

Abstract
This study aims to investigate the pre- and postconditioning effects of lipoxin A4 (LXA4) on myocardial damage caused by ischemia/reperfusion (I/R) injury. Seventy-two rats were divided into 6 groups: sham groups (C1 and C2), I/R groups (I/R1 and I/R2), and I/R plus LXA4 preconditioning and postconditioning groups (LX1 and LX2). The serum levels of IL-1β, IL-6, IL-8, IL-10, TNF-α, and cardiac troponin I (cTnI) were measured. The content and the activity of Na+-K+-ATPase as well as the superoxide dismutase (SOD), and malondialdehyde (MDA) levels were determined. Along with the examination of myocardium ultrastructure and ventricular arrhythmia scores (VAS), connexin 43 (Cx43) expression were also detected. Lower levels of IL-1β, IL-6, IL-8, TNF-α, cTnI, MDA content, and VAS and higher levels of IL-10, SOD activity, Na+-K+-ATPase content and activity, and Cx43 expression appeared in LX groups than I/R groups. Besides, H&E staining, TEM examination as well as analysis of gene, and protein confirmed that LXA4 preconditioning was more effective than postconditioning in preventing arrhythmogenesis via the upregulation of Cx43. That is, LXA4 postconditioning had better protective effect on Na+-K+-ATPase and myocardial ultrastructure.
1. Introduction
Myocardial ischemia/reperfusion (I/R) injury, a general health problem, is due to blood restoration after a critical period of coronary artery obstruction, which is associated with clinical interventions such as thrombolysis, angioplasty, and coronary bypass surgery. This reperfusion injury involves the activation of an inflammatory cascade and is manifested as functional impairment, arrhythmia, and accelerated progression of cell death in certain critically injured myocytes. The major mediators of reperfusion injury are oxygen radicals, calcium loading, and neutrophils [1–3]. Currently, great progress has been made in the basic research about the mechanism of myocardial I/R, but symptomatic treatment still remains as the primary therapeutic plan. With the development of molecular biology, pharmacy, and other related disciplines, it will be an important task for scientists in the future to find a safer, more effective therapeutic method.
Lipoxins (LXs) are the most recent addition to the family of bioactive products generated from arachidonic acid [4]. These molecules have both proinflammatory and anti-inflammatory actions [5] and inhibit neutrophil/granulocyte infiltration and reactive oxygen species (ROS) production [6, 7]. Lipoxins are short-lived endogenously produced nonclassic eicosanoids, whose appearance in inflammation signals the resolution of inflammation [8, 9]. Recent studies showed that LX could protect several organs such as heart, brain, lung, kidney, and stomach from I/R injury [6, 10–13]. However, there has been little research about their effect on myocardial systemic inflammation, ultrastructure changes, and arrhythmia caused by I/R injury.
Therefore, we aimed to investigate the effects of LXA4 preconditioning and postconditioning on myocardial I/R injury in rats. In particular, we studied the effects of LXA4 on oxidative stress, inflammatory reaction, Na+-K+-ATPase, and connexin 43 (Cx43) alternations caused by I/R injury.
2. Materials and Methods
2.1. Rat Model of Myocardial Ischemia/Reperfusion Injury
All animal procedures were approved by Wenzhou Medical College Animal Care and Use Committee, which is certified by the Chinese Association of Accreditation of Laboratory Animal Care (SYXK, Zhejiang 2010-0150). Sprague-Dawley (SD) male rats (eight weeks old, 200 to 250 g) were fed a standard diet and maintained in controlled environment at 25 ± 1°C under a 12 h light-dark cycle. Briefly, rats were anesthetized by an intraperitoneal injection of 10% chloral hydrate (300 mg·kg−1 body weight) and placed in a supine position. Blood was collected by femoral venipuncture. Next, the animals were intubated for artificial ventilation with 100% oxygen using a small animal breathing machine (tidal volume 5 mL, frequency 70 per min) and monitored by ECG. Thoracotomy was performed between the sternum and left costa, and then the pericardium was gently opened. Myocardial ischemia was induced by ligating the left anterior descending coronary artery (LAD) using a 3–0 silk suture with a section of PE tubing placed over the LAD and 1 mm from the tip of the normally positioned left atrium, and the coronary artery was occluded by pulling on the suture tightly 10 min later. After 30 min of myocardial ischemia, reperfusion started by releasing the ligature and removing the tube for 120 min. The chest wall was closed, the animal extubated, and body temperature maintained using a 37°C warm plate.
The indications of successful LAD occlusion included elevated ST segment (0.1 mv) or sharp rise of T wave, wider and higher QRS wave on ECG, and visual cyanosis of myocardial discoloration. The indications of successful reperfusion were ST segment depression (≥1/2) of the ECG, gradual falling of T wave, and QRS wave, and myocardial color turning to pink [14–16].
2.2. Animal Treatment and Grouping
LXA4 (C20H32O5, see Supplementary Figure 1 in the Supplementary Material available online at http://dx.doi.org/10.1155/2013/231351) was purchased from Cayman Chemical Company, Ann Arbor, USA (cat. number 90415). Seventy-two SD rats were used in the study and were randomly divided into 6 groups as follows: (1) applying LXA4 before I/R group 1 (LX1): femoral vein injection of LXA4 (100 μg/kg, dissolved in normal saline 2 mL/kg) before the I/R procedure; (2) applying LXA4 after I/R group 2 (LX2): after 30 min of myocardial ischemia, reperfusion lasted for 30 min before LXA4 (100 μg/kg, dissolved in normal saline 2 mL/kg) was injected through femoral vein. After that, reperfusion was resumed for an additional 90 min; (3) ischemia/reperfusion group 1 (I/R1): femoral vein injection of normal saline (2 mL/kg) before I/R; (4) ischemia/reperfusion group 2 (I/R2): after 30 min of myocardial ischemia, reperfusion lasted for 30 min, and then normal saline (2 mL/kg) was injected through femoral vein. After that, reperfusion was resumed for an additional 90 min; (5) sham group 1 (C1): rats underwent a similar operation without myocardial I/R, and other treatment was the same as I/R1 group; (6) sham group 2 (C2 group): rats underwent a similar operation without myocardial I/R, and other treatment was the same as I/R2 group.
2.3. Blood Collection and Tissue Harvest
Blood samples were collected in each group immediately before thoracotomy and after anaesthetization (T1) or after experiments (T2). In groups C1 and C2, T2 was obtained after 150 min of placing surgical suture under LAD. For all other groups, T2 blood samples were obtained after 120 min of reperfusion. The heart was removed after obtaining blood samples (T2), and a portion of myocardial tissue was fixed in 4% formalin. Pathological examination of paraffin-embedded sections was performed. A separate portion of myocardial tissue was frozen in liquid nitrogen and was kept in the freezer at −70°C.
2.4. Assessment of Arrhythmias
ECG was recorded throughout the experiment and at a fast speed throughout ischemia and reperfusion. Ventricular arrhythmias during ligation of LAD to reperfusion (T3) were determined in accordance with the guidelines of the Lambeth Convention [17]. Ventricular tachycardia (VT) was defined as a run of four or more consecutive ventricular premature beats. Ventricular fibrillation (VF) was defined as a ventricular rhythm without recognizable QRS complex, and the signal morphology changed from cycle to cycle. The ECG records for the incidence and total duration of VT and VF during reperfusion were analyzed. In order to evaluate the severity of arrhythmias, ventricular arrhythmia scores (VAS) were used by giving a certain grade to each rat as follows: 0 = less than 5 times VT; 1 = more than 5 times VT or other arrhythmias, no VF; 2 = 11~30 s VT or other arrhythmias, no VF; 3 = 31~90 s VT or other arrhythmias, no VF; 4 = 91~180 s VT or other arrhythmias, and/or less than 10 s reversible VF; 5 = more than 180 s VT or other arrhythmias, and/or more than 10 s reversible VF; 6 = irreversible VF [18].
