

Abstract
Animal development is complex yet surprisingly robust. Animals may develop alternative phenotypes conditional on environmental changes. Under unfavorable conditions, Caenorhabditis elegans larvae enter the dauer stage, a developmentally arrested, long-lived, and stress-resistant state. Dauer larvae of free-living nematodes and infective larvae of parasitic nematodes share many traits including a conserved endocrine signaling module (DA/DAF-12), which is essential for the formation of dauer and infective larvae. We speculated that conserved post-transcriptional regulatory mechanism might also be involved in executing the dauer and infective larvae fate. We used an unbiased sequencing strategy to characterize the microRNA (miRNA) gene complement in C. elegans, Pristionchus pacificus, and Strongyloides ratti. Our study raised the number of described miRNA genes to 257 for C. elegans, tripled the known gene set for P. pacificus to 362 miRNAs, and is the first to describe miRNAs in a Strongyloides parasite. Moreover, we found a limited core set of 24 conserved miRNA families in all three species. Interestingly, our estimated expression fold changes between dauer versus nondauer stages and infective larvae versus free-living stages reveal that despite the speed of miRNA gene set evolution in nematodes, homologous gene families with conserved “dauer-infective” expression signatures are present. These findings suggest that common post-transcriptional regulatory mechanisms are at work and that the same miRNA families play important roles in developmental arrest and long-term survival in free-living and parasitic nematodes.
Keywords: microRNA, nematodes, dauer larvae, post-transcriptional regulation, parasites
Introduction
Nematodes inhabit a wide range of ecological niches encompassing free-living species and obligate parasites of plants and animals. Early reports of human parasitic nematodes date back to Egypt (Poinar 2011). Intriguingly, the basic life cycle of nematodes is well conserved across nematode clades and typically involves four larval molts. The free-living nematode Caenorhabditis elegans has become the reference model to study developmental responses in the context of environmental changes. Under optimal conditions, C. elegans develops rapidly through four larval stages (L1–L4) to reproductive adults (fig. 1A; Sulston and Horvitz 1977). Under unfavorable conditions, such as starvation and crowding, worms enter an alternative third larval stage, the dauer stage. Dauer larvae are developmentally arrested, nonfeeding, stress-resistant, long-lived, and prevail in nature (Riddle and Albert 1997). Dauer larvae share many traits with infective larvae of true parasitic species: Dauer and infective larvae are the third larval stage, both have a slender appearance, a constricted esophagus, and a closed mouth. Moreover, the dauer and infective larvae fate is determined by a conserved endocrine signaling mechanism. The steroid hormone dafachronic acid (DA) interacts with the nuclear hormone receptor DAF-12 to suppress dauer formation. The DA/DAF-12 module is required for dauer formation in C. elegans and is conserved in the necromenic nematode Pristionchus pacificus and the parasitic nematode Strongyloides papillosus (Ogawa et al. 2009). Ogawa et al. showed that Δ7-DA strongly suppresses dauer formation in P. pacificus. Furthermore, they demonstrated that in the presence of Δ7-DA, the progeny of parasitic females of S. papillosus developed into free-living animals and that the formation of infective third-stage larvae (iL3s) was completely inhibited. A different study observed similar results in the human parasite S. stercoralis and in the hookworm Ancylostoma caninum (Wang and Li 2009).
Fig. 1.—

Experimental setup and computational workflow. Life cycles of Caenorhabditis elegans, Pristionchus pacificus, and Strongyloides ratti. (A) Under favorable conditions for reproduction, larvae of C. elegans and P. pacificus develop through four larval stages. Under unfavorable environmental conditions, L2 larvae enter dauer diapause. (B) Infective larvae of S. ratti develop either directly or after facultative sexual free-living adult generation. Multiplatform small RNA deep sequencing was performed on mixed and dauer stage samples of C. elegans and P. pacificus. Illumina high-throughput profiling was carried out on mixed and infective stages of S. ratti. (C) Schematic overview of in house developed miRNA prediction pipeline. (D) miRNA gene complement in all three species, including novel gene candidates (note: several genes occur multiple times in the genome).
The homologous control of dauer formation in free-living and necromenic nematodes as well as infective larvae formation in parasitic nematodes strongly supports the hypothesis that the ability to form dauer larvae represents an important step in the evolution toward parasitism (Anderson 1984; Weischer and Brown 2000; Kiontke and Sudhaus 2006; Poulin 2007; Dieterich and Sommer 2009; Ogawa et al. 2009). In particular, we are interested in the post-transcriptional regulation of the dauer/infective larval fate and in the role of microRNAs (miRNAs) in this context. Several lines of evidence suggest that post-transcriptional regulatory mechanisms dominate the transition from dauer back into the reproductive life cycle. Intriguingly, RNA polymerase II transcription appears to be reduced in dauer larvae relative to other stages based on run-on transcription assays with isolated nuclei (Dalley and Golomb 1992). Moreover, the process of dauer exit is impaired by translational repression with cycloheximide but not by the inhibition of mRNA synthesis with either amanitin or actinomycin D (Reape and Burnell 1991b, 1992), suggesting that mRNA synthesis is not necessary for dauer recovery (Reape and Burnell 1991a). Taken together, these results suggest that transcripts might be accumulated before dauer diapause or during dauer entry and that their activity is regulated during dauer and exit from dauer by post-transcriptional regulators.
In animals, miRNAs have been found to play important roles in virtually all aspects of development and differentiation (Abbott 2011; van Kouwenhove et al. 2011). miRNA precursors originate from stem loop containing primary transcripts (pri-miRNAs) and are processed into short double-stranded hairpin RNAs (pre-miRNAs) by Drosha, a RNAse III enzyme. The pre-miRNA is then cleaved by Dicer, another RNAse III enzyme, into an RNA duplex. In a complex with proteins from the Argonaut family, the 5′- or 3′-arm of the RNA duplex binds primarily to the 3′-untranslated region (3′-UTR) of partially complementary target messenger RNAs (mRNAs), causing translational repression and/or mRNA destabilization (Hammell 2008; Holtz and Pasquinelli 2009; Zhang and Su 2009; Stadler et al. 2012).
Several recent studies provide evidence that miRNA genes are involved in the regulation of lifespan as well as L1 and dauer diapause (Ibáñez-Ventoso et al. 2006; Bethke et al. 2009; Hammell et al. 2009; de Lencastre et al. 2010; Karp et al. 2011; Zhang et al. 2011; Karp and Ambros 2012). A recent study identified 17 microRNAs whose expression profiles are altered by dauer life history in comparison with continuous development (Karp et al. 2011). More specifically, it has been shown that miRNAs play critical roles in the survival and recovery from starvation-induced L1 diapause (Zhang et al. 2011) and that a feedback loop involving DAF-12 and let-7 family miRNA members coordinates cell fate decisions with starvation-induced dauer arrest (Bethke et al. 2009; Hammell et al. 2009; Karp and Ambros 2012).
Within this article, we compare the miRNA complement and its expression changes in dauer/infective versus mixed-stage samples of three nematodes species with three different life styles: the free-living nematode C. elegans, the necromenic nematode P. pacificus, and the true parasite S. ratti. Pristionchus species live in a nearly species-specific association with beetles and thus occupy a completely different ecological niche from that of C. elegans. Pristionchus pacificus dauer larvae and no other larval stages have been observed on beetles yet do not harm their host. Upon death of the host, dauer larvae resume development by feeding on the beetle’s carcass (Hong and Sommer 2006). Strongyloides ratti is a true parasite of the rat with a direct and an indirect life cycle (fig. 1B). It is unlikely that a species evolves directly from a fully free-living to a parasitic life style (Dieterich and Sommer 2009). We hypothesized that S. ratti still maintains the ancestral free-living life cycle along with the newly acquired parasitic life cycle.
In this study, we address the role of miRNAs in the dauer/infective larvae fate by comprehensive profiling of known and novel miRNA genes in the aforementioned three species. We sequenced small RNAs using a multiplatform sequencing approach (ABI SOLiD, Illumina GA II and HiSeq) (fig. 1A and B). We first developed a pipeline that identifies novel miRNAs from letter space (Illumina) and color space (SOLiD) data sets (fig. 1C) to identify and extend miRNA gene sets in these species. We then grouped miRNAs into families by sequence similarity and observed that miRNA gene sets diverge rapidly in nematodes. However, a small core set of conserved miRNA families exists, and some families even show conserved expression patterns. Our comparison of miRNA genes expressed in dauer and infective stages yielded candidate miRNAs that might serve as conserved post-transcriptional regulators of the dauer and infective larvae fate. Additionally, our study constitutes a valuable recourse for studying miRNA evolution in nematodes with emphasis on developmental arrested stages.
