.
Before calculation of intergenic sizes, filtering to remove dubious open reading frames (ORFs) was conducted based on the following criteria: all predicted proteins <200 amino acids in length and lacking a predicted Pfam domain were queried by BLASTP (Altschul et al. 1997) against a combined database consisting of the predicted proteomes for all of the above species and all other Ascomycete species and strains sequenced by the Broad Institute as of August 2012. Proteins with no blast hit <1e−5 were discarded. For A. niger CBS 513.88, for which gene predictions are particularly generous (relative to other Aspergilli and also to a more recently sequenced A. niger strain, ATCC 1015), the above filtering was carried out for all proteins <300 amino acids with no Pfam domains, and homology searching included strain ATCC 1015. Although this filtering may have removed some entirely novel genes, manual examination against pooled mixed condition RNA-seq data for A. nidulans and A. oryzae (AspGD) supports the existence of many transcriptionally silent small ORFs with unusual gene structure in at least these species. Calculation of intergenic size and other gene statistics was carried out with custom Python scripts (available on request) using gff/gtf gene annotations and Pfam prediction files. In brief, an UIR was assigned in full to each flanking gene, with the assumption that disruption of gene order or changes in UIR size across species would often lead to exclusion of genes without constraint on UIR size. In rare cases of genes within genes, or overlapping genes on the same strand, UIR was not calculated. A ceiling of 30 kb was applied for cases where genes flank large N-gaps in a genome (e.g., for unassembled regions such as centromeres).
Expression Data Sets and Analysis
Microarray and RNAseq data sets for A. nidulans, A. fumigatus, A. niger, and A. oryzae were downloaded from NCBI GEO (GSE4578, GSE38131, GSE37796, GSE37795, GSE36440, GSE32123, GSE30384, GSE27610, GSE25266, GSE24466, GSE23605, GSE22442, GSE22052, GSE21752, GSE2085, GSE19952, GSE19430, GSE18851, GSE16617, GSE16566, GSE16509, GSE15702, GSE14285, GSE12953, GSE11930, GSE11725, GSE10712, GSE10475, and GSE38694), EBI ArrayExpress (E.MEXP.3218), sourced from supplementary data (Supplementary Material online) (Sheppard et al. 2005; Twumasi-Boateng et al. 2008) or from unpublished microarray data sets for A. nidulans (AN1060 overexpression, AN1060, AN8211, and AN0888 deletion mutants, vegetative growth time-course (Noble LM and Andrianopoulos A, in preparation) and RNAseq data sets for P. marneffei (yeast and hyphal growth, asexual development, macrophage phagocytosis, hgrA and pakB deletion mutants (Andrianopoulos A, Boyce KJ, Bugeja HE, Weerasinghe H, in preparation). Expression values were generated using the NCBI GEO2R tool, the R packages limma (Oshlack et al. 2007; Ritchie et al. 2007), and edgeR (Robinson et al. 2010), or by parsing of SOFT formatted data with custom Python scripts.
Expression samples were clustered hierarchically using all raw data (average linkage, uncentred Pearson correlation). Testing for significant differential expression of UIR quintiles was performed in R, against sample size- and mean-matched control groups with variable size across species. Significance was measured for mean expression (against all shared orthologs) by two-sided t test, and for the number of genes up or downregulated at an absolute log 2 ratio of 0.5 by Fisher’s exact test (in both cases, at a threshold of P < 0.05 after adjustment for multiple testing by the false discovery rate method of Benjamini and Hochberg).
Functional Enrichment and Statistical Tests
GO term enrichments for orthologous groups across varying taxonomic ranks were calculated using GoTermFinder, hosted by Princeton University (http://go.princeton.edu/) (Boyle et al. 2004) using AspGD GO annotations against the appropriate ortholog background for each rank, with P < 0.01 after Bonferroni correction. Significance was calculated for upper and lower UIR quintiles for orthologs with maintained size, and was normalized to significance for all the equivalent quintiles for all orthologs ignoring UIR conservation. Hierarchical clustering used algorithms implemented in MeV (Saeed et al. 2003, 2006). Pfam domain co-occurrence networks were generated using a custom Python script to parse Pfam predictions and generate files for Cytoscape network analysis (Smoot et al. 2011). Networks represent pairwise associations of all domains in UIR groups for the studied Aspergillus species.
Motif, Base Frequency, and Synteny Analysis
Custom Python scripts were used to extract UIR sequence and analyze base frequency characteristics. The MEME suite (Bailey et al. 2009) was used to determine enriched motif position weight matrices, using the “any number of repetitions” (anr) model and default settings, against orthologous UIR groups across the Aspergilli and UIR groups as a whole. MAST was used to map genome-wide occurrence of the TACRGAGTA motif with a match score >1,000.
We screened large UIR sequence against all known Aspergillus transposable elements in RepBase (Jurka 2000) by blastn (Altschul et al. 1997) with an expect value cutoff of 1e−10. For A. nidulans, 15 cases of large UIR (maintained across the Aspergilli) containing transposable element fragments were found, predominantly matching Mariner/Tc1-type elements. Gene orientation and orthologous gene order among the Aspergilli was measured with a custom Python script, generating an “orthology score” representing the proportion of orthologs within a 5-gene window relative to A. nidulans in upstream or downstream directions. Results were qualitatively similar for a metric based on the length of broadly orthologous segments (defined as the total window length with greater than 50% orthology between species pairs).
Results
Upstream Intergenic Size Distribution and Conservation
UIR size was surveyed for 16 diverse Ascomycetes, representing the largest fungal phylum, and the basal Zygomycete species R. oryzae as an outgroup (fig. 1). UIR size, calculated between ORFs, varied from a median of 490 bp for S. cerevisiae to 1,510 bp for the plant pathogenic filamentous fungus M. oryzae. This correlates with the proportion of coding sequence (CDS) in the genome (Pearson’s r = −0.92, P = 7.6e−8), total genome size (r = 0.81, P = 4.2e−5) and, to a lesser extent, mean number of introns per gene (r = 0.45, P = 0.03). Genome wide, UIR size distribution does not match phylogeny closely, although at the extremes the two Sordariomycetes, F. oxysporum and M. oryzae, possess the largest intergenic regions and the two Saccharomycete yeasts, S. cerevisiae and C. albicans, the smallest (fig. 1). Phylogenetic clustering based on UIR size for the set of core orthologs across all species recapitulated known relationships substantially better than the genome-wide approach (supplementary fig. S1, Supplementary Material online), supporting the expectation that local architecture of nonconserved genes is more variable.
Fig. 1.—

Fungal phylogeny and distribution of UIR size. Maximum likelihood phylogeny based on concatenated amino acid sequence for 100 single copy orthologs ranked by UIR invariance. Taxonomic groupings used throughout the study are highlighted, ranging from phylum Ascomycota (darkest blue) to subdivision Pezizomycotina, class Eurotiomycete, order Eurotiale, and genus Aspergillus (lightest blue). Upstream intergenic size distribution for each species is shown at right. Box plots span first and third quartiles (whiskers extend to 1.5× these values), median values are indicated by a black bar.
UIR size varies over a wide range with genomic noncoding sequence, whereas the coding size of genes is relatively invariant across species. The 90% UIR percentile for S. cerevisiae, for example, is 1,558 bp, only slightly larger than the median UIR size for M. oryzae (1,503 bp), whereas for gene CDS 90% the equivalent values are 2,864 and 2,773 bp, respectively. For this reason, we took a within species rank approach to examining UIR size maintenance, rather than using fixed size limits. A corollary of this approach is that percentiles are not equal in range, so that maintenance within central UIR bins represents strict size conservation relative to relaxed bounds for the upper percentile bin. We see only if UIR size tends to remain similar, relatively, against the varying forces of selection encountered along lineages.
For each species, orthologous genes were binned by intergenic size quintile (one-fifth of all genes per species per bin), and length was examined across taxonomic ranks. At all levels, the distribution of UIR size was J-shaped (skewed toward the extremes, particularly toward large regions, fig. 2A). This largely reflects the inequality of quintile ranges, but the distribution remains skewed toward extremes when normalized to range (fig. 2B). Large UIRs could arise through “passive” indirect forces of variation generation, such as replication slippage, transposable element insertion, and chromosomal rearrangement. Assuming selection acts generally to streamline genomes, UIR would be pruned where possible and would therefore vary across lineages, according to function and the efficacy of selection. Large UIR maintenance would tend to decrease with increasing evolutionary distance, relative to other quintiles near the minimal limit of regulatory control. However, the proportion of maintained large UIR genes increases, rather than decreases, with evolutionary distance (fig. 2B). Furthermore, although a clear relationship between the size of UIR and local conservation of orthologous gene order is apparent genome-wide, in that evolutionary instability is associated with larger UIR, this trend is weak (large UIR) or reversed entirely (small UIR) for genes with maintained UIR size (fig. 2C). In all, this suggests that some large UIR may be under selection, against a general inverse association between the conservation of gene order and gene density.