2.5. Cytokine and Cardiac Troponin I (cTnI) Levels
For cytokine immunoassay, blood samples were collected by femoral venipuncture at set time points, before thoracotomy (T1) and after reperfusion (T2), and serum levels of IL-1β, IL-6, IL-8, IL-10, TNF-α, and cTnI were measured using a rat ELISA kit (Shanghai Boyun Biotech, China) in accordance with the manufacturer instructions. Cytokine levels were expressed as ng/L.
2.6. Malondialdehyde (MDA) Content Determination
MDA content was determined on frozen myocardial tissue by thiobarbituric acid assay kit (Nanjing Jiancheng Bioengineering Institute, China). The optical density at 532 nm was measured using a spectrophotometer (UV-2000, UNICO, Shanghai). 1,1,3,3-Tetramethoxypropane was used as an external standard. The level of MDA was expressed as nanomoles per milligram protein.
2.7. Superoxide Dismutase (SOD) Activity
Myocardial SOD activity was determined on frozen myocardial tissue using Xanthine Oxidase Assay Kits (Nanjing Jiancheng Bioengineering Institute, China) and expressed as units per milligram protein.
2.8. Na+-K+-ATPase Activity
The activity of Na+-K+-ATPase was assessed by measuring the release of inorganic phosphate (Pi) from ATP according to the kit protocol (Nanjing Jiancheng Bioengineering Institute, China). Briefly, frozen myocardial tissue was weighted and normal saline was added at a ratio of 1 : 9. Then, the homogenates were centrifuged to obtain the supernatant, followed by the measurement of protein concentration using Bradford method. Finally, Na+-K+-ATPase activity was determined by measuring the amount of inorganic phosphate with malachite green dye method and expressed as micromoles per milligram protein.
2.9. Histology and Immunohistochemistry
The myocardial tissue 2 mm inferior to the ligation of LAD was fixed in paraformaldehyde and embedded in paraffin. Heart tissues were cut into 5 μm sections for histopathological examination by hematoxylin-eosin (H&E) staining and for immunohistochemical detection of Cx43. Briefly, rehydrated sections were pretreated with pepsin solution for 15 min at 37°C. The following protocols were according to the manual of the S-P kit (ZSGB-BIO, Beijing). The myocardium near LAD was incubated for 10 min with a peroxidase-blocking solution and goat serum solution for 30 min and then incubated with primary antibody (rabbit anti-rat Cx43) (Cell Signaling Technology, USA) overnight at 4°C. After three rinses with PBS, the sections were incubated with biotinylated secondary antibody (goat anti-rabbit IgG, included in the S-P Kit) for 30 min and further developed with 3,30-diaminobenzidine tetrahydrochloride (DAB).
2.10. Transmission Electron Microscopy (TEM)
For TEM examination, samples containing a 2 mm portion from the edge of the incision were immediately fixed in 0.1 M phosphate buffer containing 2.5% glutaraldehyde and 2% paraformaldehyde for 4 h. The samples were then fixed with 1% osmium tetroxide for 2 h, dehydrated through a graded ethanol series, and embedded in epoxy resin. Resin-embedded blocks were cut into 60~80 nm ultrathin sections with an ultramicrotome (PT-XL, RMC, USA). The ultrathin sections were placed on carbon coated nickel grids and examined with an H-7500 transmission electron microscope (H-7500, Tokyo, Japan) operating at 80 kV.
2.11. Real-Time Quantitative PCR (RT-qPCR)
Total RNA was extracted using TRIzol reagent (Invitrogen, USA) according to the manufacturer's instructions. RNA (1 ng) was reversed with the cDNA synthesis Kit (Invitrogen, USA). Real-time polymerase chain reaction (PCR) was performed using the SYBR green system (Bio-Rad, USA). Primer sequences were as shown in Supplementary Table 1. The relative quantification of a target gene was normalized to GAPDH and calculated using the absolute quantification standard curve method. Melting curve profiles were produced at the end of each PC to confirm the specificity amplifications. Each sample was analyzed in triplicate.
2.12. Western Blotting
Equal amounts of protein (50 μg) were subjected to SDS-PAGE. Proteins were then transferred to polyvinylidene fluoride (PVDF) membrane. Membrane was blocked with 5% nonfat dry milk in Tris-buffered saline, 0.1% Tween 20 (Sigma, USA), and immunoblotting was performed using rabbit anti-rat Na+-K+-ATPase (1 : 500, Cell Signaling Technology, USA) as described by the manufacturer. Anti-β-Actin antibody (1 : 200, Santa Cruz, USA) was used as loading control. Blots were then developed by incubation with biotinylated anti-rabbit antibody (1 : 2000; Vector Laboratories), followed by incubation with ABC reagent (GE, USA). Signal was detected using an ECL luminescence kit (GE, USA) and X-ray film.
2.13. Statistical Analysis
Data were expressed as mean ± standard deviation. Statistical analysis was performed by one-way ANOVA to compare more than two groups or with paired t-test to compare two groups. All statistical analyses were performed using SPSS version 17.0 (SPSS Inc., Chicago, IL, USA). The significance level was set at P < 0.05.
3. Results
3.1. The Effects of LXA4 Preconditioning and Postconditioning on I/R-Induced Arrhythmias
The effects of different treatments on I/R-induced arrhythmias are shown in Table 1. In C1 and C2 groups, neither VT nor VF was observed and only few ventricular premature beats appeared during the whole procedure. In contrast, almost all rats in I/R1 and I/R2 groups experienced obvious ST segment elevation, occurring VT, and high frequency of VF. Furthermore, the frequency of arrhythmias was significantly reduced in LX1 and LX2 groups with the exception of 2 rats showing VF in LX2 group. Most of the arrhythmias observed occurred in the ischemia (30 min) and in the first 30 min of the reperfusion periods.
Table 1.
The ventricular arrhythmia score (VAS), the duration time of VT (VTt) and VF (VFt) in different groups at T3 (n = 12, mean ± SD).
| Group | VAS | VTt (s) | VFt (s) |
|---|---|---|---|
| C1 | 0.25 ± 0.45 | 0 | 0 |
| I/R1 | 4.67 ± 0.49* | 73.50 ± 28.40* | 16.67 ± 14.62* |
| LX1 | 2.33 ± 0.49∗# | 22.67 ± 7.51∗# | 0 |
| C2 | 0.33 ± 0.49 | 0 | 0 |
| I/R2 | 4.83 ± 0.39* | 69.67 ± 24.83* | 22.00 ± 16.03* |
| LX2 | 3.50 ± 0.80∗#Δ | 35.42 ± 14.92∗#Δ | 3.58 ± 8.50# |
Note: T3: during ligation of the left anterior descending coronary artery to reperfusion.
I/R1 and LX1 groups were compared with C1 group, and I/R2 and LX2 were compared with C2 group, *P < 0.05.
LX1 goup was compared with I/R1 group, and LX2 group was compared with I/R2 group, # P < 0.05.
LX2 group was compared with LX1 group, Δ P < 0.05.
In addition, the durations of VT and VAS were markedly higher in I/R1 and LX1 compared with C1 group, and the same was seen for I/R2 and LX2 groups when compared with C2 group, (P < 0.05 in both cases). Finally, VAS and the VT durations of LX1 and LX2 groups were significantly (P < 0.05) lower than those in I/R1 and I/R2 groups. Interestingly, these parameters in LX1 group were significantly lower than those in LX2 group (P < 0.05), indicating that LXA4 preconditioning was more effective in preventing I/R-induced arrhythmias than LXA4 postconditioning.
3.2. The Effect of LXA4 Preconditioning and Postconditioning on Serum Levels of IL-1β, IL-6, IL-8, IL-10, TNF-α, and cTnI
Serum levels of IL-1β, IL-6, IL-8, IL-10, TNF-α, and cTnI were determined using ELISA kits. We found that there was no statistical difference at T1 among all the groups. In contrast, cytokines levels at T2 in I/R1 and LX1 groups were significantly (P < 0.05) elevated compared to C1 controls, and similar results were seen for I/R2 and LX2 groups compared with C2 controls. In the meantime, reduced levels of proinflammatory cytokines such as IL-1β, IL-6, IL-8, and TNF-α and elevated level of the anti-inflammatory cytokine IL-10 were observed in LX1 and LX2 groups compared with I/R1 and I/R2 groups, respectively. These findings indicate that both LXA4 preconditioning and postconditioning could induce the expressions of anti-inflammatory cytokines and inhibit the induction of proinflammatory cytokines. Moreover, these changes were more significant in LX2 compared with LX1 group (P < 0.05), further confirming that LXA4 postconditioning was more effective in preventing I/R injury than LXA4 preconditioning (Figure 1).