Materials and Methods
Nematode Strains and Culture
We used wild-type strains of three distinct species in all our experiments. Caenorhabditis elegans and P. pacificus were grown on nematode growth medium (NGM) plates with a lawn of Escherichia coli strain OP50 (Brenner 1974). The S. ratti animals were maintained using Wistar rats by serial passage as described previously (Viney and Lok 2007; Keiser et al. 2008). Approval was obtained from the Animal Protection Board of the City of Hamburg. The strains used in this study are as follows: C. elegans N2, P. pacificus RS2333 (formerly known as PS312), and S. ratti Basel (Swiss Tropical Institute; provided by Dr G. Pluschke).
For mixed-stage cultures of C. elegans and P. pacificus (supplementary table S1, Supplementary Material online; data sets 1, 2, 5, and 6), 10–15 early adults were spotted on NGM plates, allowed to grow at 22 °C for 5 days, and washed off with M9 for RNA extractions. Nondauer (mixed stage) and dauer samples for both species (supplementary table S1, Supplementary Material online; data sets 3, 4, 7, and 8) were obtained from liquid cultures grown at 22 °C starting with synchronized L1 larvae. Synchronized L1 larvae were sampled as follows: Gravid adult worms were treated with bleach to collect embryos (Lewis and Fleming 1995). Embryos were incubated in M9 buffer overnight at 22 °C to hatch without food, causing the larvae to arrest at the L1 stage. To obtain nondauer stages in liquid culture, we suspended 100 synchronized L1 larvae in 500 ml S-medium and added 26 nM (25S) Δ7-DA on day 4 to prevent dauer formation. We added 0.5 g OP50 per 100 ml worm culture on days 1, 4, and 7 as food source. Nondauer mixed stages were then purified on day 8 or 9 and used for RNA extraction. Dauer liquid culture was obtained from 10,000 synchronized L1 larvae suspended in 500 ml S-medium. On days 1, 5, and 8 of culture, 0.5 g OP50 per 100 ml worm culture was added. Dauer larvae were purified from liquid cultures on days 11 and 12. The culture was centrifuged to obtain a worm pellet. The dauer larvae were then collected and used for RNA extraction.
To isolate iL3s of S. ratti, we collected fecal pellets on days 6–16 after subcutaneous infection of male Wistar rats with 1,800–2,500 iL3s. Charcoal coprocultures were established, incubated at 26 °C, and assessed for vitality and sterility before further processing (Lok 2007; Soblik et al. 2011). After 5–7 days incubation time, the Baermann method was used for the recovery of iL3s. Free-living stages were prepared in a similar way by reducing the incubation time to 24 h.
Total RNA Isolation and Small RNA Library Generation
Total RNA was obtained from worm pellets using standard Trizol protocols, and quality and quantity of extracted product was assessed using Nanodrop and Bioanalyzer according to the manufacturer’s protocol.
Libraries for deep sequencing were prepared from total RNA according to the manufacturer’s protocol (small RNA Expression Kit), Applied Biosystems, Foster City, CA (SOLiD sequencing), Illumina v1.5 protocol for small RNA sequencing, and the NEXTflex small RNA sequencing kit (Bioo Scientific; multiplexed libraries).
Small RNA Sequencing and Read Preprocessing
We sequenced 10 small RNA libraries on Illumina and Applied Bioscience SOLiD sequencers (see supplementary table S1, Supplementary Material online, for details). Short read processing, mapping, and miRNA gene inference were implemented in a custom bioinformatics pipeline (fig. 1C; supplementary methods, Supplementary Material online). Our pipeline involves the following steps: read quality filtering, error correction, barcode detection, adapter removal, and mapping to target genome sequences. Reads with less than 18 nt were discarded from further analysis.
miRNA Identification
We used the miRDeep2 software (Friedländer et al. 2012) to infer novel miRNA gene candidates. MiRDeep2 uses the Bowtie read mapper internally to map letter space data (Illumina) to genome sequences (Langmead et al. 2009). To predict novel miRNA genes from color space data (SOLiD), we modified the miRDeep2 pipeline and utilized the read mapper SHRiMP 2.1.1 (David et al. 2011). Illumina reads that matched with at least 18 nt to the genome and SOLiD reads with at least 16 color space matches were retained. We filtered our initial list of all candidate miRNA loci (miRDeep2 score with signal-to-noise ratio ≥10 (except data set 6: signal-to-noise ratio = 9.4) against a database of other small noncoding RNAs, including rRNAs, tRNAs, snoRNAs, snRNAs, 21U-RNAs, sbRNAs, and repeats to exclude false positives. Because of the absence of small ncRNA annotations in S. ratti, the initial miRNA candidate list was filtered against ribosomal RNAs and compared with the Rfam database (version 11; Burge et al. 2013). The remaining set of candidate miRNAs were manually inspected and curated to yield the final set of novel miRNA loci. miRNA genes were grouped into five different age classes based on miRBase v.18 (Kozomara and Griffiths-Jones 2011; supplementary methods, Supplementary Material online).
Inference of miRNA Families and Phylogenetic Trees
To identify miRNA families, we computed all-against-all pairwise sequence alignments of miRNA 5′- and 3′-arms using USEARCH 6.0.307 (Edgar 2010). We built an undirected graph of pairwise sequence similarities to group miRNA arms into families (supplementary methods, Supplementary Material online).
For each individual miRNA family with at least two precursors, we computed a multiple sequence alignment (MSA) using LocARNA 1.6.1 (Will et al. 2007). All alignments were constrained to align at the seed sequence (positions 2–8) of each miRNA.
We built simple phylogenetic trees for each gene group with the UPGMA method using the distance matrix information from LocARNA. We used custom R scripts plus two R packages (R4RNA 0.1.4 and Phangorn 1.6.0) and the RNAalifold webserver (http://rna.tbi.univie.ac.at/cgi-bin/RNAalifold.cgi, last accessed June 18, 2013) (Bernhart et al. 2008; Schliep 2011; Lai et al. 2012) to visualize and annotate MSAs (supplementary methods, Supplementary Material online).
Quantification of miRNA Expression
Preprocessed short reads from two biological conditions (dauer/infective larvae vs. mixed-stage worms) were mapped to the respective genome sequences with SHRiMP 2.1.1 (David et al. 2011; supplementary methods, Supplementary Material online). We counted the number of reads mapping to each miRNA precursor by summing up the read counts of the 5′- and 3′-arm. Reads that mapped to less than five loci were excluded from further analysis, and reads that mapped to multiple loci were added to the count of all target loci.
Raw read counts were then normalized by reference-based quantile normalization (Schulte et al. 2010). We computed log2 fold changes from normalized read counts between mixed-stage and dauer/iL3 samples. We defined the final set of differentially expressed genes by two criteria: 1) absolute log2 fold change >1 and 2) exact two-sided binomial test with a P-value cutoff that corresponds to a false discovery rate (FDR) of less than 5%.
We combined our results from the differential expression analysis with phylogenetic information from sequence alignments. Heatmaps are used to represent miRNA gene expression by color where heatmap rows are ordered by the inferred phylogeny (R packages ape 3.0.4 [Paradis et al. 2004] and NeatMap 0.3.5 [Rajaram and Oono 2010]).
Comparison of Our C. elegans miRNA Expression Data to Published Quantitative Reverse Transcriptase-Polymerase Chain Reaction Data
We compared our expression data to published data obtained by TaqMan miRNA quantitative PCR (qRT-PCR) assays (Karp et al. 2011). Accordingly, we compared small RNA-seq log2 fold changes of dauer versus mixed stage in C. elegans with −ΔΔCT values calculated from dauer versus L2m (late L2 – mid L3 stage) samples by assigning genes to expression classes with the following procedure. We grouped all miRNA genes that were present in both data sets into a 3 × 3 contingency table having three expression categories: 1) upregulated, 2) downregulated, and 3) unaffected (supplementary methods, Supplementary Material online). We tested for statistical independence of the two data sets by performing a χ2 test.
Single-Mutation Seed Network
We inferred a single-mutation seed network from all seed sequences from the miRNA sets in our three species. For this, we built an undirected graph with nodes representing seeds. Seed sequences that differ in one nucleotide were connected by edges (R package igraph 0.6.2 [Csardi and Nepusz 2006]). We visualized this network using Cytoscape 2.8.2 (Shannon et al. 2003).
Results
Sequencing of Small RNAs from Three Nematodes
We generated small RNA deep sequencing libraries from dauer and mixed-stage samples of C. elegans and P. pacificus and infective L3s and mixed-stage samples of S. ratti (fig. 1A and B). We obtained more than 196 million sequencing reads for 10 small RNA libraries. After preprocessing, approximately 120 million small RNAs (≥18 nt) mapped to their respective genomes (supplementary table S1, Supplementary Material online).