Fig. 2.—

Extremes of UIR size are maintained through evolution. (A) Bar plots of the number of orthologous gene clusters with UIR size restricted to quintiles (relative to the distribution for each species), across narrowing taxonomic ranks from subdivision Peziomycotina (all species excluding Rhizopus oryzae and the yeasts Saccharomyces cerevisiae and Candida albicans) to genus Aspergillus. Distributions largely reflect quintile ranges. (B) The same plots normalized to the standard deviation of gene UIR within each quintile to account for range inequality. Although small UIR genes are clearly most conserved across all ranks by this measure, the proportion of genes with large, maintained UIR increases with evolutionary distance. (C) The relationship between UIR length and conservation of gene order (see Materials and Methods) in the Aspergilli (synteny), across maintained UIR length quintiles. Orthology score represents the proportion of orthologs with conserved gene order relative to Aspergillus nidulans over a five gene window, calculated for each ortholog. Raw data (expanded vertically for clarity) is plotted over a smoothed generalized additive model with standard error (see Materials and Methods).
Although distant orthology predictions for transcription factors, which often share homology only within a small DNA-binding domain, are problematic, we found a single gene (from a conservative set of 1,062 core orthologs) with a large UIR maintained across all 17 species. This APSES family morphogenetic transcriptional regulator, known as stunted (stuA) in the Aspergilli (Miller et al. 1991), EFG1 in C. albicans (Stoldt et al. 1997) and represented by the paralogs PHD1 and SOK2 in S. cerevisiae (Gimeno and Fink 1994), is exceptionally complex in structure and regulation by fungal standards. The stuA locus comprises two transcriptional units (stuAα and stuAβ) from four exons, a small upstream ORF associated with translational regulation of stuAα, and a large 5′-UTR (Miller et al. 1992; Dutton et al. 1997; Wu and Miller 1997). Many of these features are conserved within the Pezizomycotina. Added to this is a UIR of mean size 10,920 bp (±1,060 SE) maintained across the studied species (supplementary fig. S2, Supplementary Material online).
UIRs and UTRs Are Associated with Transcriptional Promiscuity
The size of dedicated upstream regulatory sequence is correlated with regulatory complexity in plants and metazoans (Nelson et al. 2004; Walther et al. 2007; Colinas et al. 2008; Kristiansson et al. 2009a) and this also appears to hold true for fungi. The dimorphic yeast C. albicans undergoes substantial transcriptional rewiring in switching between white and opaque cell types, controlling niche exploitation and mating (Zordan et al. 2007; Lohse and Johnson 2009; Sasse et al. 2012). Regions bound by the switching regulator Wor1 are significantly larger than expected, and associated transcripts differentially expressed between cell types have long UTRs (Tuch et al. 2010). To test the generality of these associations in fungi, we collated expression data (42 microarray and RNAseq experiments spanning 132 conditions and five Eurotiomycete species) and measured expression differences for genes in conserved UIR length quintiles. We considered significant differences in mean expression, as well as the number of genes regulated at a fixed differential expression threshold (absolute log 2 ratio = 0.5). Conserved length UIR genes were compared against control clusters with nonconserved UIR size, against data for all shared orthologs.
Genes with UIR size maintained across species in the lower quintile (hereafter referred to as small UIR genes) or upper quintile (large UIR) were differentially regulated more often and at far higher significance than those with variable length UIR, for both mean expression (Benjamini and Hochberg adjusted t test P < 0.05) and for the number of differentially expressed genes per experiment (adjusted Fisher exact test P < 0.05; fig. 3 and supplementary fig. S3, Supplementary Material online). This was most obvious for large UIR genes, which were upregulated across the diverse experimental test conditions more often than downregulated (upregulated for 57 of 132 conditions versus 42 downregulated). Orthologs with nonconserved small UIR were still significantly different for many experiments (e.g., by mean expression for 73 conditions) and hierarchical clustering of conditions showed they often responded similarly to small, conserved length UIR genes, suggesting related gene function.
Fig. 3.—
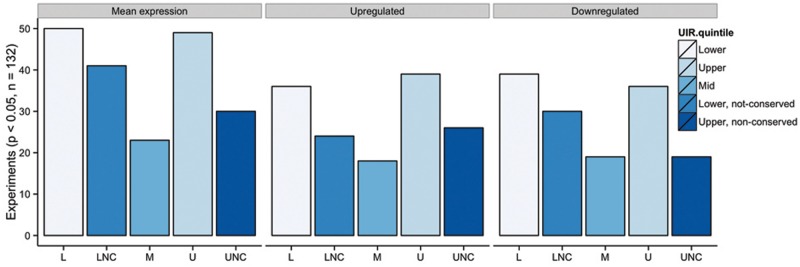
Large and small UIR genes are transcriptionally promiscuous. Histogram of genome-wide gene expression samples (42 experiments across five species, 132 samples in total) showing significantly different expression for orthologs with conserved lower (L), mid (M), and upper (U) UIR quintiles, against matched control groups with variable size across species (LNC, low not conserved with small mean size across orthologous genes; UNC, upper not conserved with high mean size). Significance is measured for mean expression (against all shared orthologs), and for the number of genes up- or downregulated at an absolute log 2 ratio of 0.5 (see Materials and Methods).
For a number of experiments, modest but highly significant correlation between gene expression and intergenic size was apparent without regard to conservation of genes or UIR size across species (e.g., maximum Spearman’s ρ for A. nidulans and A. fumigatus vegetative growth data sets of 0.33 and 0.32). Asexual development, starvation, and other stress data sets all showed correlation with UIR size; typically spanning both up- and downregulated expression (supplementary fig. S4, Supplementary Material online).
Expression promiscuity of genes with large UIR might be due in part to regulation via UTRs, because UIR was calculated based on ORFs and 5′-UTR size therefore contributes to and might correlate with UIR size. The correlation between UIR and UTR size is weak but generally significant across four Aspergilli for which genome-wide data are available (Pearson’s r = 0.15 for the 47% of genes with a predicted 5′-UTR of any length across all species, fig. 4A). Most 5′-UTRs physically represent around 25% of UIR size (median 154 bp, against a median UIR size of 627 bp), and so need not substantially influence calculation of UIR size. However, large UIR genes are approximately 1.5 times more likely than other genes to have an annotated 5′- and 3′-UTR, and the extremes of conserved UIR size positively correlate with both 3′- and, particularly, 5′-UTR size; that is, small UIR genes have small 5′-UTRs (relative to genes with small but nonconserved UIR size), whereas large genes have large 5′- and 3′-UTRs (relative to genes with large but nonconserved UIR size, fig. 4B). This suggests that for the subset of orthologs with conserved UIR and UTR, similar forces shape both intergenic and transcribed sequence.
Fig. 4.—

Large UTRs are associated with a subset of large UIR genes. (A) Relationship between 5′-UTR and UIR size for four Aspergilli based on RNAseq annotation (data from the Aspergillus Genome Database, see Materials and Methods). Correlations are weak but highly significant for Aspergillus niger and A. oryzae (P < 1e−10) for the full data set, whereas equivalent significance is only seen for A. nidulans and A. fumigatus over lower UIR ranges. (B) Mean UTR length for orthologs with conserved UIR length, relative to the equivalent quintiles for all data regardless of conservation across species. Error bars represent standard error of the mean.
It might also be predicted that genes in a divergent orientation, with independent regulatory controls, might be associated with larger UIR, and that a bias in gene orientation might therefore influence UIR calculation (Chang, Wu, et al. 2012). Gene upstream regions are indeed larger than downstream regions across the Aspergilli on average (data not shown). However, although there was a clear, general trend toward larger UIR for divergent genes within the Aspergilli (P = 4.6e−18 by two-sided t test), the reverse relationship held for genes with evolutionarily maintained large UIR (P = 1e-3, supplementary fig. S5, Supplementary Material online). A general bias in gene orientation was apparent across all Aspergillus genomes. Divergent genes are significantly more common than serially arranged genes, and this is particularly stark for small UIR genes (10:1 ratio, supplementary fig. S5, Supplementary Material online). However, the ratio between divergent and serial genes for large, maintained UIR and for nonconserved genes is very similar (1.15 and 1.19). In sum, these data suggest genes with large, maintained UIR are not subject to a bias either in physical arrangement or in increased UIR size as a function of gene arrangement.