Figure 1.
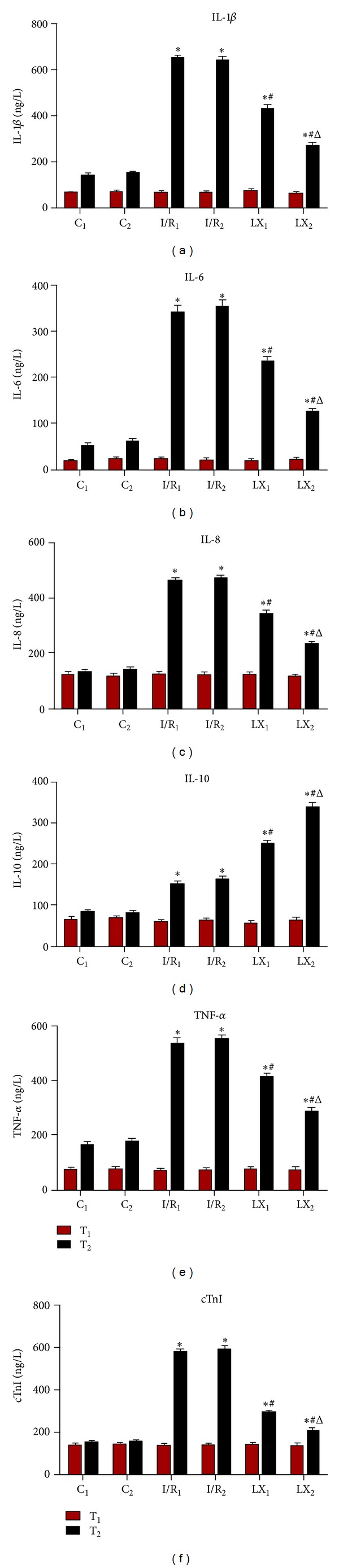
Comparison of concentrations of serum IL-1β, IL-6, IL-8, IL-10; TNF-α;, and cTnI at T1 and T2 among all groups (n = 12 for each group). At time point T1, blood was collected immediately before thoracotomy. At time point T2, blood was collected right after the I/R procedure was over. (a) IL-1β; (b) IL-6; (c) IL-8; (d) IL-10; (e) TNF-α; (f) cTnI. *P < 0.05 for comparisons of I/R1 and LX1 groups with C1 group, P < 0.05 for comparisons of I/R2 and LX2 with C2 group; # P < 0.05 for comparisons of LX1 group with I/R1 group, P < 0.05 for comparisons of LX2 group with I/R2 group; Δ P < 0.05 for comparisons of LX2 group with LX1 group.
3.3. The Effect of LXA4 Preconditioning and Postconditioning on MDA Levels and SOD and Na+-K+-ATPase Activities
The production of MDA, SOD, and Na+-K+-ATPase activities in myocardial tissues in response to I/R injury were investigated. Compared with C1 group, MDA production and SOD activity were increased and Na+-K+-ATPase activity was reduced in I/R1 and LX1 groups, and the same results were obtained in I/R2 and LX2 groups compared to C2 group (P < 0.05). Furthermore, decreased MDA production and increased activities of SOD and Na+-K+-ATPase in LX1 and LX2 groups were observed compared with I/R1 and I/R2 groups, respectively. Similar results were observed when LX2 group was compared with LX1 group, indicating that LXA4 postconditioning was more protective against I/R injury than LXA4 preconditioning (Figure 2).
Figure 2.
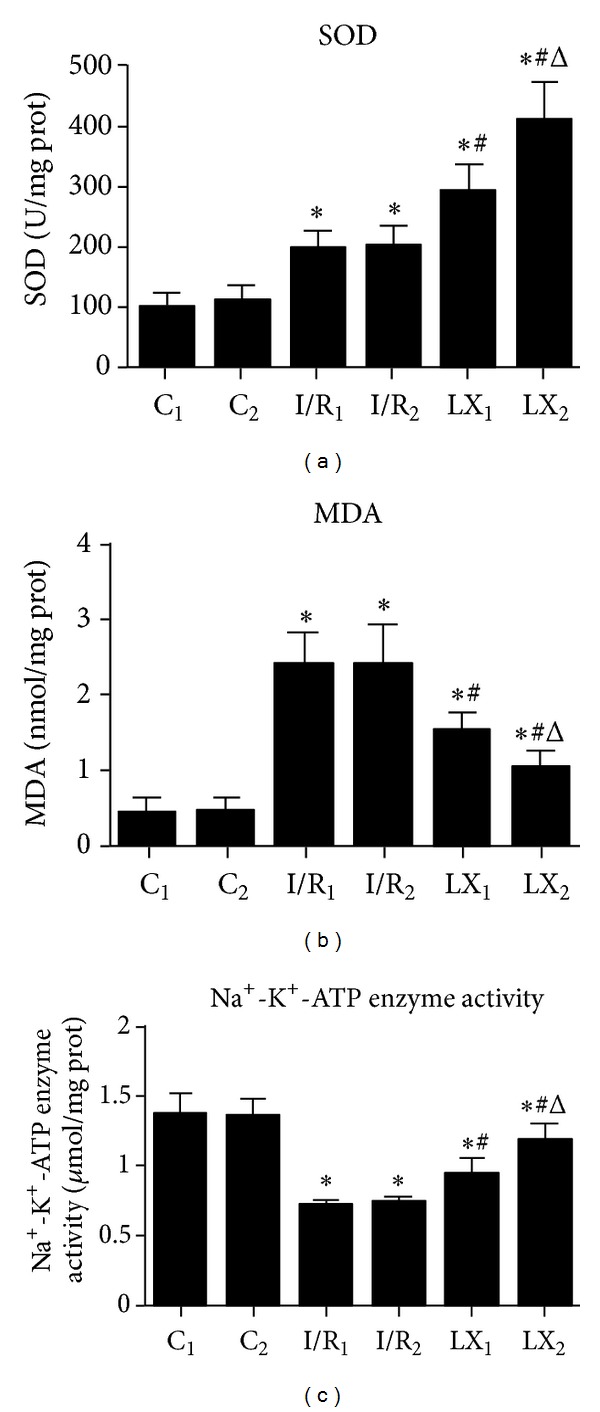
Comparison of cardiac MDA content and SOD and Na+-K+-ATPase activities at T2 among all groups (n = 12 for each group). At time point T2, myocardial tissue was collected right after the I/R procedure was over and kept frozen in liquid nitrogen. MDA content and SOD and Na+-K+-ATPase activities were measured as described in the section of Materials and Methods. (a) SOD activity; (b) MDA content; (c) Na+-K+-ATPase activity. *P < 0.05 for comparisons of I/R1 and LX1 groups with C1 group, P < 0.05 for comparisons of I/R2 and LX2 with C2 group; # P < 0.05 for comparisons of LX1 group with I/R1 group, P < 0.05 for comparisons of LX2 group with I/R2 group; Δ P < 0.05 for comparisons of LX2 group with LX1 group.
3.4. The Effect of LXA4 Preconditioning and Postconditioning on the Ultrastructural Changes of the Myocardium
TEM images of ultrathin sections of myocardial tissue are shown in Figure 3. Cardiomyocytes were clearly seen in a well-arranged myofilament and intercalated disc manner in sham groups (C1 and C2), as well as abundant normal mitochondria with no swelling, normal matrix density, and intact cristae. The round endoplasmic reticulum can also be seen between the myofilaments and the cytoplasm (Figures 3(a) and 3(b)). However, in I/R1 and I/R2 groups, myocardial I/R produced remarkable ultrastructural damages associated with irregularities and edematous separation of myofilaments, hypercontraction, and shortening of sarcomeres. Large area of cytoplasmic vacuolization and mitochondrial swelling were evident with decreased matrix density and distortion of cristae (Figures 3(c) and 3(d)). Treatment with LXA4 showed clear protection with relatively parallel arrangement of myofilaments and normal sarcomeres. Mitochondria were normal with mild swelling and normal matrix density but slightly damaged cristae. However, mild cytoplasmic rarefaction with mild edema could still be seen (Figures 3(e) and 3(f)).