Our multiplatform small RNA-seq approach highly enriches for miRNAs relative to other types of small RNAs. For example, 88% of reads obtained by SOLiD sequencing (data set 1 in supplementary table S1, Supplementary Material online) and 79% of reads obtained by Illumina sequencing (data set 2 in supplementary table S1, Supplementary Material online) mapped to mature miRNAs in C. elegans (miRBase v.18). In total, we could identify 193 of 223 (87%) previously annotated C. elegans miRNAs (data set 1). In the remainder of reads, we detected sense and antisense hits to other small noncoding RNAs, including 21U-RNAs (the so-called “pi-RNAs” in C. elegans) and protein-coding regions, 5′-UTRs, and 3′-UTRs (supplementary table S1, Supplementary Material online). In P. pacificus, we were able to detect 123 of 124 previously annotated miRNA genes (99%) in our small RNA data sets. No miRNA genes have been annotated for S. ratti at the time of writing of this article. On the basis of the apparent quality of our data, we could exploit our high sequencing depth to extend, refine, and define the miRNA gene complements of C. elegans, P. pacificus, and S. ratti. Furthermore, we corrected or complemented sequence annotations for 5′- or 3′-arms of known miRNAs in both C. elegans and P. pacificus (a list of all miRNA sequences including sequence corrections are tabulated in supplementary table S2, Supplementary Material online).
Our multiplatform deep sequencing approach is comprehensive enough to identify tissue- and stage-specific miRNAs, such as lsy-6, a very rare miRNA, which is only expressed in less than 10 cells (Johnston and Hobert 2003) and is hardly detected by qRT-PCR (Karp et al. 2011).
Unbiased Identification of Novel miRNA Genes
We applied the miRDeep2 program (Friedländer et al. 2012) to predict novel miRNA genes. This program uses a probabilistic model to discriminate miRNA candidate loci consistent with the expected processing of miRNA precursors by Dicer from other spurious candidate loci. We used individual small RNA data sets to predict miRNA genes for C. elegans, P. pacificus, and S. ratti in developmentally arrested (data sets 4, 8, and 10) and mixed-stage (data sets 1, 2, 5, 6, and 9) samples as summarized in supplementary figure S1A, Supplementary Material online. To predict novel miRNAs based on the SOLiD data, we modified the miRDeep2 prediction pipeline as described in Materials and Methods (fig. 1C). In total, we identified 33 novel C. elegans miRNA candidates (24 in mixed stage, 8 in dauer, and 1 in both), 230 novel P. pacificus miRNAs (91 in mixed stage, 26 in dauer, and 113 in both), and 106 miRNAs in S. ratti (18 in mixed stage, 8 in iL3, and 80 in both) (supplementary table S3, Supplementary Material online).
These results augment and complement the set of annotated miRNAs in all three species (fig. 1D). Although miRNAs in C. elegans have been extensively studied with 223 annotated to date (miRBase v.18), we used our multiplatform approach to expand the set to 257 miRNA genes (one gene was duplicated on the genome). In contrast to C. elegans, only 124 miRNAs were annotated in P. pacificus (miRBase v.18) based on a Roche 454 FLX sequencing run with a low sequencing depth of approximately 160,000 reads (de Wit et al. 2009). Because P. pacificus has a significantly larger genome size compared with C. elegans and contains a higher number of protein-coding genes (Dieterich et al. 2008), we speculated that the sequencing depth to profile miRNAs was not sufficient to capture the full complement of miRNAs. Indeed, our data nearly triples the set of empirically supported miRNAs in P. pacificus, bringing the total to 362 (six genes occurred multiple times on the genome). Our data provide the first miRNA gene set annotation in S. ratti, which is the first annotation for any Strongyloides parasite. The size of the predicted miRNA gene complements correlates well with the species genome sizes (supplementary fig. S1B, Supplementary Material online). In addition, we were able to resolve the 5′- or 3′-arms of known miRNA genes in C. elegans and P. pacificus that have not been annotated so far (supplementary table S2, Supplementary Material online).
These observations, together with the fact that we found both arms of most miRNAs covered by reads (up to 95% S. ratti), suggest that these candidates are bona fide miRNA genes.
Most miRNA Genes Are Not Conserved among Distantly Related Nematodes
Our deep miRNA profiling, subsequent identification of novel miRNA candidates, and the revision of previous miRNA annotations in C. elegans, P. pacificus, and S. ratti provided the basis for a comprehensive phylogenetic investigation of miRNAs across these three nematode species. Typically, gene phylogenies are derived from sequence similarity (i.e., MSAs) and a phylogenetic reconstruction method. For miRNAs, the seed region (positions 2–8 from the 5′-end) is the major determinant of target specificity and is widely used to group miRNA genes into families (Liu et al. 2008; Krol et al. 2010). However, the seed region is too small to distinguish cases of homoplasy from cases of common decent. To pinpoint potential cases of convergent evolution, we inferred miRNA families based on sequence similarity of the full miRNA 5′- or 3′-arm and contrasted them with miRNA sets, which were solely defined by seed identity.
Every precursor of a miRNA gene has the potential to generate two distinct regulatory RNAs derived from opposite strands of the stem (fig. 2A). It is widely assumed that the more abundant sequence in small RNA sequencing data exclusively functions to suppress target transcripts (miRNA “mature” product), whereas its counterpart, the partially complementary sequence produced from the duplex stem, is nonfunctional (miRNA “star” product) (Ambros et al. 2003). Nevertheless, experiments in Drosophila melanogaster have demonstrated that miRNA star species can be loaded into RNA-induced silencing complex (RISC) and show regulatory activity (Bender 2008; Okamura et al. 2008; Stark et al. 2008; Tyler et al. 2008). Moreover, because miRNA cloning involves amplification steps, sequence-specific biases arising from small RNA library preparation and sequencing technology cannot be excluded (Hafner et al. 2011). Therefore, we reannotated all miRNA arms as “5p” or “3p” (supplementary table S2 and S3, Supplementary Material online; van Kouwenhove et al. 2011) for subsequent analyses and investigated 5p/3p read count ratios for C. elegans and P. pacificus miRNAs profiled from mixed stages by different sequencing platforms (supplementary table S4, Supplementary Material online). We observed large variations in miRNA 5′- to 3′-arm read count ratios across sequencing platforms.
Fig. 2.—

miRNA homology and seed conservation. (A) miRNA preprocessing gives rise to two potential seed sequences (modified from Friedländer et al. 2008). (B) A total of 725 precursors were stratified into 399 gene families by sequence similarity of the full miRNA arm based on the most conserved arm. If both arms of a miRNA were annotated, the arm contained in the largest group (inferred from all 1,335 miRNA 5′- and 3′-arms) was considered. If group sizes were equal, the arm with the highest degree of conservation was considered (see definition of miRNA ages in supplementary methods, Supplementary Material online, for details). In case of equal conservation level, a miRNA arm was randomly chosen. (C) A total of 725 precursors were stratified into 374 seed groups by perfect seed sequence identity (positions 2–8) considering seeds based on the most conserved miRNA arm of a precursor. The miRNA arm was selected as explained earlier.
Because of the above arguments, we initially derived miRNA conservation levels by all 1,335 miRNA 5′- and 3′-arms (corresponding to 725 precursors) based on sequence similarity of the full arm. We then inferred gene families for free-living, necromenic, and parasitic nematodes based on the most conserved arm of each precursor. In short, we retained the miRNA arm that belongs to the largest family or exhibited the highest seed conservation in the phyla Nematoda, Arthropoda, Lophotrochozoa, and Vertebrata and discarded the other arm (see supplementary methods, Supplementary Material online, for details). By this method, we grouped 725 precursors into 399 different gene families (fig. 2B; see supplementary fig. S2, Supplementary Material online, for multiple alignments of each family with at least two precursors). This analysis indicates that 63 (24.5%) precursors in C. elegans, 88 (24.3%) in P. pacificus, and 37 (34.9%) in S. ratti are conserved among all three species represented by 24 (6%) distinct families (supplementary table S5, Supplementary Material online). Moreover, 286 (39.4%) precursors were conserved between at least two species. Evidently, miRNA families could comprise multiple precursor sequences. Our analysis revealed that only a small fraction of miRNA families represent approximately one-quarter of all precursors (188/725, 25.9%). Nevertheless, the majority of miRNA genes from three distantly related nematodes are not conserved. This finding is consistent with a previous study from de Wit et al. (2009). The authors presented the first experimental study on the evolution of miRNA genes in C. elegans, C. briggsae, C. remanei, and P. pacificus and concluded that the majority of miRNAs are conserved within the Caenorhabditis genus, with the notable exception of P. pacificus miRNAs.