Conserved UIR Size Is Highly Predictive of Function in Ascomycetes
The number of orthologs with conserved UIR rises steeply as genetic distance falls, but much of this may be due to chance conservation. To test this, we looked for patterns of functional enrichment in orthologous UIR quintiles, relative to nonconserved quintiles, across the narrowing taxonomic ranks of subdivision Pezizomycotina (14 species), class Eurotiomycete (12 species), order Eurotiale (8 species), and genus Aspergillus (6 species). At all levels, functional enrichments for genes of small, conserved UIR size were predominantly associated with “housekeeping” processes such as ribosomal biogenesis (fig. 5A and B), as has been observed in other organisms (Walther et al. 2007; Kristiansson et al. 2009a). Although enrichments were highly significant, genes annotated to these processes accounted for only 21% of the small UIR cluster, indicating a far more diverse membership. Overall, relative to genes with nonconserved small UIR, these genes comprised fewer introns and were approximately 1.3 times more likely to encode Pfam domains (fig. 6).
Fig. 5.—

Functional enrichments in differentially conserved small and large UIR orthologs. (A) Significantly enriched Gene Ontology (GO) terms (P < 0.01 by hypergeometric testing after Bonferroni correction) for genes with conserved UIR size (upper or lower quintile) across the taxonomic ranks of subdivision Pezizomycotina (14 species), class Eurotiomycete (12 species), order Eurotiale (8 species), and genus Aspergillus (6 species). Heatmap color scale represents both term significance (log 10 P value after subtraction of significance for nonconserved UIR quintiles) and UIR size; positive values (red) for large UIR orthologs, negative values (blue) for small UIR orthologs. Term dendrogram is based on hierarchical clustering (average linkage, Euclidean distance), selected annotations are shown at right. (B, C) Pfam protein domain co-occurrence networks for genes of small (lower quintile, B) and large (upper quintile, C) UIR size maintained across the Aspergilli, arranged by sub-network connectivity. Two nodes (domains) are connected by an edge if they occur within the same protein. Node size and colour are scaled by domain frequency within each conserved UIR group (repeats in a single protein are counted once), edge thickness is scaled by the number of co-occurrences for each domain pair within each conserved UIR group. Common motifs in small UIR genes are WD40, HAT (half a TPR), Heat, TPR_2 and Helicase_C domains, which are predominantly involved in protein–protein and protein–RNA interaction and RNA processing. Relative to large UIR orthologs, motifs are not highly connected within small UIR orthologs indicating functional stability (clustering co-efficient = 0.35, average number of neighbors = 2.12). Common motifs in large UIR genes are C2H2 and Zn2C6 zinc finger DNA-binding domains, protein kinase, LRR_1 (leucine rich repeat), WD40, PUF (Pumilio-family RNA binding repeat) and RRM_1 repeat motifs, C2 (calcium binding), and MFS and ABC transporter domains. Although the Zn2C6 zinc finger domain is the dominant DNA binding domain in fungi (found on average 4.2 times more frequently than C2H2 domains in the six Aspergilli), C2H2 domains are far more common among large UIR genes. Relative to small UIR genes, common motifs in large UIR genes are typically highly connected locally and globally (clustering coefficient = 4.27, average number of neighbors = 2.55), indicative of greater functional complexity and versatility (Yang et al. 2012).
Fig. 6.—
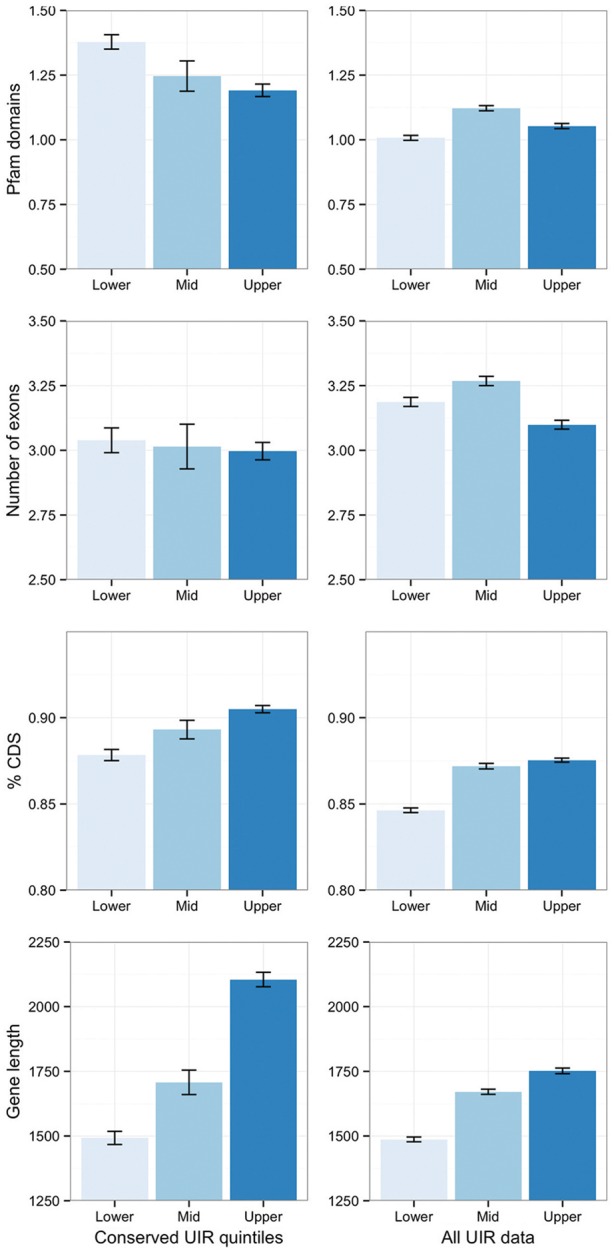
General characteristics of conserved UIR genes. Mean Pfam domains and exons per gene, % coding sequence and gene length (genomic sequence) for orthologs with small (lower quintile) and large (upper quintile) intergenic sequence for the genus Aspergillus, against the equivalent quintiles for all data regardless of conservation across species. Similar trends are seen for all other taxonomic ranks. Error bars are standard error.
Large UIR genes showed more dynamic functional enrichments over taxonomic ranks (fig. 5A), although this measurement is biased by the use of Aspergillus and S. cerevisiae-centric functional associations. These genes were also of larger size (mean ∼1.2 times genomic sequence), encoded more Pfam domains and, against the trend of expanded coding, UTR and UIR size, tended to have fewer and smaller introns than average across all taxonomic levels (fig. 6). At the most distant level, the Pezizomycotina, all functional enrichments were related to gene expression, metabolic regulation, and transcription factor activity. Within Eurotiales and the Aspergilli, the general term of biological regulation was maximally significant; encompassing transcription factor, protein kinase and G-protein coupled receptor genes (accounting for more than a third of large UIR genes, P = 2.4e−7 over genes with large, nonconserved UIR), along with filamentous growth and cell wall biogenesis activity (fig. 5A). Notably, although the Zn2C6 zinc finger domain is by far the most common DNA-binding domain in fungi (found on average 4.2 times more frequently than C2H2 domains in the six Aspergilli), C2H2 domains predominate among large UIR genes (fig. 5C). Annotated targets of regulation were asexual and, to a lesser extent, sexual development, and both primary and secondary metabolism.
In fungi, genes involved in secondary metabolite production are often clustered, allowing co-regulation by cluster-specific transcription factors and local chromatin modification (Yu and Keller 2005; Palmer and Keller 2010; Reyes-Dominguez et al. 2010; Strauss and Reyes-Dominguez 2011). If UIR reflects regulatory complexity then this economy of regulation would be expected to be associated with small UIR, and indeed we found that gene clusters required for biosynthesis of sterigmatocystin, penicillin, monodictyphenone, terrequinone, and orsellenic acid in A. nidulans are extremely compact relative to the genome as a whole (P < 0.01 for every cluster, P = 2.5e−29 for all 50 genes by two-sided t test, data not shown).