Figure 3.

Transmission electron microscopy ((a)–(f)) and histological study ((g)–(l), He ×400) of cardiac tissues from rats of different groups at T2 time point. (a) Group C1 and (b) group C2: regular myocardial myofilament arrangement, clear intercalated disk structure, normal mitochondrial morphology, and structure with endoplasmic reticulum embedded in myofilament matrix; (c) group I/R1 and (d) group I/R2: disordered arrangement of myofilament, a large number of myofilament fractured, intercalated disk structure unclear, and abnormal mitochondrial morphology (a high degree of swelling, membrane lysis, fuzzy ridge structure, formation of vacuoles, and endoplasmic reticulum vacuolization); (e) group LX1 and (f) group LX2: myofilament arrangement is a little sparse, the intercalated disk structure is slightly fuzzy, and slightly swollen mitochondria; (g) group C1 and (h) group C2: normal size cardiomyocyte, no hemorrhage, or neutrophil granulocyte infiltration; (i) group I/R1 and (j) group I/R2: Cardiomyocyte degeneration, hemorrhage, edema, and significant interstitial neutrophil granulocyte infiltration; (k) group LX1 and (l) group LX2: no significant cardiomyocyte degeneration, no obvious bleeding, slight edema between the myocardial fibers, and mild neutrophil granulocyte infiltration.
3.5. The Effect of LXA4 Preconditioning and Postconditioning on Pathological Changes
To further assess the effect of LXA4 on I/R injury, we analyzed the pathological changes it induced (Figure 3). In the sham groups (C1 and C2), myocardial fibers were normal with clear striations. No epimorphosis, necrosis, or neutrophil infiltration was observed (Figures 3(g) and 3(h)). In contrast, nontreated I/R groups (I/R1 and I/R2) showed local swelling, myocardial necrosis, disorganized myocardial fibers, and ruptured cells with large number of inflammatory cells in the cytoplasm (Figures 3(i) and 3(j)). In the LXA4-treated groups (LX1 and LX2), myocardial cells with normal structure and shape can be seen, though the myocardial fibers were mildly swollen or partially ruptured and slight edema can be seen in the interstitial tissues with small amount of inflammatory cells (Figures 3(k) and 3(l)).
3.6. The Effect of LXA4 Preconditioning and Postconditioning on Cx43 Expression
The gap junction protein Cx43 is the major component of gap junctions between cardiomyocytes of mammalian ventricular myocardium, including human and rodent hearts. Using an immunohistochemical approach, we wanted to know whether LXA4 preconditioning and postconditioning affected Cx43 expression. As shown in Figure 4, Cx43 expression, demonstrated by a pervasive brown/yellow color in cardiac muscle cells, was observed in myocardial tissue samples from all the groups. The areas of Cx43 expression in myocardial tissue samples from sham groups (C1 and C2) were larger than those detected in the I/R groups, which was also confirmed by the quantitative analysis of integral optical density (IOD) (Figure 4). Cx43 was well arranged together at both ends of the intact cardiac myocyte in I/R1 and I/R2 groups, and Cx43 staining was significantly reduced when compared with the sham groups. However, in LXA4-treated LX1 and LX2 groups, most Cx43 staining was located at the ends of intact cardiac myocytes (intercalate disc) and redistributed in both sides of the cell. Compared with nontreated I/R1 and I/R2 groups, the IOD value was statistically higher in LX1 and LX2 groups, respectively. Additionally, Cx43 expression of LXA4 preconditioning (LX1 group) was significantly (P < 0.05) higher than LXA4 postconditioning (LX2 group) (Figure 4).
Figure 4.
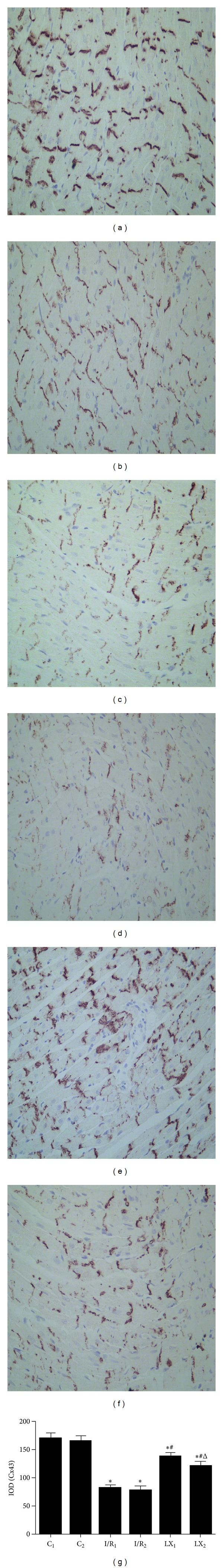
Immunohistochemical staining and IOD of Cx43 in cardiac tissues from rats of different groups at T2. (a) Group C1 and (b) group C2: the Cx43 positive particle intensively and regularly was distributed and was clustered in the intercalated disk; (c) group I/R1 and (d) group I/R2: sparsely distributed Cx43 positive particles; (e) group LX1 and (f) group LX2: Slightly decreased Cx43 positive particles, mostly still clustered in the intercalated disk. (g) For IOD of Cx43. *P < 0.05 for comparisons of I/R1 and LX1 groups with C1 group, P < 0.05 for comparisons of I/R2 and LX2 with C2 group; # P < 0.05 for comparisons of LX1 group with I/R1 group, P < 0.05 for comparisons of LX2 group with I/R2 group; Δ P < 0.05 for comparisons of LX2 group with LX1 group.
3.7. The Effects of I/R and LXA4 Treatment on Na+-K+-ATPase and Cx43 at mRNA Level
The Na+-K+-ATPase and Cx43 mRNA levels were examined using real-time quantitative PCR to determine if I/R injury influences the Na+-K+-ATPase and Cx43 expressions and whether this effect is blocked by the different LXA4 treatment (Figure 5). Compared with the sham groups (C1 and C2), mRNA levels of Na+-K+-ATPase and Cx43 in I/R1, LX1, I/R2, and LX2 were remarkably reduced. Meanwhile, Na+-K+-ATPase and Cx43 expressions in LXA4-treated LX1 and LX2 groups were higher than the saline-treated I/R1 and I/R2 groups, respectively (P < 0.05). Furthermore, Na+-K+-ATPase mRNA level of LXA4 preconditioning (LX1) was lower than that of LXA4 postconditioning (LX2). However, Cx43 expression was higher in LX1 compared to LX2 group (P < 0.05).
Figure 5.