The seed sequence of a miRNA is the major determinant of target specificity and represents the functional entity of a miRNA. Seed sequences are short (7 nt) and identical, or almost identical seed sequences may have evolved through convergent evolution and are not conserved by descent. To investigate the impact of convergent evolution on miRNAs in our setting, we selected the most conserved arm of each miRNA as previously explained and classified these into 374 distinct groups based on perfect seed sequence identity (fig. 2C). Twenty-nine seed sequences (29/374, 7.8%) were conserved among all species representing 225 (31%) precursors. Thus, comparing the results of both classification procedures, that is, full miRNA arm identity versus seed identity, revealed that five seed sequences are shared among all species exclusively using perfect seed similarity as classification criteria. These seed groups (1, 7, 12, 16, and 18) correspond to the following C. elegans precursors: mir-34/-59/-228/-790/-791/-1820 (multiple alignments of precursors contained in seed groups are illustrated in supplementary fig. S3, Supplementary Material online). Previous studies suggested that some of these genes are involved in developmental timing, embryogenesis, gonad migration, adult viability, and DNA damage response (Kato et al. 2009; Brenner et al. 2010). Interestingly, within all these five seed groups, the location of the seed sequence alternates between 5′ and 3′. This suggests that some precursors most likely acquired a shared set of possible gene targets through convergent evolution.
miRNA Expression Changes from Sequencing Data Agree with Published qRT-PCR Results
With the updated miRNA gene set at hand, we performed a stage-wise comparison of miRNA expression levels across species to identify miRNA genes that are not only conserved in sequence but also in expression pattern. To this end, we measured expression changes of miRNAs in dauer/iL3 relative to mixed-stage samples in C. elegans, P. pacificus, and S. ratti (data sets 3, 4, and 7–10; supplementary table S1, Supplementary Material online). As a quality control, we directly compared our data to previously published miRNA expression changes in C. elegans measured by qRT-PCR (Karp et al. 2011).
To quantify miRNA expression changes, we normalized miRNA library read counts by reference-based quantile normalization (Schulte et al. 2010), where mixed-stage libraries were selected as the reference. We estimated expression changes for 177 (69%) miRNAs between normalized dauer and mixed-stage read counts in C. elegans (supplementary table S6, Supplementary Material online). As a result, we detected 71 (40%) C. elegans miRNAs that exhibited differences in expression in the developmentally arrested stage compared with mixed-stage samples. Most miRNAs were downregulated (53, or 30%), whereas 18 (10%) miRNAs showed a relative increase in dauer expression (table 1).
Table 1.
Significantly Upregulated miRNAs in Caenorhabditis elegans Dauer Larvae (FDR < 0.05)
| Rank | miRNA Gene | miRNA Family ID | Seed | Seed Conservationa | Seed Conservation Profilea | Log2 Fold Changes | Observed Function |
|---|---|---|---|---|---|---|---|
| 1 | miR-797 | miRNA family 50 | AUCACAG | mir-2/-43/-250 | +/+/+/− | 4.44 | Gonad migration (Brenner et al. 2010) |
| 2 | miR-4809 | miRNA family 37 | UAAGUUC | mir-1018/-4809/-4810 | −/−/−/− | 3.52 | — |
| 3 | miR-2210 | — | GGCAGAU | mir-72 | +/−/−/+ | 3.34 | — |
| 4 | miR-1824 | — | GGCAGUG | mir-34 | +/+/+/+ | 3.16 | DNA damage response (Kato et al. 2009) |
| 5 | miR-4807 | — | UGAGUUC | mir-983 | −/+/−/− | 2.76 | — |
| 6 | miR-248 | — | UACACGU | mir-248 | +/−/−/− | 2.75 | — |
| 7 | novel-miR-V_24974 | — | GGCUCAA | — | −/−/−/− | 2.19 | — |
| 8 | novel-miR-I_285 | — | GCGGGAC | — | −/−/−/− | 2.10 | — |
| 9 | miR-247 | miRNA family 26 | GACUAGA | mir-44/-61/-247/-279 | +/+/+/− | 1.94 | Gonad migration (Brenner et al. 2010) |
| 10 | miR-34 | miRNA family 31 | GGCAGUG | mir-34/-1824/-2227/-2239/-4933 | +/+/+/+ | 1.89 | DNA damage response (Kato et al. 2009) |
| 11 | miR-1 | miRNA family 4 | GGAAUGU | mir-1/-796 | +/+/+/+ | 1.85 | Synaptic transmission (Simon et al. 2008) |
| 12 | miR-1820 | — | UUUGAUU | mir-315 | +/+/+/+ | 1.59 | — |
| 13 | miR-791 | — | UUGGCAC | mir-791 | +/+/+/+ | 1.56 | — |
| 14 | miR-54 | miRNA family 40 | ACCCGUA | mir-51/-52/-53/-54/-55/-56/- 2233/-2237/-2271/-2274 | +/+/+/+ | 1.35 | Embryogenesis, pharynx attachment, developmental timing (Alvarez-Saavedra and Horvitz 2010; Brenner et al. 2010; Shaw et al. 2010) |
| 15 | miR-254 | — | GCAAAUC | mir-254 | +/−/−/− | 1.32 | — |
| 16 | miR-71 | miRNA family 30 | AUCACUA | mir-34/-71/-2953 | +/−/−/+ | 1.26 | Lifespan, AWC L/R neuron fate specification (de Lencastre et al. 2010; Boulias and Horvitz 2012; Hsieh et al. 2012) |
| 17 | miR-84 | miRNA family 1 | GAGGUAG | let-7, mir-48/-241/-795 | +/+/+/+ | 1.23 | Developmental timing, vulval cell fate specification (Reinhart et al. 2000; Abrahante et al. 2003; Lin et al. 2003; Abbott et al. 2005; Grosshans et al. 2005; Johnson et al. 2005) |
| 18 | miR-794 | miRNA family 1 | GAGGUAA | — | −/+/−/− | 1.14 | — |
Note.—Seed conservation column displays IDs of miRNAs that share a common seed within the phyla Nematoda. If the seed is not conserved in Nematoda, miRNA IDs from the subsequent phyla are displayed (in the order of Nematoda, Arthropoda, Lophotrochozoa, and Vertebrata). miRNA family ID is assigned if a miRNA is conserved at least once within C. elegans, P. pacificus, or S. ratti.
aNematoda/Arthropoda/Lophotrochozoa/Vertebrata.
Karp et al. monitored life-history-related expression level changes for 107 miRNAs in C. elegans using qRT-PCR. We compared our dauer versus mixed-stage expression change data with their dauer versus L2m (late L2 – mid-L3) expression changes. For a direct comparison, we discretized miRNA expression changes into three categories: 1) upregulated, 2) downregulated, and 3) unaffected. This comparison was performed for 93 miRNA genes. Thirteen miRNAs were not measured in the qRT-PCR experiment (L2m or dauer), and we did not detect miR-798 in our small RNA-seq data. Both methods indicate a good agreement of expression change classes (fig. 3A; P = 1.1 × 10−5, χ2 test). However, 34% of miRNAs were classified into different expression categories. In particular, a few individual miRNAs were downregulated in dauer in our small RNA-seq data but unaffected in the qRT-PCR data, including members of the cotranscribed mir-35-41 cluster and mir-246, which are known to be specifically enriched in C. elegans embryos (Lau et al. 2001; Brenner et al. 2010) (mir-41 was exclusively detected in our deep sequencing experiment). Such discrepancies could be explained by the specific developmental expression of these miRNAs, because we compared dauer with mixed-stage samples instead of L2m. Figure 3B depicts our small RNA-seq log2 ratios plotted against −ΔΔCT values of the qRT-PCR experiment from Karp et al. (2011). Only three miRNAs (mir-34/-71/-248) are reported as upregulated in dauer relative to L2m. We observed the same expression pattern for all these genes in our C. elegans dauer to mixed-stage comparison. Four genes, mir-230/-241/-788/-795, are consistently downregulated in both studies. Note that all miRNAs in the upper left quadrant that appear to be upregulated in dauer in the qRT-PCR experiment were classified as unaffected due to a nonsignificant t-test or inability to reproduce the results.
Fig. 3.—

Small RNA-seq expression profiles in Caenorhabditis elegans agree with qRT-PCR data. (A) Contingency table of expression fold changes of C. elegans dauer versus mixed stage obtained by Illumina small RNA deep sequencing compared with qRT-PCR data of dauer versus L2m from Karp et al. (2011) classified according to three categories (upregulated, downregulated, and unaffected). Expression fold changes of both data sets are significantly correlated (P = 1.1 × 10−5, χ2 test). (B) Quantitative comparison of expression fold changes obtained by small RNA-seq and qRT-PCR experiments in C. elegans. Names of all miRNAs with a significant expression change of at least 2-fold in both experiments are displayed. Significance of differential miRNA levels in small RNA-seq data between mixed stage and dauer/iL3 was determined by a two-sided binomial test constrained on the total library sizes followed by correction for multiple testing (FDR < 0.05).