Large UIR Genes Are Enriched in Polypyrimidine Tracts and a Novel Putative Regulatory Motif with Relaxed Gene Proximity Bias
Given the high level of functional coherence in conserved UIR classes, particularly large UIR genes, we sought to identify cis-regulatory sequences shared across the Aspergilli. The presence of evolutionarily conserved motifs far upstream of ORFs would bolster the case for regulatory relevance of large UIR. We also considered general characteristics of sequence composition that might influence nucleosome positioning or other aspects of chromatin structure.
Putative transcription factor binding sites conserved across the Aspergilli were detected by MEME analysis for most large UIR orthologous genes (Bailey et al. 2009), including some of the few experimentally defined Aspergillus transcription factor sites such as HGATAR (AreA binding) and CCAAT (HapB/C/D binding) boxes, and many short CGG/CCG-based motifs that are common targets of fungal Zn(II)2Cys6 cluster transcription factors. Overwhelmingly, however, large tracts of repetitive sequence were identified as the most significantly enriched “motifs” upstream of genes with large, conserved UIR size. Strong composition, strand and gene proximity biases were evident: most common were gene proximal CT-rich tracts on the plus strand (PPPTs, for plus-strand polypyrimidine tracts), although strand bias decayed with distance so that AG tracts were also common further upstream (fig. 7A and B). AT tracts, known to be involved in nucleosome positioning and DNA replication initiation in other fungi (Ioshikhes et al. 2006), were also present at lower frequency; however, these were not enriched relative to the control population of large, nonconserved UIR genes.
Fig. 7.—

Sequence composition biases and enriched motifs in conserved, large UIR. (A) Counts of low complexity sequence tracts within conserved or nonconserved large UIR in the Aspergilli (sliding window of 30 bp, step size 15 bp, with two-base composition >0.75, counts binned by 50 bp). CT and, to a lesser extent, AG-rich tracts are enriched relative to the matched control population of large mean, variable UIR size orthologs. (B) Plots of repetitive sequence tract location (kb upstream of the translation start site) for conserved large UIR orthologs (upper) and nonconserved large UIR orthologs (lower). Polypyrimidine tracts are overrepresented on the plus strand within the first 2 kb of upstream sequence. (C) Mean orthology score (proportion of orthologs across Aspergilli relative to Aspergillus nidulans within a 5 gene window) upstream or downstream of genes with maintained UIR size, (lower, mid, and upper quintile) or for the remainder of orthologs with nonconserved UIR size. Stability of gene order is generally higher upstream than downstream of genes, but large UIR is associated with reduced stability. (D) Motifs detected as significantly enriched within the first 2 kb upstream of genes with conserved, large UIR in the Aspergilli are predominately low-complexity sequence, with the exception of the consensus motif TACTCYGTA/TACRGAGTA.
Although gene-proximally enriched, PPPTs were also distributed throughout extended upstream sequence in many orthologs (supplementary fig. S6, Supplementary Material online), although positional conservation generally fell with increasing distance. Notably, of the top 35 large UIR orthologs ranked by extended conservation in polypyrimidine tract positioning, 27 are regulatory factors (15 transcription, 8 signal transduction, and 4 translation regulators). All characterized proteins play pivotal roles in either metabolic regulation—including carbon catabolite repression (C2H2 transcription factor CreA and ubiquitin hydrolase CreB), acetyl-CoA metabolism (synthase FacA and carboxylase AccA), fatty acid utilization (Zn(II)2Cys6 transcription factor FarB), and nitrogen metabolism (glutamate synthase GltA)—or development, such as C2H2 transcription factors BrlA, MsnA, and SteA, and the bHLH transcription factor DevR. However, most genes are uncharacterized (outside of S. cerevisiae or S. pombe, in cases where clear homologs exist), and are of interest by association. In particular, we note the presence of the ortholog of S. cerevisiae SWI1, an ARID/BRIGHT domain DNA binding component of the nucleosome remodeling SWI/SNF complex, and a HMG box protein (AN3667), both of which are themselves predicted to bind low-complexity sequence. Also of interest, given the long-suspected role for translational regulation of development in Aspergilli (Han et al. 1993; Timberlake 1993; Marhoul and Adams 1996; Navarro and Aguirre 1998; Scherer et al. 2002; Galagan, Calvo, et al. 2005; Busch and Braus 2007; Nowrousian et al. 2007), are multiple genes for predicted RNA-binding proteins AN1288 (La domain), AN8038 (RRM, R3H, SUZ-C domains), AN1158 (ortholog of S. cerevisiae repressor SSD1), and AN7480 (multi-RRM domain ortholog of S. pombe negative regulator of differentiation, nrd1).
Once potential cause of both UIR expansion and repetitive sequence tracts is transposable element activity; however, we identified few cases of relic elements (e.g., 15 for A. nidulans, see Materials and Methods), suggesting recent insertions are not a major driver. Recombination between large intergenic regions or replication slippage could also contribute to expansion of low complexity sequence (Bzymek and Lovett 2001; Viguera et al. 2001; Yang et al. 2008). To examine the relationship between gene rearrangement and UIR size, we calculated ortholog collinearity across the Aspergilli, considering synteny relative to gene orientation separately (upstream or downstream). Notably, although orthology upstream of genes was significantly higher than downstream, both at the genome-wide level and for smaller UIR quintiles, there was no difference for large UIR genes (fig. 7C). Recombination upstream of large UIR genes during evolution is therefore elevated, in proportion to target size, and may contribute to expansion of repetitive tracts.
Finally, while a clear majority of conserved, putative regulatory motifs detected across large UIR orthologs showed a strong gene proximity bias, suggesting distal upstream sequence may not be important for regulation by classical sequence-specific transcription factors, we found a single prominent contrary case. A novel semi-palindromic, information-rich motif, consensus TACRGAGTA/TACTCYGTA, was enriched upstream of these genes (fig. 8D), typically in multiple copies dispersed throughout the entire intergenic region so that motif count correlated with UIR size (Pearson’s r = 0.78). Mapping of the motif position weight matrix (PWM) across the genomes of three Aspergilli showed a depletion of consensus motifs within ORFs (P = 1.4e−16, χ2 test) and a 7-fold overrepresentation in intergenic regions (P = 0). Although a modest but significant gene proximity bias was evident for the 504 orthologous genes with one or more motifs, orthologs with multiple motifs (mean > 5, n = 54 orthologs) did not show this bias, suggesting distal and proximal motifs may be similarly important for these genes (supplementary fig. S4, Supplementary Material online). Known developmental regulators were significantly enriched among orthologs with conserved motifs (e.g., devR, flbA, flbC, flbD, imeB, medA, msnA, nosA, stuA, rasA, rasB, veA, and velB), other characterized genes were involved in regulation of core metabolic, mitogenic, stress response, and signal transduction pathways.
Fig. 8.—

Broadly conserved polypyrimidine tracts are enriched upstream of large UIR regulatory genes. Six examples of orthologous, large UIR genes in the Aspergilli with semi-conserved repetitive sequence tracts. CT-, AT-, and AG-rich tracts were counted (sliding window of 30 bp, step size 15 bp, with a two-base composition >0.75. Points are plotted for both bases where a window passes the threshold) and plotted by location (bp upstream of the translation start site). Shown are UIR for the pivotal fungal developmental regulators C2H2 transcription factor BrlA, APSES domain transcription factor StuA and kinase ImeB, the carbon catabolite repression factor CreA (C2H2 transcription factor), and uncharacterized genes in Aspergilli AN6578 (ortholog of Candida albicans transcriptional regulator of white-opaque switching, WOR1 and Schizosaccharomyces pombe gti1) and AN8741 (a conserved C2H2 transcription factor with weak homology to fungal developmental regulator FlbC).
Discussion
We have extended the typical functional comparative genomic analysis approach with the inclusion of conserved genomic structure surrounding orthologous genes in fungi. The evolutionary relevance of this factor is supported by the skewed distributions of UIR size conservation, strong functional (GO) and expression biases that vary with UIR size, and a number of exaggerated gene structure and sequence characteristics. We stress, however, that the within species percentile approach to classifying UIR size means that upper quintile genes are not subject to strict maintenance of UIR size, but are simply consistently large relative to the within species distribution. Despite great variation in genome architecture, conservation of UIR size for at least one orthologous gene (Aspergillus stuA) is detectable across all studied species, maintained for approximately 600 Myr (Stajich et al. 2009). This gene is subject to exceptionally complex regulatory control, both transcriptional and posttranslational, in a number of species, and exceptionally oversized intergenic sequence is likely to be important for this (Dutton et al. 1997; Lachke et al. 2003; IpCho et al. 2010; Lysoe et al. 2011).