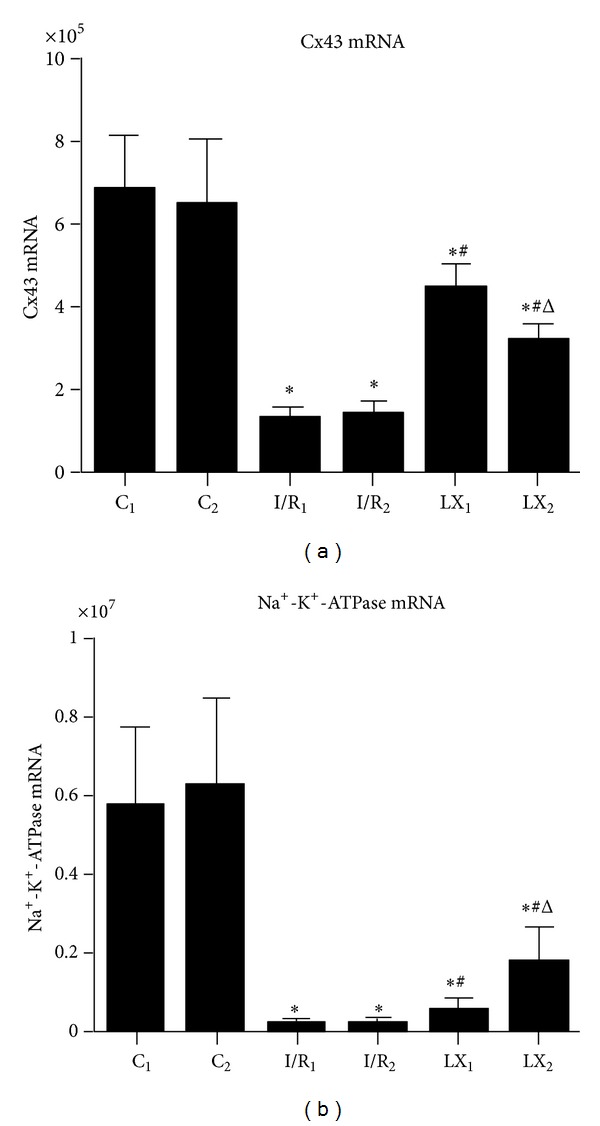
mRNA levels of Cx43 and Na+-K+-ATPase of cardiac tissues from rats of different groups at T2. Samples were collected right after I/R procedure. (a) Cx43 mRNA; (b) Na+-K+-ATPase mRNA. *P < 0.05 for comparisons of I/R1 and LX1 groups with C1 group, P < 0.05 for comparisons of I/R2 and LX2 with C2 group; # P < 0.05 for comparisons of LX1 group with I/R1 group, P < 0.05 for comparisons of LX2 group with I/R2 group; Δ P < 0.05 for comparisons of LX2 group with LX1 group.
3.8. Further Analysis of the Effects of LXA4 Treatment on Na+-K+-ATPase Isoform Expression
The results of Na+-K+-ATPase isoforms analyzed by western blotting are shown in Figure 6. Compared to the sham C1 group, the expressions of Na+-K+-ATPase isoforms α 1, α 2, α 3, and β 1 were markedly reduced in I/R1 and LX1 groups and similar results were obtained in I/R2 and LX2 groups, compared to C2 group (P < 0.05). However, Na+-K+-ATPase isoform levels in LXA4-treated LX1 and LX2 groups were higher than the saline-treated I/R1 and I/R2 groups respectively (P < 0.05). Additionally, protein levels of Na+-K+-ATPase in LXA4 preconditioning (LX1) were significantly (P < 0.05) lower than those measured in LXA4 postconditioning (LX2).
Figure 6.
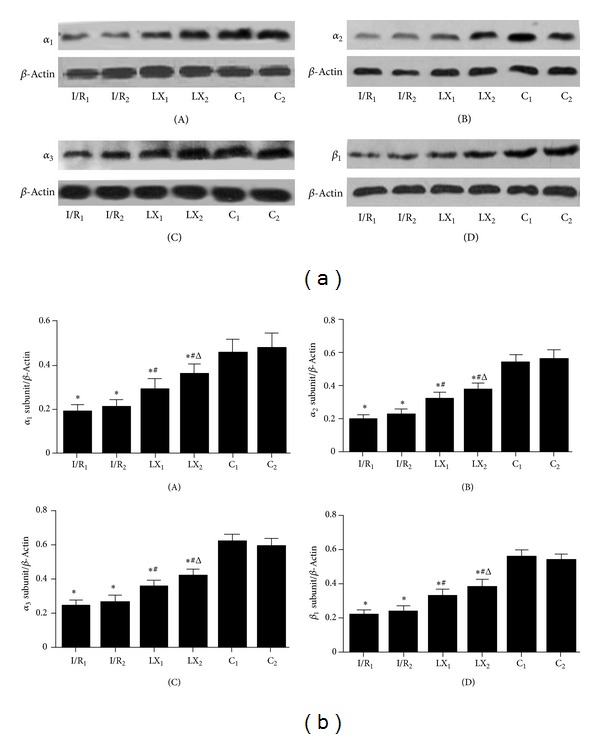
Protein levels of α 1, α 2, and α 3 and β 1 subunits of Na+-K+-ATPase of cardiac tissues from rats of different groups at T2. Samples were collected right after I/R procedure was over and western blot was performed as described in the section of Materials and Methods, and β-Actin was used as a loading control. Representative blots of three independent experiments were shown. (a) Western blotting: (A) α 1 subunit of Na+-K+-ATPase; (B) α 2 subunit; (C) α 3 subunit; (D) β 1 subunit. (b) Quantitative data of western blot: (A) α 1 subunit of Na+-K+-ATPase; (B) α 2 subunit; (C) α 3 subunit; (D) β 1 subunit. *P < 0.05 for comparisons of I/R1 and LX1 groups with C1 group, P < 0.05 for comparisons of I/R2 and LX2 with C2 group; # P < 0.05 for comparisons of LX1 group with I/R1 group, P < 0.05 for comparisons of LX2 group with I/R2 group; Δ P < 0.05 for comparisons of LX2 group with LX1 group.
4. Discussion
With the widespread use of cardiac surgery for congenital heart disease and coronary artery bypass grafting under extracorporeal circulation, thrombolytic therapy, percutaneous transluminal coronary intervention, organ transplantation, and cardiopulmonary resuscitation, it has currently become a hot topic for cardiovascular scientists to find safe and effective drugs to treat myocardial I/R injury [10]. In this study, we used rats to induce I/R injury because of the little variation in their cardiovascular system, similarity to human coronary artery territory, less myocardial collateral circulation, and the high probability of occurrence of I/R injury. Due to the smaller size of coronary artery in rats, more than 30 min of blocking time can easily lead to permanent occlusion of coronary artery [14, 19, 20]. Therefore, we chose 30 min of myocardial ischemia followed by 120 min reperfusion. LXA4 is usually dissolved in ethanol. However, we found that ethanol affected the health of experimental animals. Therefore, LXA4 was dissolved in saline as suggested by the manufacturer's manual.
Currently, the effect of I/R injury on myocardial ultrastructure is not entirely clear. El Kebir et al. [21] reported that LX can inhibit neutrophil aggregation at inflammatory sites and regulate the balance of proinflammatory/anti-inflammatory cytokines. In addition, LXA4 can suppress vascular-endothelial-growth-factor- (VEGF-) induced inflammatory reaction through the regulation of IL-6, TNF-α, IL-8, ICAM-1, and IL-10 expressions in human umbilical vein endothelial cells [22]. Souza et al. [13] found that reduced production of TNF-α following LXA4 treatment of intestinal I/R injury may be due to the increased IL-10 expression, and LXA4 could not prevent reperfusion injury in IL-10-deficient mice. In our study, we confirmed that I/R resulted in myocardial inflammatory injury through analyzing serum components before and after I/R. Although both proinflammatory/anti-inflammatory cytokines were upregulated, the expressions of proinflammatory cytokines were much higher than the anti-inflammatory ones. However, with LXA4 treatment, the expressions of proinflammatory cytokines (IL-1β, IL-6, IL-8, and TNF-α) were reduced while the anti-inflammatory cytokine IL-10 was upregulated, leading to an anti-inflammatory state (Figure 1).
The results of H&E staining confirmed that LXA4 could alleviate I/R injury as evidenced by reduced swelling, well-organized cardiac muscle, clear striations, and less inflammatory cell infiltration. In accordance with the pathological changes, we found that LXA4 protected the ultrastructures of myocardial tissue as shown by normal matrix density, relatively parallel arrangement of myofilaments, and normal sarcomere mitochondria by TEM examination (Figure 3).