Although Karp et al. chose a targeted approach to measure expression changes of 107 selected miRNA genes, we applied an unbiased strategy that allowed us to detect an unrestricted set of differentially expressed miRNA genes. In addition, we detected 35 differentially expressed miRNAs that were not monitored in the qRT-PCR experiment. Eight of those were upregulated in dauer, including mir-1820 and mir-1824, which seeds are identical to the highly conserved mir-315 and mir-34 family, respectively; 27 were downregulated, including lsy-6. Moreover, two of our novel miRNA candidates (cel-novel-mir-I_285 and cel-novel-mir-V_24974) were upregulated in dauer and three novel miRNAs (cel-novel-mir-IV_24134, cel-novel-mir-IV_3136, and cel-novel-mir-I_1422) downregulated.
Overall, our data are in good agreement with reported qRT-PCR fold changes from Karp et al. The observed discrepancies could be explained by: 1) differences in experimental design (dauer/mixed stages and dauer/L2m), 2) differences in assay biases (Willenbrock et al. 2009; Git et al. 2010), and 3) asynchronous sampling across experiments (Karp et al. 2011). Notably, we applied an unbiased approach and were able to detect additional miRNA gene candidates that might be involved in regulatory pathways important for developmental arrest and long-term survival.
Differential Expression Analysis Identifies Cross-Species Candidate Regulators
To begin to understand whether the set of deeply conserved miRNAs may control aspects of developmental arrest in free-living and parasitic nematodes, we examined relative expression changes of developmentally arrested stages (dauer/iL3) to mixed-stage populations in the necromenic nematode P. pacificus and the parasite S. ratti (supplementary table S6, Supplementary Material online). For 40% (71/177) of miRNA genes in C. elegans, 60% (198/331) in P. pacificus, and 35% (37/106) in S. ratti, we observed significant changes in expression levels. The majority of miRNAs that were differentially expressed in P. pacificus and S. ratti demonstrated an increase in expression in developmentally arrested stages (113/331 [34.1%] and 21/106 [19.8%], respectively). In contrast, most miRNAs in C. elegans for which we observed expression changes were downregulated in C. elegans dauer larvae (53/177, 29.9%) (18 miRNAs upregulated). Furthermore, one-quarter of P. pacificus miRNAs and 15% of S. ratti miRNAs were detected to be downregulated.
We wanted to address the long-standing hypothesis that dauer and infective larvae share a common origin. We demonstrated that 190 (26%) miRNAs are shared among C. elegans (63/257, 24.5%), P. pacificus (89/362, 24.6%), and S. ratti (37/106, 34.9%) based on sequence similarity of the miRNA 5′- or 3′-arm, respectively. If dauer and infective larvae have a common origin, we would expect to find conserved miRNAs in dauer and iL3 that show a coherent expression signature. We tested this hypothesis by constructing seed-constrained MSAs for each individual miRNA family as defined by sequence identity (supplementary fig. S2, Supplementary Material online). We used these alignments to infer phylogenetic trees and combined this phylogenetic information with our miRNA log2 expression fold changes (supplementary fig. S3, Supplementary Material online).
Overall, we discovered four miRNA gene families with homologs in all three species that show a coherent expression pattern (i.e., at least one family member from each species is differentially expressed as the majority of family members): two families are upregulated (the mir-1 and mir-71 families) and two families are downregulated (the mir-240 and mir-35 families; supplementary table S7, Supplementary Material online).
In the following, we consider mir-71 and mir-34, two miRNA candidates that were upregulated in our small RNA-seq data and also in the published qRT-PCR data. The mir-71 family is conserved across all three species and shows a coherent expression pattern, whereas the mir-34 family could represent a case of convergent evolution in P. pacificus.
The mir-71 family includes one S. ratti gene (mir-contig_74965_17996) and two genes in C. elegans (mir-71/-2953) and P. pacificus (mir-71/-Contig59.74_29947) (fig. 4A). Interestingly, the majority of miRNA genes of the mir-71 family were increased in expression in dauer and iL3. This conserved expression signature indicates the potential importance of mir-71 family members for developmentally arrested stages in free-living and parasitic nematodes.
Fig. 4.—

mir-71 and mir-34 family miRNAs as cross-species candidate regulators in developmental arrest. MSAs for two candidate miRNA families mir-71 (A) and mir-34 (B) computed by LocARNA (Will et al. 2007). Multiple alignments were constrained to align at the seed sequence position of each individual miRNA. The seed (position 2-8) of miR-71 and miR-34 is marked with a red line. Arcs above the alignment represent secondary structure information. Arc colors encode the fraction of canonical paired bases. Alignment colors are annotated according to their agreement with the predicted secondary structure. Nucleotides that are base-paired according to the structure are colored in green and unpaired bases in red. If mutations have occurred but base-pairing potential is preserved, nucleotides are displayed in blue (dark blue for mutations in both bases and light blue for single-sided mutations). Unpaired nucleotides are colored in black and gaps in gray. Heatmaps represent miRNA gene expression by color where heatmap rows are ordered by the inferred phylogeny from the alignments. Arrows next to the miRNA in the heatmap plot denote significantly up- (↑) or downregulation (↓) in dauer/iL3.
Investigating the mir-34 family revealed that this family contains one miRNA gene from C. elegans (mir-34) and three S. ratti genes that are clustered on the genome (located within 10 kb of distance on the same contig) demonstrating an expansion of the mir-34 miRNA repertoire in S. ratti (fig. 4B). Strikingly, we did not find a mir-34 precursor in P. pacificus, despite mir-34 being highly conserved from nematodes to humans. For the identified family members, we observed a conserved expression signature: All mir-34 family miRNAs are upregulated in dauer and iL3, suggesting that they may be important in developmental arrest in free-living and parasitic nematodes. It is rather unlikely that we have missed mir-34 in P. pacificus given the high sequencing depth. Therefore, we conclude that mir-34 was lost in the lineage leading to P. pacificus.
Interestingly, our seed conservation analysis detected two miRNA genes in P. pacificus (mir-2239-1/-2) that give rise to miRNA arms with seed sequences identical to the miR-34 seed “GGCAGUG.” Both miRNAs could potentially regulate similar target sets (miRNA family 58; supplementary fig. S2, Supplementary Material online). However, these miRNA genes did not show an upregulation in dauer larvae (supplementary table S6, Supplementary Material online).
Moreover, we considered additional miRNA candidates in P. pacificus that could compensate for the “loss” of mir-34. We identified likely candidates by collecting miRNA genes whose seed sequence differs by one nucleotide from the miR-34 seed “GGCAGUG” and examined their expression in dauer larvae.
P. pacificus miR-34 Seed Neighbors Are Upregulated in Dauer Larvae
To assign functional conservation by 7-nt seed sequence identity is a conservative strategy. Bartel (2009) discusses different modes of canonical target recognition: 7mer-A1 sites, 7mer-m8 sites (our seed classification), and 8mer sites. Other miRNA genes might exist that regulate similar target sets like mir-34 but have been missed by our stringent seed classification method. To overcome this problem, we examined seed changes of miRNA arms annotated in all three species. To do so in a systematic way, we generated a network in which nodes represent seed sequences. Nodes are connected if the corresponding seed sequences differ by one nucleotide. The resulting network consists of 742 nodes connected with 837 edges and 200 singletons (seeds not connected to any other seed). It contains 71 connected components with a maximum of 534 seeds (57%) in the largest component (supplementary fig. S5, Supplementary Material online).
We examined the neighborhood of the mir-34 family in the seed network (fig. 5) to identify substitutes for mir-34 with an identical expression pattern. Three of the four neighbors of the conserved miR-34 seed node (“GGCAGUG”) originate from P. pacificus miRNA genes. All those genes were upregulated in dauer. One seed neighbor (“GCCAGUG”) that did not change its expression in developmental arrest originated from a miRNA in S. ratti. However, none of the three identified P. pacificus seed neighbors bear any sequence resemblance to the mir-34 family. This finding corroborates our hypothesis of mir-34 being lost in the lineage to P. pacificus. Three miRNA genes with conserved expression yet distinct seed sequences could act compensatory in the context of dauer development.