UIR Maintenance and Regulatory Complexity
UIR size is correlated with regulatory complexity in Neurospora crassa, C. elegans and Arabidopsis (Nelson et al. 2004; Walther et al. 2007; Colinas et al. 2008). In yeast, genes annotated to GO categories such as “cell wall” and “transport” and the common environmental response are associated with longer UIR, while the opposite relationship holds for essential genes (Zhou et al. 2008; Kristiansson et al. 2009b). In the dimorphic fungus C. albicans, intergenic regions bound by the master regulator of white-opaque switching Wor1 are 5-fold larger than average, and bound genes are associated with large, variable UTRs (including EFG1, another dimorphism regulator and ortholog of Aspergillus stuA), suggesting alternative promoter use (Zordan et al. 2007; Tuch et al. 2010).
These data have given rise to the simple theory that intergenic size directly reflects regulatory complexity (Nelson et al. 2004), that is, the number of modular or combinatorial transcription factor binding sites, at least in streamlined genomes where forces of genome expansion are counterbalanced by selection. Although transcription factor sites have not been systematically determined for fungi outside the model yeasts, it seems likely this hypothesis stands for the diverse Ascomycetes analyzed here. Although we found large UIR size to be associated with frequent transcriptional regulation across diverse conditions, this was also the case, though to a lesser extent, for small UIR genes. This may simply reflect the nature of the genes with conserved UIR size and of the expression data surveyed; ribosomal biogenesis genes are highly responsive to metabolic changes, and are strongly differentially expressed across various growth conditions and developmental stages in Aspergilli. Consistent with our expression correlation analysis, not all ribosome biogenesis genes are found within our (arbitrarily defined) small UIR quintile group across all species, suggesting differential selection for genome minimization or gene regulation across lineages. However, a general association between core metabolic activities and reduced UIR size is clear.
Extremes of UTR size were also associated with frequent transcriptional regulation, although in most cases the overlap in response between UIR and UTR size was not strong (not shown). Correlation between 5′-UTR and UIR size was generally significant across four Aspergilli with genome-wide annotations, and therefore contributes to calculation of UIR size. However, the contribution of UTR to UIR size is modest and so is unlikely to confound UIR measurement, instead 5′-UTR length may further reflect the complexity of transcriptional controls. Although the length distribution of 5′-UTRs is near phylogenetically constant, suggesting general independence with organismal complexity (Lynch et al. 2005), exceptions exist at smaller scales; both 5′-UTR and UIR size are significantly larger in the regulatory circuits of Wor1 in C. albicans and Oct4 in mouse (Tuch et al. 2010), and 5′-UTR length is associated with variable, inducible expression in S. cerevisiae and C. albicans (Lin and Li 2012).
Initiation of development may be the most complex decision in the life history of filamentous fungi (Martinelli and Clutterbuck 1971; Timberlake 1980). With limited ability to evade environmental fluctuations, metabolic economics and basic biophysical constraints determine the capacity to protect, reproduce and disseminate the genome appropriately for the conditions as some combination of multinucleate mycelium or differentiated spores. This requires precise spatial regulation of high-level metabolic processes, such as catabolite repression systems, and developmental genetic networks. Accordingly, genes with large UIR maintained across all taxonomic ranks from Pezizomycetes to the Aspergilli were strongly enriched in information processing functions, principally sequence-specific DNA binding and signal transduction, a correlate of organismal complexity (Vogel and Chothia 2006). Encoded protein domains were more evolutionarily mobile than those of small UIR genes, but showed a strong bias away from the Zn(II)2Cys6 cluster DNA-binding domain predominant in fungal transcription factors and toward the more evolutionarily conserved C2H2 domain (Weirauch and Hughes 2011). This predicts that Zn(II)2Cys6 transcription factors are more likely to be restricted in their target domains, such as secondary metabolite gene clusters or non-preferred “alternative” primary metabolic pathways associated with fungal heterotrophism.
Many well-characterized metabolic and developmental regulators have large, maintained UIR—31% of these genes in the Aspergilli, for example, have been experimentally studied in A. nidulans against 18% for all orthologs. This suggests either a characterization or mutational bias (characterized genes, most of which have been identified through mutant alleles, are significantly longer in both ORF and UIR than uncharacterized genes, data not shown), or, more likely in our view, genuine enrichment for mutant phenotypes. Characterized genes encode many more conserved Pfam domains than uncharacterized genes, and genes with maintained large UIRs (and often large UTRs) may represent a specific subset of regulators with wide domain activity. The presence of many predicted regulators with large UIR that are currently uncharacterized in filamentous fungi, either without clear orthologs in the model yeasts or with orthologs of reduced UIR, implies these genes may be of similar functional importance in these organisms. However, this hypothesis awaits comprehensive experimental validation.
In this regard, evidence that mutants for at least some characterized large UIR genes can be complemented without the entire intergenic region argues against a strict functional requirement (Boylan et al. 1987; Miller et al. 1991; Bok and Keller 2004; Katz et al. 2006; Zhao et al. 2006; Chung et al. 2011; Park et al. 2012). Even in genomes as divergent as those of S. cerevisiae and C. elegans, most transcription factor binding sites are closely gene proximal, typically within 500 bp upstream of ORFs (Harbison et al. 2004; Niu et al. 2011). However, a lack of obvious phenotypes in the laboratory does not rule out the potential for subtle quantitative differences that may be far from selectively neutral in a competitive, heterogeneous environment, a point we return to later.
Polypyrimidine Tracts Are Enriched in Large, Conserved UIR
Analysis of conserved putative functional motifs in large UIR genes maintained across the Aspergilli found significantly enriched tracts of low-complexity sequence and at least one highly conserved, information-rich motif preferentially located in large UIR regulatory genes. Although some low-complexity tracts were AT-rich, a known factor influencing intrinsic nucleosome affinity (Jiang and Pugh 2009; Tsankov et al. 2010; Jansen and Verstrepen 2011), plus strand CT-rich tracts were far more frequent, were enriched over orthologs with more variable UIR size, and in many cases were broadly conserved in position across the Aspergilli. We are not aware of any data implicating polypyrimidine sequence in nucleosome positioning, although related poly-T tracts have recently been found to demarcate gene transcription start sites in the AT-rich genome of Dictyostelium discoideum (Chang et al. 2012). Although recent transposable element insertion does not appear to be a major driver of UIR and repetitive sequence expansion, replication slippage is a potential cause and consequence (Hancock 1995).
Presumably, large UIR must in many cases accommodate noncoding sequence important for chromosome maintenance and control such as replication origins, which number in the hundreds in S. cerevisiae and S. pombe (Segurado et al. 2003; Nieduszynski et al. 2006; Feng et al. 2007). Eukaryotic replication initiation origin sequences are best understood in S. cerevisiae and its close relatives, but recent comparative study with S. pombe and D. melanogaster suggests primary sequence determinants, size and position are highly plastic through evolution. In the yeasts, degenerate AT-rich sequence motifs in nucleosome depleted regions are recognized (Givens et al. 2012). In N. crassa, sequence is thought to be less predictive than the presence of chromatin remodeling proteins and activating histone modifications (Eaton et al. 2011), though a study using different methods found replication origins in both mouse and N. crassa are significantly associated with G- and GC-rich sequence upstream of actively transcribed genes (Cayrou et al. 2011). As for replication origins, matrix-scaffold attachments regions (MARs) required for attachment of chromatin to the nuclear matrix are also found in intergenic regions (Glazko et al. 2003). Among a number of features associated with MARs are polypurine/polypyrimidine tracts, which are capable of forming triple-helical structures (hydrogen bond stabilized H-DNA) in a pH dependent manner (Mirkin and Frank-Kamenetskii 1994; Mariappan et al. 1999; Glazko et al. 2003; Buske et al. 2011).