There has been report showing that LXA4 can inhibit the generation of ROS [7]. We found that I/R groups had increased MDA levels and SOD activity, which indicated enhanced oxidative stress. After LXA4 treatment, SOD activity was significantly improved and MDA levels were markedly reduced (Figure 2), suggesting that the alleviated I/R injury seen after LXA4 treatment might be due to decreased ROS generation. Mitochondria are the main organelle maintaining the cell respiration and oxidation. Close to 90% of cellular oxygen intake is used in oxidative phosphorylation reactions in the mitochondria, but 1% to 2% of oxygen can escape to form ROS. ROS can decrease the efficiency of mitochondrial performance and cause swelling of mitochondria by attacking the mitochondrial inner membrane. Indeed, mitochondria are both a primary source and target of ROS [23]. As we can see from the TEM images (Figure 3), the swelling condition of mitochondria has been remarkably alleviated after LXA4 treatment, with clear cristae and well-arranged cardiac fiber ultrastructure. This agrees with the MDA levels and the SOD activity data and further confirms that reduced I/R injury may be due to the inhibition of oxygen-free radical targeting of mitochondrial membranes.
The Na+-K+-ATPase is a heterodimer that plays an important role in the maintenance of cell homeostasis by regulating membrane potential and cation transport across the sarcolemmal membrane, which is a reliable indicator of metabolic disorders and tissue injury. I/R injury could affect mitochondrial function and inhibit Na+-K+-ATPase activity, leading to intracellular calcium overload, activation of proteases, and release of oxygen-free radicals, which in turn aggravate I/R injury. Belliard et al. [24] reported that the maintenance of Na+-K+-ATPase cell surface abundance was crucial in myocyte survival after an ischemic attack treated by ouabain preconditioning, and the protection conferred by increased surface expression of Na+-K+-ATPase may be independent of ion transport. Meanwhile, Zheng et al. [25] found that the protection from cardiac injury may be determined by the Na+-K+-ATPase activity, involving ERK1/2 and PI3 K/Akt signal pathways. Furthermore, continued intermittent hypobaric hypoxia can reduce the reperfusion injury in guinea pig by increasing myocardial Na+-K+-ATPase activity [26]. The Na+-K+-ATPase consists of α and β subunits; the α subunit is responsible for the catalytic activity of the enzyme and includes three isoforms: α 1, α 2, and α 3 in the rat heart. The β subunit is responsible for the proper localization and insertion of Na+-K+-ATPase in the sarcolemmal membrane. In this study, the expressions of α 1, α 2, and α 3 proteins decreased after ischemia/reperfusion injury, which affected the active center and further reduced the activity of the ATPase enzyme. The expression of β subunit also declined after I/R injury, and this could cause damage to the cytoskeleton, lead to detachment of Na+-K+-ATPase from the cell membrane, and further reduce its activity [27]. The effect of ischemia/reperfusion on the expressions of Na+-K+-ATPase α 1, α 2, and α 3 subunits were slightly different from earlier report [28], which may be due to differences in experimental and detection methods. The content and the activity of Na+-K+-ATPase can reflect myocardial injury to a certain extent, and I/R injury can also be alleviated by increasing its content and activity. Figtree et al. [29] suggested that cardiomyocytes could be protected potentially by reversing the oxidation inhibition of Na+-K+-ATPase. And from the our study, we know that LXA4 enhanced Na+-K+-ATPase subunits expressions (Figures 5 and 6) and reduced cTnI concentration, indicating that the cardioprotective effect of LXA4 could be due to increased Na+-K+-ATPase content and activity.
The gap junction (GJ) plays key roles not only in electrical coupling of cardiomyocytes but also in intercellular transport of biologically active substances. GJ is mainly located in the intercalated disc of the cardiac tissue [30] and participates in decision making for cell survival versus cell death in various types of cells. It has been suggested that reperfusion injury to the heart is partially mediated by gap junctions. Cx43 forms gap junctions that facilitate electrical cell-cell coupling and unopposed/nonjunction hemichannels which provide a pathway for the exchange of ions and metabolites between cytoplasm and extracellular milieu [31]. It has also been known that Cx43 participates in the process of myocardiac ischemia/reperfusion injury [32, 33]. During I/R injury, alterations in Cx43 expression, its localization and its phosphorylation status, and changes in GJ properties collectively contribute to myocardial infarction and arrhythmogenesis [34–36]. From our research, we found that Cx43 was well arranged together in both ends of intact cardiac myocytes (intercalate disc) in the sham group, while in the I/R group, Cx43 expression was reduced which was associated with damaged cellular ultrastructures (Figures 4 and 5). In addition, increased VAS, VTt, and VFt duration and reduced Cx43 mRNA and protein expressions after injury strongly indicate that I/R injury could induce arrhythmia through Cx43 pathway.
The mechanism of arrhythmogenesis caused by I/R injury has not yet been illustrated clearly. Cabo et al. found that this may be due to the structural and functional alternations of Cx43 after injury [37], and Danik reported that Cx43 knockout mice showed complicated tachyarrhythmia [38]. Recently, two lipoxins have been identified, lipoxin A4 (LXA4) and lipoxin B4 (LXB4). LXA4 treatment in our study showed that this molecule can alleviate Cx43 degradation and protect its distribution that limited the damage to the ultrastructure of cardiac tissue caused by I/R injury. At the same time, the mRNA transcriptional levels of Cx43 were notably increased, which was in accordance with the immunohistochemical results. This indicates that LXA4 plays a protective role in regulating the expressive levels of Cx43 protein and gene. In addition, reduced arrhythmia frequency and VAS were observed, indicating that the protective effect of LXA4 may be associated with Cx43 remodeling, an observation consistent with a previous study showing that cardiac function could be preserved through Cx43 remodeling [39].
The pharmacological preconditioning and postconditioning with LXA4 can both attenuate myocardial I/R injury in rats, with LXA4 preconditioning being more effective in preventing the arrhythmogenesis than the LXA4 postconditioning. An effect associated with higher Cx43 expression and lower VAS. However, LXA4 postconditioning had a better protective effect on Na+-K+-ATPase and the ultrastructure of the myocardium, which may be related to the physicochemical property, duration of the treatment, and the dose of LXA4. We also observed that most arrhythmias happened in the ischemia (30 min) and the first 30 min of reperfusion, which may be due to the activated compensatory mechanism of the injured body and increased tolerance to the drug. Cx43 is easy to be degraded when I/R injury happens [40]. Cx43 is also involved in pathological and physiological processes other than arrhythmias during I/R injury [41, 42], and it may cause uncontrolled opening of hemichannels in the plasma membrane [43] and may be deleterious to the myocardium [44]. Blocking hemichannels may confer cardioprotection by preventing ionic imbalance, excessive entry of Na+ and Ca+, ATP leakage, cell swelling, and loss of critical metabolites [40, 45, 46]. Therefore, Cx43-mediated intercellular communication of the injured tissue can be enlarged into other cells, and this may explain that LXA4 preconditioning induced higher Cx43 expression but caused more serious damage to myocardium. At the same time, it indicated that the up-regulation of Cx43 is helpless in the myocardial energy metabolism, systemic inflammation, and ultrastructure. The application of pharmacological preconditioning has been limited largely by the unpredictability of I/R injury, and this implicates that LXA4 postconditioning could have a great potential for clinical application in I/R injury in the future.
Although there have been reports showing that LXA4 treatment was protective in cerebral I/R injury in rats [12] and myocardial I/R injury in mice [47] and rabbits [48], ours is the first study to report a protective effect of LXA4 preconditioning and postconditioning on myocardial I/R injury in rats. Further studies are needed to optimize the dose, the timing, and the route of administration of LXA4.
5. Conclusions
LXA4 preconditioning and postconditioning in myocardial I/R injury can attenuate the metabolic disturbance in the myocardium through upregulating Na+-K+-ATPase expression. Meanwhile, both LXA4 preconditioning and postconditioning protected the ultrastructure of the myocardium by inhibiting the inflammatory reaction and oxidative stress. In addition, the upregulation of Cx43 expression had a positive effect in preventing arrhythmogenesis caused by I/R injury. All the results showed that LXA4 has great potential for protecting the myocardium from myocardial I/R injury.
Supplementary Material
Supplementary Figure 1: Chemical structure of LXA4. Lipid-derived lipoxins are produced at the site of vascular and mucosal inflammation where they down-regulate polymorphonuclear leukocyte recruitment and function. 5(S),6(R),15(R)-LipoxinA4 (5(S),6(R),15(R)-LXA4) is derived from the aspirin- triggered formation of 15(R)-HETE from arachidonic acid. Formula Weight: 352.5.