Fig. 5.—
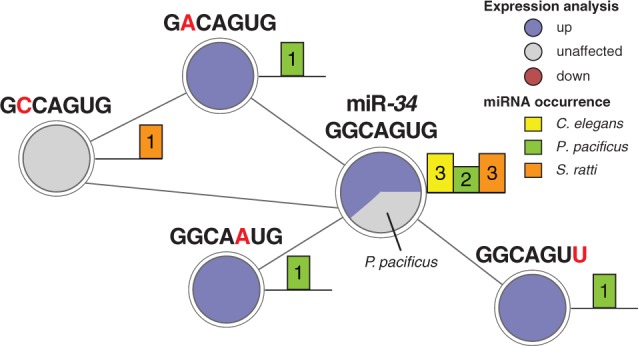
Expression conservation of miR-34 seed neighbors. The neighborhood of miRNA seeds was analyzed regarding expression changes in dauer and infective larvae. The neighborhood subnetwork of the mir-34 seed reveals conserved upregulation of all Pristionchus pacificus seed neighbors. Node color represents expression changes classified into upregulated (blue), downregulated (red), and unaffected (light gray). Barplot next to a node represents the number of times a miRNA seed was identified in a specific nematode. Note that mir-4933 of Caenorhabditis elegans was not measured in our data and is represented in the light gray pie.
Discussion
In harsh conditions, such as low food supply and stress, many nematodes are able to form dauer larvae, a developmentally arrested, stress-resistant, and long-lived state (Cassada and Russell 1975; Klass and Hirsh 1976). Infective larvae of parasitic nematodes share many morphological, behavioral, and physiological traits with dauer larvae of free-living nematodes (Blaxter and Bird 1997; Buerglin et al. 1998; Sommer and Ogawa 2011). Accordingly, dauer larvae have been suggested as an evolutionary precursor of infective larvae that facilitated the repeated evolution of parasitism (a preadaptation; Dieterich and Sommer 2009). We hypothesized that regulatory modules exist that play similar roles in regulating environmentally triggered alternative lifestyles across distantly related species in the nematode phylum. Consistent with this hypothesis, our results demonstrate that conserved “dauer-infective” miRNA expression signatures are present.
miRNA genes have been associated with signaling pathways that regulate the dauer fate (Bethke et al. 2009; Hammell et al. 2009; Karp and Ambros 2012). However, a systematic assessment of the roles of miRNA genes as post-transcriptional regulators in dauer fate decisions and their conservation in parasitic nematodes has been elusive. Our miRNA gene discovery and expression profiling in developmentally arrested stages of three representative species (C. elegans, P. pacificus, and S. ratti) reveal substantially regulation of miRNA genes in developmental arrest in free-living and parasitic nematodes. Our analyses of these data provide several implications.
First, our study of expression changes in C. elegans demonstrates that 71 miRNAs exhibit differences in expression in dauer compared with nondauer stages. A subset of 18 miRNAs is significantly upregulated in this comparison (table 1). We intersected our data with recently published qRT-PCR data (comparison of dauer and L2 larvae; Karp et al. 2011) and identified mir-34/-71/-248 to be upregulated in both data sets. So far, mir-34 and mir-71 have been assigned roles in longevity and stress response (Ibáñez-Ventoso et al. 2006; Kato et al. 2009; de Lencastre et al. 2010; Pincus et al. 2011; Zhang et al. 2011; Boulias and Horvitz 2012). Additionally, using an undirected sequencing approach allowed us to identify a number of differentially expressed miRNAs (8 up- and 27 downregulated in dauer) that were not reported in the qRT-PCR experiment.
Second, we present the first profiling and comparison of miRNA genes in nematodes from different life styles with emphasis on developmental arrested stages. We conclude that our reported miRNA gene complements in C. elegans, P. pacificus, and S. ratti are likely to be complete due to the high recovery rate of known miRNA genes in C. elegans (92%) and P. pacificus (99%) by our multiplatform sequencing strategy. Furthermore, the size of predicted miRNA gene sets correlates well with the species genome sizes (supplementary fig. S1B, Supplementary Material online). Moreover, it is frequently assumed that the mature miRNA, the sequences that is loaded into the RISC complex, is more abundant in sequencing data than the star sequence. However, we observed large variations in 5′-arm to 3′-arm read count ratios depending on the sequencing platform employed (supplementary table S4, Supplementary Material online). In essence, we see a strong platform dependency of read count patterns across miRNA arm and refrain from assigning mature and star sequences.
Finally, by examining sequence identity of miRNAs among free-living and parasitic nematodes, we identified a small core set of 24 miRNA families that are conserved among all three species. Importantly, despite rapid miRNA evolution in nematodes, homologous gene families with conserved “dauer-infective” expression signatures are present. In particular, we find two miRNA gene families with homologs in all three species that demonstrate coherent upregulation and two families with coherent downregulation in developmental arrest (supplementary table S7, Supplementary Material online). Consistent with qRT-PCR data, we detected three miRNA genes (mir-34/-71/-248) to be upregulated in C. elegans dauer. Although we did not detect any miRNA in P. pacificus or S. ratti that is homologous to mir-248, we find mir-34 to be conserved in S. ratti and mir-71 in both species. Although mir-34 is not conserved in P. pacificus, the same seed sequence (“GGCAGUG”; positions 2–8) is found in two apparently nonconserved P. pacificus miRNAs: mir-2239-1 and mir-2239-2. Both miRNAs are nondifferential in the dauer fate. A careful inspection of our single-mutation seed network uncovered expression conservation of all P. pacificus miR-34 seed neighbors (i.e., upregulation in the dauer fate; fig. 5).
Taken together, our in-depth assessment of miRNA genes in free-living and parasitic nematodes revealed conserved post-transcriptional regulators with similar expression signatures in dauer versus nondauer fates. We highlighted the case of mir-34 and mir-71, which are both important regulators of stress response and aging not only in worms but also in flies and mammals (Li et al. 2009; Kosik 2010; Leung and Sharp 2010; Liu et al. 2012). On the one hand, the mir-71 family is a well-conserved post-transcriptional regulator with coherent expression across all three species. On the other hand, the mir-34 family could constitute a case of convergent gene evolution in P. pacificus. Herein, unrelated miRNA precursors with identical or almost identical (off by one substitution) seed sequences show similar expression patterns in the dauer fate as the reference family.
In conclusion, our study provides a valuable resource to researchers for studying miRNA genes and their evolution in general and specifically aspects in developmental arrest in free-living and parasitic nematodes.
Supplementary Material
Supplementary tables S1–S7 and figures S1–S5 are available at Genome Biology and Evolution online (http://www.gbe.oxfordjournals.org/).
Acknowledgments
The authors wish to express their gratitude for numerous discussions with members of the Berlin Institute for Medical Systems Biology at the Max Delbrück Centre. They especially thank Adrian Streit (MPI for Developmental Biology) and Richard Lucius (HU Berlin) for stimulating discussions. R.A. and C.D. thank Kris Gunsalus for her help in drafting this manuscript. This work was supported by the MDC-NYU PhD exchange program, the Federal Ministry for Education and Research (BMBF), and the Senate of Berlin, Berlin, Germany, for the Berlin Institute for Medical Systems Biology at the Max Delbrück Centre (0315362A).