Such features of chromosome and genome maintenance might influence gene expression dynamics, though it is not obvious why pronounced yet broad functional biases in associated genes would be observed. An alternative theory is that both the size and sequence composition of UIR may subtly influence regulatory kinetics of genes in a manner that is relatively inconsequential in the laboratory but still evolutionarily relevant. One such factor may be transcription factor search time, which is dependent on target site accessibility and therefore on factors such as DNA sequence, chromatin structure and localization (Halford and Marko 2004; Wunderlich and Mirny 2008; Lomholt et al. 2009).
A Novel Motif Conserved among Pezizomycotina May Coregulate Key Developmental and Metabolic Genes
The single information-rich motif (consensus TACRGAGTA) enriched upstream of large UIR genes shows only a weak gene proximity bias, and none at all in multiple motif genes. This suggests that, at least in some cases, distant intergenic motifs have been maintained and may therefore be important in gene regulation. Either TACRGAGTA motif genes are coregulated in a classical transcription factor network or their downstream regulation by other specific factors is enabled generally by a TACRGAGTA-binding factor, for example, through chromatin accessibility, in a manner similar to “pioneer” factors that establish transcriptional competence (Harrison et al. 2011; Garber et al. 2012; Maston et al. 2012). Although we did see significantly correlated differential expression for these genes across a number of conditions, given correlation between large UIR, the presence of TACRGAGTA motifs and a functional bias toward developmental and other regulators, it is impossible to disentangle these factors without targeted experimental manipulation. We note that intergenic motif enrichment was significant well beyond the Aspergilli (more so than conservation upstream of orthologous genes; consensus motifs were found upstream of only 26 orthologs across the Pezizomycotina), suggesting strong conservation of the putative binding factor but relaxed selection on motif location over the several hundred million years since divergence (Taylor and Berbee 2006; Li and Johnson 2010).
In summary, we have found that conserved local architecture of genetic loci is a surprisingly accurate predictor of gene function in diverse fungi. The ability to detect this relationship may be due in part to the relatively streamlined genomes of such species. Although large UIR is not strictly required under laboratory conditions for many genes studied experimentally, unique structural and sequence characteristics, and strong biases in expression and function suggest clear evolutionary relevance. With genome-wide gene characterization projects underway or in sight for a number of filamentous fungi, it will be interesting to test this estimate of regulatory complexity against the revealed topologies of genetic networks.
Supplementary Material
Supplementary figures S1–S6 are available at Genome Biology and Evolution online (http://www.gbe.oxfordjournals.org/).
Acknowledgments
This work was supported by Australian Research Council and National Health and Medical Research Council grants to A.A. and an Australian Postgraduate Award to L.M.N. The authors thank AspGD, JGI, JCVI, and Broad Institute for provision of data, and also thank Tom Harrop and reviewers for helpful comments on the manuscript.
Literature Cited
- Altschul SF, et al. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 1997;25:3389–3402. doi: 10.1093/nar/25.17.3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arnaud MB, et al. The Aspergillus genome database, a curated comparative genomics resource for gene, protein and sequence information for the Aspergillus research community. Nucleic Acids Res. 2010;38:D420–D427. doi: 10.1093/nar/gkp751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bailey TL, et al. MEME SUITE: tools for motif discovery and searching. Nucleic Acids Res. 2009;37:W202–W208. doi: 10.1093/nar/gkp335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basu MK, Carmel L, Rogozin IB, Koonin EV. Evolution of protein domain promiscuity in eukaryotes. Genome Res. 2008;18:449–461. doi: 10.1101/gr.6943508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bok JW, Keller NP. LaeA, a regulator of secondary metabolism in Aspergillus spp. Eukaryot Cell. 2004;3:527–535. doi: 10.1128/EC.3.2.527-535.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boylan MT, Mirabito PM, Willett CE, Zimmerman CR, Timberlake WE. Isolation and physical characterization of three essential conidiation genes from Aspergillus nidulans. Mol Cell Biol. 1987;7:3113–3118. doi: 10.1128/mcb.7.9.3113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boyle EI, et al. GO::TermFinder—open source software for accessing Gene Ontology information and finding significantly enriched Gene Ontology terms associated with a list of genes. Bioinformatics. 2004;20:3710–3715. doi: 10.1093/bioinformatics/bth456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Busch S, Braus GH. How to build a fungal fruit body: from uniform cells to specialized tissue. Mol Microbiol. 2007;64:873–876. doi: 10.1111/j.1365-2958.2007.05711.x. [DOI] [PubMed] [Google Scholar]
- Buske FA, Mattick JS, Bailey TL. Potential in vivo roles of nucleic acid triple-helices. RNA Biol. 2011;8:427–439. doi: 10.4161/rna.8.3.14999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bzymek M, Lovett ST. Instability of repetitive DNA sequences: the role of replication in multiple mechanisms. Proc Natl Acad Sci U S A. 2001;98:8319–8325. doi: 10.1073/pnas.111008398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cayrou C, et al. Genome-scale analysis of metazoan replication origins reveals their organization in specific but flexible sites defined by conserved features. Genome Res. 2011;21:1438–1449. doi: 10.1101/gr.121830.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang DT, Wu CY, Fan CY. A study on promoter characteristics of head-to-head genes in Saccharomyces cerevisiae. BMC Genomics. 2012;13(1 Suppl):S11. doi: 10.1186/1471-2164-13-S1-S11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang GS, et al. Unusual combinatorial involvement of poly-A/T tracts in organizing genes and chromatin in Dictyostelium. Genome Res. 2012;22:1098–1106. doi: 10.1101/gr.131649.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung D-W, et al. Acon-3, the Neurospora crassa ortholog of the developmental modifier, medA, complements the conidiation defect of the Aspergillus nidulans mutant. Fungal Genet Biol. 2011;48:370–376. doi: 10.1016/j.fgb.2010.12.008. [DOI] [PubMed] [Google Scholar]
- Colinas J, Schmidler SC, Bohrer G, Iordanov B, Benfey PN. Intergenic and genic sequence lengths have opposite relationships with respect to gene expression. PLoS One. 2008;3:e3670. doi: 10.1371/journal.pone.0003670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cornell MJ, et al. Comparative genome analysis across a kingdom of eukaryotic organisms: specialization and diversification in the fungi. Genome Res. 2007;17:1809–1822. doi: 10.1101/gr.6531807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dutton JR, Johns S, Miller BL. StuAp is a sequence-specific transcription factor that regulates developmental complexity in Aspergillus nidulans. EMBO J. 1997;16:5710–5721. doi: 10.1093/emboj/16.18.5710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eaton ML, et al. Chromatin signatures of the Neurospora crassa replication program. Genome Res. 2011;21:164–174. doi: 10.1101/gr.116038.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng W, Raghuraman MK, Brewer BJ. Mapping yeast origins of replication via single-stranded DNA detection. Methods. 2007;41:151–157. doi: 10.1016/j.ymeth.2006.07.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galagan JE, et al. Sequencing of Aspergillus nidulans and comparative analysis with A. fumigatus and A. oryzae. Nature. 2005;438:1105–1115. doi: 10.1038/nature04341. [DOI] [PubMed] [Google Scholar]
- Galagan JE, Henn MR, Ma LJ, Cuomo CA, Birren B. Genomics of the fungal kingdom: insights into eukaryotic biology. Genome Res. 2005;15:1620–1631. doi: 10.1101/gr.3767105. [DOI] [PubMed] [Google Scholar]