According to DNA sequence of various indexes, Primer 3.0 software was used to design Na+-K+-ATPase and Cx43 primer. GAPDH is control gene. Primer sequences of Na+-K+-ATPase and Cx43 were as shown in Supplementary Table 1.
Conflict of Interests
The authors declare no conflict of interests.
Acknowledgments
This study was supported by Science and Technology Planning Project of Education Bureau of Zhejiang province, China (no. Y200804950), the Opening Foundation of Zhejiang Provincial Top Key Discipline of Surgery (no. 2011MZ005), and the Key Discipline Program of Pediatric Surgery of Health Bureau of Zhejiang province (no. 11-ZC27).
References
- 1.Hoffman JW, Jr., Gilbert TB, Poston RS, Silldorff EP. Myocardial reperfusion injury: etiology, mechanisms, and therapies. Journal of Extra-Corporeal Technology. 2004;36(4):391–411. [PubMed] [Google Scholar]
- 2.Maxwell SRJ, Lip GYH. Reperfusion injury: a review of the pathophysiology, clinical manifestations and therapeutic options. International Journal of Cardiology. 1997;58(2):95–117. doi: 10.1016/s0167-5273(96)02854-9. [DOI] [PubMed] [Google Scholar]
- 3.Park JL, Lucchesi BR. Mechanisms of myocardial reperfusion injury. Annals of Thoracic Surgery. 1999;68(5):1905–1912. doi: 10.1016/s0003-4975(99)01073-5. [DOI] [PubMed] [Google Scholar]
- 4.Serhan CN, Hamberg M, Samuelsson B. Lipoxins: novel series of biologically active compounds formed from arachidonic acid in human leukocytes. Proceedings of the National Academy of Sciences of the United States of America. 1984;81(17):5335–5339. doi: 10.1073/pnas.81.17.5335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Serhan CN, Chiang N, van Dyke TE. Resolving inflammation: dual anti-inflammatory and pro-resolution lipid mediators. Nature Reviews Immunology. 2008;8(5):349–361. doi: 10.1038/nri2294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Scully M, Gang C, Condron C, Bouchier-Hayes D, Cunningham AJ. Protective role of cyclooxygenase (COX)-2 in experimental lung injury: evidence of a lipoxin A4-mediated effect. Journal of Surgical Research. 2012;175(1):176–184. doi: 10.1016/j.jss.2011.02.020. [DOI] [PubMed] [Google Scholar]
- 7.Zhou X-Y, Wu P, Zhang L, et al. Effects of lipoxin A4 on lipopolysaccharide induced proliferation and reactive oxygen species production in RAW264.7 macrophages through modulation of G-CSF secretion. Inflammation Research. 2007;56(8):324–333. doi: 10.1007/s00011-007-7012-7. [DOI] [PubMed] [Google Scholar]
- 8.Lee HN, Na HK, Surh YJ. Resolution of inflammation as a novel chemopreventive strategy. Seminars in Immunopathology. 2013;35(2):151–161. doi: 10.1007/s00281-013-0363-y. [DOI] [PubMed] [Google Scholar]
- 9.Isobe Y, Kato T, Arita M. Emerging roles of eosinophils and eosinophil-derived lipid mediators in the resolution of inflammation. Frontiers in Immunology. 2012;3, article 270 doi: 10.3389/fimmu.2012.00270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ye Y, Perez-Polo JR, Aguilar D, Birnbaum Y. The potential effects of anti-diabetic medications on myocardial ischemia-reperfusion injury. Basic Research in Cardiology. 2011;106(6):925–952. doi: 10.1007/s00395-011-0216-6. [DOI] [PubMed] [Google Scholar]
- 11.Peskar BM, Ehrlich K, Schuligoi R, Peskar BA. Role of lipoxygenases and the lipoxin A4/annexin 1 receptor in ischemia-reperfusion-induced gastric mucosal damage in rats. Pharmacology. 2009;84(5):294–299. doi: 10.1159/000244017. [DOI] [PubMed] [Google Scholar]
- 12.Wu L, Miao S, Zou LB, et al. Lipoxin A4 inhibits 5-lipoxygenase translocation and leukotrienes biosynthesis to exert a neuroprotective effect in cerebral ischemia/reperfusion injury. Journal of Molecular Neuroscience. 2012;48(1):185–200. doi: 10.1007/s12031-012-9807-4. [DOI] [PubMed] [Google Scholar]
- 13.Souza DG, Fagundes CT, Amaral FA, et al. The required role of endogenously produced lipoxin A4 and annexin-1 for the production of IL-10 and inflammatory hyporesponsiveness in mice. Journal of Immunology. 2007;179(12):8533–8543. doi: 10.4049/jimmunol.179.12.8533. [DOI] [PubMed] [Google Scholar]
- 14.Testai L, Martelli A, Marino A, et al. The activation of mitochondrial BK potassium channels contributes to the protective effects of naringenin against myocardial ischemia/reperfusion injury. Biochemical Pharmacology. 2013;85(11):1634–1643. doi: 10.1016/j.bcp.2013.03.018. [DOI] [PubMed] [Google Scholar]
- 15.Zhang J, Gong E. The protective effect of ginkgo flavonoids phospholipid complex on vascular endothelial in myocardial reperfusion injury in rats. Journal of Mathematical Medicine. 2009;22(1):15–18. [Google Scholar]
- 16.Wen JY, Tan N, Yang DH. Electrocardiogram changes on myocardial ischemia-reperfusion model in rats. South China Journal of Cardiovascular Diseases. 2011;17(6):503–506. [Google Scholar]
- 17.Walker MJA, Curtis MJ, Hearse DJ, et al. The Lambeth conventions: guidelines for the study of arrhythmias in ischaemia, infarction, and reperfusion. Cardiovascular Research. 1988;2(7):447–455. doi: 10.1093/cvr/22.7.447. [DOI] [PubMed] [Google Scholar]
- 18.Bruce AF, Rothery S, Dupont E, Severs NJ. Gap junction remodelling in human heart failure is associated with increased interaction of connexin43 with ZO-1. Cardiovascular Research. 2008;77(4):757–765. doi: 10.1093/cvr/cvm083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tratsiakovich Y, Gonon AT, Krook A, et al. Arginase inhibition reduces infarct size via nitric oxide, protein kinase C epsilon and mitochondrial ATP-dependent K+ channels. European Journal of Pharmacology. 2013;712(1–3):16–21. doi: 10.1016/j.ejphar.2013.04.044. [DOI] [PubMed] [Google Scholar]
- 20.Zhao MM, Yang JY, Wang XB, Tang CS, Du JB, Jin HF. The PI3K/Akt pathway mediates the protection of SO(2) preconditioning against myocardial ischemia/reperfusion injury in rats. Acta Pharmacologica Sinica. 2013;34(4):501–506. doi: 10.1038/aps.2012.204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.El Kebir D, József L, Pan W, et al. 15-epi-lipoxin A4 inhibits myeloperoxidase signaling and enhances resolution of acute lung injury. American Journal of Respiratory and Critical Care Medicine. 2009;180(4):311–319. doi: 10.1164/rccm.200810-1601OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Baker N, O’Meara SJ, Scannell M, Maderna P, Godson C. Lipoxin A4: anti-inflammatory and anti-angiogenic impact on endothelial cells. Journal of Immunology. 2009;182(6):3819–3826. doi: 10.4049/jimmunol.0803175. [DOI] [PubMed] [Google Scholar]
- 23.Krause DN, Duckles SP, Pelligrino DA. Influence of sex steroid hormones on cerebrovascular function. Journal of Applied Physiology. 2006;101(4):1252–1261. doi: 10.1152/japplphysiol.01095.2005. [DOI] [PubMed] [Google Scholar]