Literature Cited
- Abbott AL. Uncovering new functions for microRNAs in Caenorhabditis elegans. Curr Biol. 2011;21:R668–R671. doi: 10.1016/j.cub.2011.07.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abbott AL, et al. The let-7 microRNA family members mir-48, mir-84, and mir-241 function together to regulate developmental timing in Caenorhabditis elegans. Dev Cell. 2005;9:403–414. doi: 10.1016/j.devcel.2005.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abrahante JE, et al. The Caenorhabditis elegans hunchback-like gene lin-57/hbl-1 controls developmental time and is regulated by microRNAs. Dev Cell. 2003;4:625–637. doi: 10.1016/s1534-5807(03)00127-8. [DOI] [PubMed] [Google Scholar]
- Alvarez-Saavedra E, Horvitz HR. Many families of C. elegans microRNAs are not essential for development or viability. Curr Biol. 2010;20:367–373. doi: 10.1016/j.cub.2009.12.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ambros V, et al. A uniform system for microRNA annotation. RNA. 2003;9:277–279. doi: 10.1261/rna.2183803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson RC. The origins of zooparasitic nematodes. Can J Zool. 1984;62:317–328. [Google Scholar]
- Bartel DP. MicroRNAs: target recognition and regulatory functions. Cell. 2009;136:215–233. doi: 10.1016/j.cell.2009.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bender W. MicroRNAs in the Drosophila bithorax complex. Genes Dev. 2008;22:14–19. doi: 10.1101/gad.1614208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernhart SH, Hofacker IL, Will S, Gruber AR, Stadler PF. RNAalifold: improved consensus structure prediction for RNA alignments. BMC Bioinformatics. 2008;9:474. doi: 10.1186/1471-2105-9-474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bethke A, Fielenbach N, Wang Z, Mangelsdorf DJ, Antebi A. Nuclear hormone receptor regulation of microRNAs controls developmental progression. Science. 2009;324:95–98. doi: 10.1126/science.1164899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blaxter M, Bird D. Parasitic nematodes. In: Riddle DL, Blumenthal T, Meyer BJ, Priess JR, editors. C. elegans II. New York: Cold Spring Harbor Laboratory Press; 1997. pp. 851–878. [PubMed] [Google Scholar]
- Boulias K, Horvitz HR. The C. elegans microRNA mir-71 acts in neurons to promote germline-mediated longevity through regulation of DAF-16/FOXO. Cell Metab. 2012;15:439–450. doi: 10.1016/j.cmet.2012.02.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brenner JL, Jasiewicz KL, Fahley AF, Kemp BJ, Abbott AL. Loss of individual microRNAs causes mutant phenotypes in sensitized genetic backgrounds in C. elegans. Curr Biol. 2010;20:1321–1325. doi: 10.1016/j.cub.2010.05.062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brenner S. Genetics of Caenorhabditis elegans. Genetics. 1974;77:71–94. doi: 10.1093/genetics/77.1.71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buerglin TR, Lobos E, Blaxter ML. Caenorhabditis elegans as a model for parasitic nematodes. Int J Parasitol. 1998;28:395–411. doi: 10.1016/s0020-7519(97)00208-7. [DOI] [PubMed] [Google Scholar]
- Burge SW, et al. Rfam 11.0: 10 years of RNA families. Nucleic Acids Res. 2013;41:D226–D232. doi: 10.1093/nar/gks1005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cassada RC, Russell RL. The dauerlarva, a post-embryonic developmental variant of the nematode Caenorhabditis elegans. Dev Biol. 1975;46:326–342. doi: 10.1016/0012-1606(75)90109-8. [DOI] [PubMed] [Google Scholar]
- Csardi G, Nepusz T. The igraph software package for complex network research. InterJ Complex Syst. 2006;1695:1–9. [Google Scholar]
- Dalley BK, Golomb M. Gene expression in the Caenorhabditis elegans dauer larva: developmental regulation of Hsp90 and other genes. Dev Biol. 1992;151:80–90. doi: 10.1016/0012-1606(92)90215-3. [DOI] [PubMed] [Google Scholar]
- David M, Dzamba M, Lister D, Ilie L, Brudno M. SHRiMP2: sensitive yet practical SHort Read Mapping. Bioinformatics. 2011;27:1011–1012. doi: 10.1093/bioinformatics/btr046. [DOI] [PubMed] [Google Scholar]
- de Lencastre A, et al. MicroRNAs both promote and antagonize longevity in C. elegans. Curr Biol. 2010;20:2159–2168. doi: 10.1016/j.cub.2010.11.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Wit E, Linsen SEV, Cuppen E, Berezikov E. Repertoire and evolution of miRNA genes in four divergent nematode species. Genome Res. 2009;19:2064–2074. doi: 10.1101/gr.093781.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dieterich C, Sommer RJ. How to become a parasite—lessons from the genomes of nematodes. Trends Genet. 2009;25:203–209. doi: 10.1016/j.tig.2009.03.006. [DOI] [PubMed] [Google Scholar]
- Dieterich C, et al. The Pristionchus pacificus genome provides a unique perspective on nematode lifestyle and parasitism. Nat Genet. 2008;40:1193–1198. doi: 10.1038/ng.227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edgar RC. Search and clustering orders of magnitude faster than BLAST. Bioinformatics. 2010;26:2460–2461. doi: 10.1093/bioinformatics/btq461. [DOI] [PubMed] [Google Scholar]
- Friedländer MR, Mackowiak SD, Li N, Chen W, Rajewsky N. miRDeep2 accurately identifies known and hundreds of novel microRNA genes in seven animal clades. Nucleic Acids Res. 2012;40:37–52. doi: 10.1093/nar/gkr688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedländer MR, et al. Discovering microRNAs from deep sequencing data using miRDeep. Nat Biotechnol. 2008;26:407–415. doi: 10.1038/nbt1394. [DOI] [PubMed] [Google Scholar]
- Git A, et al. Systematic comparison of microarray profiling, real-time PCR, and next-generation sequencing technologies for measuring differential microRNA expression. RNA. 2010;16:991–1006. doi: 10.1261/rna.1947110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grosshans H, Johnson T, Reinert KL, Gerstein M, Slack FJ. The temporal patterning microRNA let-7 regulates several transcription factors at the larval to adult transition in C. elegans. Dev Cell. 2005;8:321–330. doi: 10.1016/j.devcel.2004.12.019. [DOI] [PubMed] [Google Scholar]
- Hafner M, et al. RNA-ligase-dependent biases in miRNA representation in deep-sequenced small RNA cDNA libraries. RNA. 2011;17:1697–1712. doi: 10.1261/rna.2799511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hammell CM. The microRNA-argonaute complex: a platform for mRNA modulation. RNA Biol. 2008;5:123–127. doi: 10.4161/rna.5.3.6570. [DOI] [PubMed] [Google Scholar]
- Hammell CM, Karp X, Ambros V. A feedback circuit involving let-7-family miRNAs and DAF-12 integrates environmental signals and developmental timing in Caenorhabditis elegans. Proc Natl Acad Sci U S A. 2009;106:18668–18673. doi: 10.1073/pnas.0908131106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holtz J, Pasquinelli AEE. Uncoupling of lin-14 mRNA and protein repression by nutrient deprivation in Caenorhabditis elegans. RNA. 2009;15:400–405. doi: 10.1261/rna.1258309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong RL, Sommer RJ. Chemoattraction in Pristionchus nematodes and implications for insect recognition. Curr Biol. 2006;16:2359–2365. doi: 10.1016/j.cub.2006.10.031. [DOI] [PubMed] [Google Scholar]
- Hsieh Y-W, Chang C, Chuang C-F. The microRNA mir-71 inhibits calcium signaling by targeting the TIR-1/Sarm1 adaptor protein to control stochastic L/R neuronal asymmetry in C. elegans. PLoS Genet. 2012;8:e1002864. doi: 10.1371/journal.pgen.1002864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ibáñez-Ventoso C, et al. Modulated microRNA expression during adult lifespan in Caenorhabditis elegans. Aging Cell. 2006;5:235–246. doi: 10.1111/j.1474-9726.2006.00210.x. [DOI] [PubMed] [Google Scholar]
- Johnson SM, et al. RAS is regulated by the let-7 microRNA family. Cell. 2005;120:635–647. doi: 10.1016/j.cell.2005.01.014. [DOI] [PubMed] [Google Scholar]
- Johnston RJ, Hobert O. A microRNA controlling left/right neuronal asymmetry in Caenorhabditis elegans. Nature. 2003;426:845–849. doi: 10.1038/nature02255. [DOI] [PubMed] [Google Scholar]
- Karp X, Ambros V. Dauer larva quiescence alters the circuitry of microRNA pathways regulating cell fate progression in C. elegans. Development. 2012;139:2177–2186. doi: 10.1242/dev.075986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karp X, Hammell M, Ow MC, Ambros V. Effect of life history on microRNA expression during C. elegans development. RNA. 2011;17:639–651. doi: 10.1261/rna.2310111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kato M, et al. The mir-34 microRNA is required for the DNA damage response in vivo in C. elegans and in vitro in human breast cancer cells. Oncogene. 2009:2419–2424. doi: 10.1038/onc.2009.106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keiser J, Thiemann K, Endriss Y, Utzinger J. Strongyloides ratti: in vitro and in vivo activity of tribendimidine. PLoS Negl Trop Dis. 2008;2:e136. doi: 10.1371/journal.pntd.0000136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiontke K, Sudhaus W. Ecology of Caenorhabditis species [Internet] 2006. WormBook, ed. The C. elegans Research Community, WormBook [cited 2013 Jun 18]. doi/10.1895/wormbook.1.37.1. Available from: http://www.wormbook.org. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klass M, Hirsh D. Non-ageing developmental variant of Caenorhabditis elegans. Nature. 1976;260:523–525. doi: 10.1038/260523a0. [DOI] [PubMed] [Google Scholar]