- Garber M, et al. A high-throughput chromatin immunoprecipitation approach reveals principles of dynamic gene regulation in mammals. Mol Cell. 2012;47:810–822. doi: 10.1016/j.molcel.2012.07.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gimeno CJ, Fink GR. Induction of pseudohyphal growth by overexpression of PHD1, a Saccharomyces cerevisiae gene related to transcriptional regulators of fungal development. Mol Cell Biol. 1994;14:2100–2112. doi: 10.1128/mcb.14.3.2100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Givens RM, et al. Chromatin architectures at fission yeast transcriptional promoters and replication origins. Nucleic Acids Res. 2012;40:7176–7189. doi: 10.1093/nar/gks351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glazko GV, Koonin EV, Rogozin IB, Shabalina SA. A significant fraction of conserved noncoding DNA in human and mouse consists of predicted matrix attachment regions. Trends Genet. 2003;19:119–124. doi: 10.1016/S0168-9525(03)00016-7. [DOI] [PubMed] [Google Scholar]
- Halford SE, Marko JF. How do site-specific DNA-binding proteins find their targets? Nucleic Acids Res. 2004;32:3040–3052. doi: 10.1093/nar/gkh624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han S, Navarro J, Greve RA, Adams TH. Translational repression of brlA expression prevents premature development in Aspergillus. EMBO J. 1993;12:2449–2457. doi: 10.1002/j.1460-2075.1993.tb05899.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hancock JM. The contribution of slippage-like processes to genome evolution. J Mol Evol. 1995;41:1038–1047. doi: 10.1007/BF00173185. [DOI] [PubMed] [Google Scholar]
- Harbison CT, et al. Transcriptional regulatory code of a eukaryotic genome. Nature. 2004;431:99–104. doi: 10.1038/nature02800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harrison MM, Li XY, Kaplan T, Botchan MR, Eisen MB. Zelda binding in the early Neurospora crassa melanogaster embryo marks regions subsequently activated at the maternal-to-zygotic transition. PLoS Genet. 2011;7:e1002266. doi: 10.1371/journal.pgen.1002266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ioshikhes IP, Albert I, Zanton SJ, Pugh BF. Nucleosome positions predicted through comparative genomics. Nat Genet. 2006;38:1210–1215. doi: 10.1038/ng1878. [DOI] [PubMed] [Google Scholar]
- IpCho SV, et al. The transcription factor StuA regulates central carbon metabolism, mycotoxin production, and effector gene expression in the wheat pathogen Stagonospora nodorum. Eukaryot Cell. 2010;9:1100–1108. doi: 10.1128/EC.00064-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jansen A, Verstrepen KJ. Nucleosome positioning in Saccharomyces cerevisiae. Microbiol Mol Biol Rev. 2011;75:301–320. doi: 10.1128/MMBR.00046-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang C, Pugh BF. Nucleosome positioning and gene regulation: advances through genomics. Nat Rev Genet. 2009;10:161. doi: 10.1038/nrg2522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jurka J. Repbase update: a database and an electronic journal of repetitive elements. Trends Genet. 2000;16:418–420. doi: 10.1016/s0168-9525(00)02093-x. [DOI] [PubMed] [Google Scholar]
- Katz M, Gray K, Cheetham B. The Aspergillus nidulans xprG (phoG) gene encodes a putative transcriptional activator involved in the response to nutrient limitation. Fungal Genet Biol. 2006;43:190–199. doi: 10.1016/j.fgb.2005.12.001. [DOI] [PubMed] [Google Scholar]
- Kelkar YD, Ochman H. Causes and consequences of genome expansion in fungi. Genome Biol Evol. 2012;4:13–23. doi: 10.1093/gbe/evr124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kristiansson E, Thorsen M, Tamas MJ, Nerman O. Evolutionary forces act on promoter length: identification of enriched cis-regulatory elements. Mol Biol Evol. 2009a;26:1299–1307. doi: 10.1093/molbev/msp040. [DOI] [PubMed] [Google Scholar]
- Kristiansson E, Thorsen M, Tamás MJ, Nerman O. Evolutionary forces act on promoter length: identification of enriched cis-regulatory elements. Mol Biol Evol. 2009b;26:1299–1307. doi: 10.1093/molbev/msp040. [DOI] [PubMed] [Google Scholar]
- Lachke SA, Srikantha T, Soll DR. The regulation of EFG1 in white-opaque switching in Candida albicans involves overlapping promoters. Mol Microbiol. 2003;48:523–536. doi: 10.1046/j.1365-2958.2003.t01-1-03448.x. [DOI] [PubMed] [Google Scholar]
- Lespinet O, Wolf YI, Koonin EV, Aravind L. The role of lineage-specific gene family expansion in the evolution of eukaryotes. Genome Res. 2002;12:1048–1059. doi: 10.1101/gr.174302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Johnson AD. Evolution of transcription networks—lessons from yeasts. Curr Biol. 2010;20:R746–R753. doi: 10.1016/j.cub.2010.06.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin Z, Li WH. Evolution of 5′ untranslated region length and gene expression reprogramming in yeasts. Mol Biol Evol. 2012;29:81–89. doi: 10.1093/molbev/msr143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lohse MB, Johnson AD. White-opaque switching in Candida albicans. Curr Opin Microbiol. 2009;12:650–654. doi: 10.1016/j.mib.2009.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lomholt MA, van den Broek B, Kalisch SM, Wuite GJ, Metzler R. Facilitated diffusion with DNA coiling. Proc Natl Acad Sci U S A. 2009;106:8204–8208. doi: 10.1073/pnas.0903293106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lynch M, Conery JS. The origins of genome complexity. Science. 2003;302:1401–1404. doi: 10.1126/science.1089370. [DOI] [PubMed] [Google Scholar]
- Lynch M, Scofield DG, Hong X. The evolution of transcription-initiation sites. Mol Biol Evol. 2005;22:1137–1146. doi: 10.1093/molbev/msi100. [DOI] [PubMed] [Google Scholar]
- Lysoe E, Pasquali M, Breakspear A, Kistler HC. The transcription factor FgStuAp influences spore development, pathogenicity, and secondary metabolism in Fusarium graminearum. Mol Plant Microb Interact. 2011;24:54–67. doi: 10.1094/MPMI-03-10-0075. [DOI] [PubMed] [Google Scholar]
- Marhoul JF, Adams TH. Aspergillus fabM encodes an essential product that is related to poly(A)-binding proteins and activates development when overexpressed. Genetics. 1996;144: 1463–1470. doi: 10.1093/genetics/144.4.1463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mariappan SV, Catasti P, Silks LA, 3rd, Bradbury EM, Gupta G. The high-resolution structure of the triplex formed by the GAA/TTC triplet repeat associated with Friedreich’s ataxia. J Mol Biol. 1999;285:2035–2052. doi: 10.1006/jmbi.1998.2435. [DOI] [PubMed] [Google Scholar]
- Martinelli SD, Clutterbuck AJ. A quantitative survey of conidiation mutants in Aspergillus nidulans. J Gen Microbiol. 1971;69:261–268. doi: 10.1099/00221287-69-2-261. [DOI] [PubMed] [Google Scholar]
- Maston GA, Landt SG, Snyder M, Green MR. Characterization of enhancer function from genome-wide analyses. Annu Rev Genomics Hum Genet. 2012;13:29–57. doi: 10.1146/annurev-genom-090711-163723. [DOI] [PubMed] [Google Scholar]
- Miller KY, Toennis TM, Adams TH, Miller BL. Isolation and transcriptional characterization of a morphological modifier: the Aspergillus nidulans stunted (stuA) gene. Mol Gen Genet. 1991;227:285–292. doi: 10.1007/BF00259682. [DOI] [PubMed] [Google Scholar]
- Miller KY, Wu J, Miller BL. StuA is required for cell pattern formation in Aspergillus. Genes Dev. 1992;6:1770–1782. doi: 10.1101/gad.6.9.1770. [DOI] [PubMed] [Google Scholar]
- Mirkin SM, Frank-Kamenetskii MD. H-DNA and related structures. Annu Rev Biophys Biomol Struct. 1994;23:541–576. doi: 10.1146/annurev.bb.23.060194.002545. [DOI] [PubMed] [Google Scholar]
- Navarro RE, Aguirre J. Posttranscriptional control mediates cell type-specific localization of catalase A during Aspergillus nidulans development. J Bacteriol. 1998;180:5733–5738. doi: 10.1128/jb.180.21.5733-5738.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson CE, Hersh BM, Carroll SB. The regulatory content of intergenic DNA shapes genome architecture. Genome Biol. 2004;5:R25. doi: 10.1186/gb-2004-5-4-r25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nieduszynski CA, Knox Y, Donaldson AD. Genome-wide identification of replication origins in yeast by comparative genomics. Genes Dev. 2006;20:1874–1879. doi: 10.1101/gad.385306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nilsen TW, Graveley BR. Expansion of the eukaryotic proteome by alternative splicing. Nature. 2010;463:457–463. doi: 10.1038/nature08909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niu W, et al. Diverse transcription factor binding features revealed by genome-wide ChIP-seq in C. elegans. Genome Res. 2011;21:245–254. doi: 10.1101/gr.114587.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nowrousian M, Piotrowski M, Kuck U. Multiple layers of temporal and spatial control regulate accumulation of the fruiting body-specific protein APP in Sordaria macrospora and Neurospora crassa. Fungal Genet Biol. 2007;44:602–614. doi: 10.1016/j.fgb.2006.09.009. [DOI] [PubMed] [Google Scholar]