- 24.Belliard A, Sottejeau Y, Duan Q, Karabin JL, Pierre SV. Modulation of cardiac Na+-K+-ATPase cell surface abundance by simulated ischemia/reperfusion and ouabain preconditioning. American Journal of Physiology. 2013;304(1):H94–H103. doi: 10.1152/ajpheart.00374.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zheng J, Koh X, Hua F, Li G, Larrick JW, Bian J. Cardioprotection induced by Na+/K+-ATPase activation involves extracellular signal-regulated kinase 1/2 and phosphoinositide 3-kinase/Akt pathway. Cardiovascular Research. 2011;89(1):51–59. doi: 10.1093/cvr/cvq263. [DOI] [PubMed] [Google Scholar]
- 26.Guo H, Guo F, Zhang L, et al. Enhancement of Na/K pump activity by chronic intermittent hypobaric hypoxia protected against reperfusion injury. American Journal of Physiology. 2011;300(6):H2280–H2287. doi: 10.1152/ajpheart.01164.2010. [DOI] [PubMed] [Google Scholar]
- 27.Khalil PN, Neuhof C, Huss R, et al. Calpain inhibition reduces infarct size and improves global hemodynamics and left ventricular contractility in a porcine myocardial ischemia/reperfusion model. European Journal of Pharmacology. 2005;528(1–3):124–131. doi: 10.1016/j.ejphar.2005.10.032. [DOI] [PubMed] [Google Scholar]
- 28.Haddock PS, Shattock MJ, Hearse DJ. Modulation of cardiac Na+-K+ pump current: role of protein and nonprotein sulfhydryl redox status. American Journal of Physiology. 1995;269(1, part 2):H297–H307. doi: 10.1152/ajpheart.1995.269.1.H297. [DOI] [PubMed] [Google Scholar]
- 29.Figtree GA, Karimi GK, Liu CC, Rasmussen HH. Oxidative regulation of the Na+-K+ pump in the cardiovascular system. Free Radical Biology and Medicine. 2012;53(12):2263–2268. doi: 10.1016/j.freeradbiomed.2012.10.539. [DOI] [PubMed] [Google Scholar]
- 30.Severs NJ, Bruce AF, Dupont E, Rothery S. Remodelling of gap junctions and connexin expression in diseased myocardium. Cardiovascular Research. 2008;80(1):9–19. doi: 10.1093/cvr/cvn133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hervé JC, Derangeon M. Gap-junction-mediated cell-to-cell communication. Cell and Tissue Research. 2013;352(1):21–31. doi: 10.1007/s00441-012-1485-6. [DOI] [PubMed] [Google Scholar]
- 32.Smyth JW, Hong T, Gao D, et al. Limited forward trafficking of connexin 43 reduces cell-cell coupling in stressed human and mouse myocardium. The Journal of Clinical Investigation. 2010;120(1):266–279. doi: 10.1172/JCI39740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Janczarska K, Kieć-Wilk B, Leszczyńska-Gołabek I, Malczewska-Malec M, Bodzioch M. The role of connexin 43 in preconditioning. Impact on mitochondrial function. Kardiologia Polska. 2010;68(1):91–96. [PubMed] [Google Scholar]
- 34.Papp R, Gönczi M, Kovács M, Seprényi G, Végh A. Gap junctional uncoupling plays a trigger role in the antiarrhythmic effect of ischaemic preconditioning. Cardiovascular Research. 2007;74(3):396–405. doi: 10.1016/j.cardiores.2007.02.021. [DOI] [PubMed] [Google Scholar]
- 35.Figueroa XF, Duling BR. Gap junctions in the control of vascular function. Antioxidants and Redox Signaling. 2009;11(2):251–266. doi: 10.1089/ars.2008.2117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Adesse D, Garzoni LR, Huang H, Tanowitz HB, Meirelles MD, Spray DC. Trypanosoma cruzi induces changes in cardiac connexin43 expression. Microbes and Infection. 2008;10(1):21–28. doi: 10.1016/j.micinf.2007.09.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Cabo C, Yao J, Boyden PA, et al. Heterogeneous gap junction remodeling in reentrant circuits in the epicardial border zone of the healing canine infarct. Cardiovascular Research. 2006;72(2):241–249. doi: 10.1016/j.cardiores.2006.07.005. [DOI] [PubMed] [Google Scholar]
- 38.Danik SB, Rosner G, Lader J, Gutstein DE, Fishman GI, Morley GE. Electrical remodeling contributes to complex tachyarrhythmias in connexin43-deficient mouse hearts. The FASEB Journal. 2008;22(4):1204–1212. doi: 10.1096/fj.07-8974com. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Fontes MSC, van Veen TAB, de Bakker JMT, van Rijen HVM. Functional consequences of abnormal Cx43 expression in the heart. Biochimica et Biophysica Acta. 2012;1818(8):2020–2029. doi: 10.1016/j.bbamem.2011.07.039. [DOI] [PubMed] [Google Scholar]
- 40.Penna C, Perrelli M, Raimondo S, et al. Postconditioning induces an anti-apoptotic effect and preserves mitochondrial integrity in isolated rat hearts. Biochimica et Biophysica Acta. 2009;1787(7):794–801. doi: 10.1016/j.bbabio.2009.03.013. [DOI] [PubMed] [Google Scholar]
- 41.Harris AL. Connexin channel permeability to cytoplasmic molecules. Progress in Biophysics and Molecular Biology. 2007;94(1-2):120–143. doi: 10.1016/j.pbiomolbio.2007.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Miura T, Ohnuma Y, Kuno A, et al. Protective role of gap junctions in preconditioning against myocardial infarction. American Journal of Physiology. 2004;286(1):H214–H221. doi: 10.1152/ajpheart.00441.2003. [DOI] [PubMed] [Google Scholar]
- 43.Scemes E, Spray DC, Meda P. Connexins, pannexins, innexins: novel roles of ‘hemi-channels’. Pflugers Archiv European Journal of Physiology. 2009;457(6):1207–1226. doi: 10.1007/s00424-008-0591-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.García-Dorado D, Rodríguez-Sinovas A, Ruiz-Meana M. Gap junction-mediated spread of cell injury and death during myocardial ischemia-reperfusion. Cardiovascular Research. 2004;61(3):386–401. doi: 10.1016/j.cardiores.2003.11.039. [DOI] [PubMed] [Google Scholar]
- 45.Kanno S, Kovacs A, Yamada KA, Saffitz JE. Connexin43 as a determinant of myocardial infarct size following coronary occlusion in mice. Journal of the American College of Cardiology. 2003;41(4):681–686. doi: 10.1016/s0735-1097(02)02893-0. [DOI] [PubMed] [Google Scholar]
- 46.Johansen D, Sanden E, Hagve M, Chu X, Sundset R, Ytrehus K. Heptanol triggers cardioprotection via mitochondrial mechanisms and mitochondrial potassium channel opening in rat hearts. Acta Physiologica. 2011;201(4):435–444. doi: 10.1111/j.1748-1716.2010.02221.x. [DOI] [PubMed] [Google Scholar]
- 47.Ye Y, Qian J, Castillo AC, et al. Phosphodiesterase-3 inhibition augments the myocardial infarct size-limiting effects of exenatide in mice with type 2 diabetes. American Journal of Physiology. 2013;304(1):H131–H141. doi: 10.1152/ajpheart.00609.2012. [DOI] [PubMed] [Google Scholar]
- 48.Chen Z, Wu Z, Huang C, et al. Effect of lipoxin A4 on myocardial ischemia reperfusion injury following cardiac arrest in a rabbit model. Inflammation. 2013;36(2):468–475. doi: 10.1007/s10753-012-9567-x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure 1: Chemical structure of LXA4. Lipid-derived lipoxins are produced at the site of vascular and mucosal inflammation where they down-regulate polymorphonuclear leukocyte recruitment and function. 5(S),6(R),15(R)-LipoxinA4 (5(S),6(R),15(R)-LXA4) is derived from the aspirin- triggered formation of 15(R)-HETE from arachidonic acid. Formula Weight: 352.5.
According to DNA sequence of various indexes, Primer 3.0 software was used to design Na+-K+-ATPase and Cx43 primer. GAPDH is control gene. Primer sequences of Na+-K+-ATPase and Cx43 were as shown in Supplementary Table 1.