- Kosik KS. MicroRNAs and cellular phenotypy. Cell. 2010;143:21–26. doi: 10.1016/j.cell.2010.09.008. [DOI] [PubMed] [Google Scholar]
- Kozomara A, Griffiths-Jones S. miRBase: integrating microRNA annotation and deep sequencing data. Nucleic Acids Res. 2011;39:D152–D157. doi: 10.1093/nar/gkq1027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krol J, Loedige I, Filipowicz W. The widespread regulation of microRNA biogenesis, function and decay. Nat Rev Genet. 2010;11:597–610. doi: 10.1038/nrg2843. [DOI] [PubMed] [Google Scholar]
- Lai D, Proctor JR, Zhu JYA, Meyer IM. R-CHIE: a web server and R package for visualizing RNA secondary structures. Nucleic Acids Res. 2012;40:e95. doi: 10.1093/nar/gks241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B, Trapnell C, Pop M, Salzberg SL. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009;10:R25. doi: 10.1186/gb-2009-10-3-r25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lau NC, Lim LP, Weinstein EG, Bartel DP. An abundant class of tiny RNAs with probable regulatory roles in Caenorhabditis elegans. Science. 2001;294:858–862. doi: 10.1126/science.1065062. [DOI] [PubMed] [Google Scholar]
- Leung AKL, Sharp PA. MicroRNA functions in stress responses. Mol Cell. 2010;40:205–215. doi: 10.1016/j.molcel.2010.09.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis JA, Fleming JT. Basic culture methods. Methods Cell Biol. 1995;48:3–29. [PubMed] [Google Scholar]
- Li X, Cassidy JJ, Reinke CA, Fischboeck S, Carthew RW. A microRNA imparts robustness against environmental fluctuation during development. Cell. 2009;137:273–282. doi: 10.1016/j.cell.2009.01.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin S-Y, et al. The C. elegans hunchback homolog, hbl-1, controls temporal patterning and is a probable microRNA target. Dev Cell. 2003;4:639–650. doi: 10.1016/s1534-5807(03)00124-2. [DOI] [PubMed] [Google Scholar]
- Liu N, et al. The evolution and functional diversification of animal microRNA genes. Cell Res. 2008;18:985–996. doi: 10.1038/cr.2008.278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu N, et al. The microRNA miR-34 modulates ageing and neurodegeneration in Drosophila. Nature. 2012;482:519–523. doi: 10.1038/nature10810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lok JB. Strongyloides stercoralis: a model for translational research on parasitic nematode biology [Internet]. Wormbook. 2007. ed. The C. elegans Research Community [cited 2013 Jun 18]. doi/10.1895/wormbook.1.134.1. Available from: http://www.wormbook.org. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogawa A, Streit A, Antebi A, Sommer RJ. A conserved endocrine mechanism controls the formation of dauer and infective larvae in nematodes. Curr Biol. 2009;19:67–71. doi: 10.1016/j.cub.2008.11.063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okamura K, et al. The regulatory activity of microRNA* species has substantial influence on microRNA and 3′ UTR evolution. Nat Struct Mol Biol. 2008;15:354–363. doi: 10.1038/nsmb.1409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paradis E, Claude J, Strimmer K. APE: analyses of phylogenetics and evolution in R language. Bioinformatics. 2004;20:289–290. doi: 10.1093/bioinformatics/btg412. [DOI] [PubMed] [Google Scholar]
- Pincus Z, Smith-Vikos T, Slack FJ. MicroRNA predictors of longevity in Caenorhabditis elegans. PLoS Genet. 2011;7:e1002306. doi: 10.1371/journal.pgen.1002306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poinar GO. The evolutionary history of nematodes: as revealed in stone, amber and mummies. Leiden (The Netherlands): Brill; 2011. [Google Scholar]
- Poulin R. Evolutionary ecology of parasites. Princeton (NJ): Princeton University Press; 2007. [Google Scholar]
- Rajaram S, Oono Y. NeatMap—non-clustering heat map alternatives in R. BMC Bioinformatics. 2010;11:45. doi: 10.1186/1471-2105-11-45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reape TJ, Burnell AM. Dauer larva recovery in Caenorhabditis elegans—I. The effect of mRNA synthesis inhibitors on recovery, growth and pharyngeal pumping. Comp Biochem Physiol. 1991a;98:239–243. [Google Scholar]
- Reape TJ, Burnell AM. Dauer larva recovery in the nematode Caenorhabditis elegans—II. The effect of inhibitors of protein synthesis on recovery, growth and pharyngeal pumping. Comp Biochem Physiol. 1991b;98:245–252. [Google Scholar]
- Reape TJ, Burnell AM. Dauer larva recovery in the nematode Caenorhabditis elegans—III. The effect of inhibitors of protein and mRNA synthesis on the activity of the enzymes of intermediary. Comp Biochem Physiol B. 1992;102:241–245. [Google Scholar]
- Reinhart BJ, et al. The 21-nucleotide let-7 RNA regulates developmental timing in Caenorhabditis elegans. Nature. 2000;403:901–906. doi: 10.1038/35002607. [DOI] [PubMed] [Google Scholar]
- Riddle DL, Albert PS. Genetic and environmental regulation of Dauer larva development. In: Riddle DL, Blumenthal T, Meyer BJ, Priess JR, editors. C. elegans II. New York: Cold Spring Harbor Laboratory Press; 1997. pp. 739–768. [PubMed] [Google Scholar]
- Schliep KP. phangorn: phylogenetic analysis in R. Bioinformatics. 2011;27:592–593. doi: 10.1093/bioinformatics/btq706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schulte JH, et al. Deep sequencing reveals differential expression of microRNAs in favorable versus unfavorable neuroblastoma. Nucleic Acids Res. 2010;38:5919–5928. doi: 10.1093/nar/gkq342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shannon P, et al. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 2003;13:2498–2504. doi: 10.1101/gr.1239303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaw WR, Armisen J, Lehrbach NJ, Miska EA. The conserved miR-51 microRNA family is redundantly required for embryonic development and pharynx attachment in Caenorhabditis elegans. Genetics. 2010;185:897–905. doi: 10.1534/genetics.110.117515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simon DJ, et al. The microRNA miR-1 regulates a MEF-2-dependent retrograde signal at neuromuscular junctions. Cell. 2008;133:903–915. doi: 10.1016/j.cell.2008.04.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soblik H, et al. Life cycle stage-resolved proteomic analysis of the excretome/secretome from Strongyloides ratti—identification of stage-specific proteases. Mol Cell Proteomics. 2011;10:M111.010157. doi: 10.1074/mcp.M111.010157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sommer RJ, Ogawa A. Hormone signaling and phenotypic plasticity in nematode development and evolution. Curr Biol. 2011;21:R758–R766. doi: 10.1016/j.cub.2011.06.034. [DOI] [PubMed] [Google Scholar]
- Stadler M, Artiles K, Pak J, Fire A. Contributions of mRNA abundance, ribosome loading, and post- or peri-translational effects to temporal repression of C. elegans heterochronic miRNA targets. Genome Res. 2012;22:2418–2426. doi: 10.1101/gr.136515.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stark A, et al. A single Hox locus in Drosophila produces functional microRNAs from opposite DNA strands. Genes Dev. 2008;22:8–13. doi: 10.1101/gad.1613108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sulston JE, Horvitz HR. Post-embryonic cell lineages of the nematode, Caenorhabditis elegans. Dev Biol. 1977;56:110–156. doi: 10.1016/0012-1606(77)90158-0. [DOI] [PubMed] [Google Scholar]
- Tyler DM, et al. Functionally distinct regulatory RNAs generated by bidirectional transcription and processing of microRNA loci. Genes Dev. 2008;22:26–36. doi: 10.1101/gad.1615208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Kouwenhove M, Kedde M, Agami R. MicroRNA regulation by RNA-binding proteins and its implications for cancer. Nat Rev Cancer. 2011;11:644–656. doi: 10.1038/nrc3107. [DOI] [PubMed] [Google Scholar]
- Viney ME, Lok JB. Strongyloides spp [Internet] 2007. WormBook, ed. The C. elegans Research Community [cited 2013 Jun 18]. doi/10.1895/wormbook.1.141.1. Available from: http://www.wormbook.org. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang YP, Li KB. Correlation of expression profiles between microRNAs and mRNA targets using NCI-60 data. BMC Genomics. 2009;10:218. doi: 10.1186/1471-2164-10-218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weischer B, Brown DJF. An introduction to nematodes. Sofia, Bulgaria: Pensoft; 2000. [Google Scholar]
- Will S, Reiche K, Hofacker IL, Stadler PF, Backofen R. Inferring noncoding RNA families and classes by means of genome-scale structure-based clustering. PLoS Comput Biol. 2007;3:e65. doi: 10.1371/journal.pcbi.0030065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willenbrock H, et al. Quantitative miRNA expression analysis: comparing microarrays with next-generation sequencing. RNA. 2009;15:2028–2034. doi: 10.1261/rna.1699809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang R, Su B. Small but influential: the role of microRNAs on gene regulatory network and 3′UTR evolution. J Genet Genomics. 2009;36:1–6. doi: 10.1016/S1673-8527(09)60001-1. [DOI] [PubMed] [Google Scholar]
- Zhang X, Zabinsky R, Teng Y, Cui M, Han M. microRNAs play critical roles in the survival and recovery of Caenorhabditis elegans from starvation-induced L1 diapause. Proc Natl Acad Sci U S A. 2011;108:17997–18002. doi: 10.1073/pnas.1105982108. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.