- Oshlack A, Emslie D, Corcoran LM, Smyth GK. Normalization of boutique two-color microarrays with a high proportion of differentially expressed probes. Genome Biol. 2007;8:R2. doi: 10.1186/gb-2007-8-1-r2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ostlund G, et al. InParanoid 7: new algorithms and tools for eukaryotic orthology analysis. Nucleic Acids Res. 2010;38:D196–D203. doi: 10.1093/nar/gkp931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palmer JM, Keller NP. Secondary metabolism in fungi: does chromosomal location matter? Curr Opin Microbiol. 2010;13:431–436. doi: 10.1016/j.mib.2010.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park HS, Bayram O, Braus GH, Kim SC, Yu JH. Characterization of the velvet regulators in Aspergillus fumigatus. Mol Microbiol. 2012;86:937–953. doi: 10.1111/mmi.12032. [DOI] [PubMed] [Google Scholar]
- Raffaele S, Kamoun S. Genome evolution in filamentous plant pathogens: why bigger can be better. Nat Rev Microbiol. 2012;10:417–430. doi: 10.1038/nrmicro2790. [DOI] [PubMed] [Google Scholar]
- Reyes–Dominguez Y, et al. Heterochromatic marks are associated with the repression of secondary metabolism clusters in Aspergillus nidulans. Mol Microbiol. 2010;76:1376–1386. doi: 10.1111/j.1365-2958.2010.07051.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ritchie ME, et al. A comparison of background correction methods for two-colour microarrays. Bioinformatics. 2007;23:2700–2707. doi: 10.1093/bioinformatics/btm412. [DOI] [PubMed] [Google Scholar]
- Robinson MD, McCarthy DJ, Smyth GK. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics. 2010;26:139–140. doi: 10.1093/bioinformatics/btp616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saeed AI, et al. TM4: a free, open-source system for microarray data management and analysis. Biotechniques. 2003;34:374–378. doi: 10.2144/03342mt01. [DOI] [PubMed] [Google Scholar]
- Saeed AI, et al. TM4 microarray software suite. Methods Enzymol. 2006;411:134–193. doi: 10.1016/S0076-6879(06)11009-5. [DOI] [PubMed] [Google Scholar]
- Sasse C, Hasenberg M, Weyler M, Gunzer M, Morschhauser J. White-opaque switching of Candida albicans allows immune evasion in an environment-dependent fashion. Eukaryot Cell. 2012;12:50–58. doi: 10.1128/EC.00266-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scherer M, Wei H, Liese R, Fischer R. Aspergillus nidulans catalase-peroxidase gene (cpeA) is transcriptionally induced during sexual development through the transcription factor StuA. Eukaryot Cell. 2002;1:725–735. doi: 10.1128/EC.1.5.725-735.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Segurado M, de Luis A, Antequera F. Genome-wide distribution of DNA replication origins at A+T-rich islands in Schizosaccharomyces pombe. EMBO Rep. 2003;4:1048–1053. doi: 10.1038/sj.embor.7400008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheppard DC, et al. The Aspergillus fumigatus StuA protein governs the up-regulation of a discrete transcriptional program during the acquisition of developmental competence. Mol Biol Cell. 2005;16:5866–5879. doi: 10.1091/mbc.E05-07-0617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smoot ME, Ono K, Ruscheinski J, Wang PL, Ideker T. Cytoscape 2.8: new features for data integration and network visualization. Bioinformatics. 2011;27:431–432. doi: 10.1093/bioinformatics/btq675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stajich JE, et al. The fungi. Curr Biol. 2009;19:R840–R845. doi: 10.1016/j.cub.2009.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stoldt VR, Sonneborn A, Leuker CE, Ernst JF. Efg1p, an essential regulator of morphogenesis of the human pathogen Candida albicans, is a member of a conserved class of bHLH proteins regulating morphogenetic processes in fungi. EMBO J. 1997;16:1982–1991. doi: 10.1093/emboj/16.8.1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strauss J, Reyes-Dominguez Y. Regulation of secondary metabolism by chromatin structure and epigenetic codes. Fungal Genet Biol. 2011;48:62–69. doi: 10.1016/j.fgb.2010.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor JW, Berbee ML. Dating divergences in the fungal tree of life: review and new analyses. Mycologia. 2006;98:838–849. doi: 10.3852/mycologia.98.6.838. [DOI] [PubMed] [Google Scholar]
- Timberlake W. Translational triggering and feedback fixation in the control of fungal development. Plant Cell. 1993;5:1453–1460. doi: 10.1105/tpc.5.10.1453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Timberlake WE. Developmental gene regulation in Aspergillus nidulans. Dev Biol. 1980;78:497–510. doi: 10.1016/0012-1606(80)90349-8. [DOI] [PubMed] [Google Scholar]
- Tordai H, Nagy A, Farkas K, Banyai L, Patthy L. Modules, multidomain proteins and organismic complexity. FEBS J. 2005;272:5064–5078. doi: 10.1111/j.1742-4658.2005.04917.x. [DOI] [PubMed] [Google Scholar]
- Tsankov AM, Thompson DA, Socha A, Regev A, Rando OJ. The role of nucleosome positioning in the evolution of gene regulation. PLoS Biol. 2010;8:e1000414. doi: 10.1371/journal.pbio.1000414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tuch BB, et al. The transcriptomes of two heritable cell types illuminate the circuit governing their differentiation. PLoS Genet. 2010;6:e1001070. doi: 10.1371/journal.pgen.1001070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Twumasi-Boateng K, et al. Transcriptional profiling identifies a role for BrlA in the response to nitrogen depletion, and for StuA in the regulation of secondary metabolite clusters in Aspergillus fumigatus. Eukaryot Cell. 2008;8:104–115. doi: 10.1128/EC.00265-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Viguera E, Canceill D, Ehrlich SD. Replication slippage involves DNA polymerase pausing and dissociation. EMBO J. 2001;20:2587–2595. doi: 10.1093/emboj/20.10.2587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vogel C, Chothia C. Protein family expansions and biological complexity. PLoS Comp Biol. 2006;2:e48. doi: 10.1371/journal.pcbi.0020048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walther D, Brunnemann R, Selbig J. The regulatory code for transcriptional response diversity and its relation to genome structural properties in A. thaliana. PLoS Genet. 2007;3:e11. doi: 10.1371/journal.pgen.0030011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weirauch MT, Hughes TR. A catalogue of eukaryotic transcription factor types, their evolutionary origin, and species distribution. Subcell Biochem. 2011;52:25–73. doi: 10.1007/978-90-481-9069-0_3. [DOI] [PubMed] [Google Scholar]
- Wu J, Miller BL. Aspergillus asexual reproduction and sexual reproduction are differentially affected by transcriptional and translational mechanisms regulating stunted gene expression. Mol Cell Biol. 1997;17:6191–6201. doi: 10.1128/mcb.17.10.6191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wunderlich Z, Mirny LA. Spatial effects on the speed and reliability of protein-DNA search. Nucleic Acids Res. 2008;36:3570–3578. doi: 10.1093/nar/gkn173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang D, et al. General trends in the utilization of structural factors contributing to biological complexity. Mol Biol Evol. 2012;29:1957–1968. doi: 10.1093/molbev/mss064. [DOI] [PubMed] [Google Scholar]
- Yang S, et al. Repetitive element-mediated recombination as a mechanism for new gene origination in Neurospora crassa. PLoS Genet. 2008;4:e3. doi: 10.1371/journal.pgen.0040003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu JH, Keller N. Regulation of secondary metabolism in filamentous fungi. Annu Rev Phytopathol. 2005;43:437–458. doi: 10.1146/annurev.phyto.43.040204.140214. [DOI] [PubMed] [Google Scholar]
- Zhao W, et al. Deletion of the regulatory subunit of protein kinase A in Aspergillus fumigatus alters morphology, sensitivity to oxidative damage, and virulence. Infect Immun. 2006;74:4865–4874. doi: 10.1128/IAI.00565-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou L, Ma X, Sun F. The effects of protein interactions, gene essentiality and regulatory regions on expression variation. BMC Syst Biol. 2008;2:54. doi: 10.1186/1752-0509-2-54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zordan RE, Miller MG, Galgoczy DJ, Tuch BB, Johnson AD. Interlocking transcriptional feedback loops control white-opaque switching in Candida albicans. PLoS Biol. 2007;5:e256. doi: 10.1371/journal.pbio.0050256. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.