

Abstract
Osteoarthritis (OA) is characterized by a loss of joint mobility and pain resulting from progressive destruction and loss of articular cartilage secondary to chondrocyte death and/ or senescence. Certain stimuli including nitric oxide (NO) and the pro-inflammatory cytokine tumor necrosis factor α (TNF-α have been implicated in this chondrocyte death and the subsequent accelerated damage to cartilage. In this study, we demonstrate that a corticotrophin releasing factor (CRF) family peptide, urocortin (Ucn), is produced by a human chondrocyte cell line, C-20/A4, and acts both as an endogenous survival signal and as a cytoprotective agent reducing the induction of apoptosis by NO but not TNF-α when added exogenously. Furthermore, treatment with the NO donor S-nitroso-N-acetyl-𝒟-ℒ-penicillamine upregulates chondrocyte Ucn expression, whereas treatment with TNF-α does not. The chondroprotective effects of Ucn are abolished by both specific ligand depletion (with an anti-Ucn antibody) and by CRF receptor blockade with the pan-CRFR antagonist α-helical CRH(9-41). CRFR expression was confirmed by reverse transcription-PCR with subsequent amplicon sequence analysis and demonstrates that C-20/A4 cells express both CRFR1 and CRFR2, specifically CRFR1α and CRFR2β. Protein expression of these receptors was confirmed by western blotting. The presence of both Ucn and its receptors in these cells, coupled with the induction of Ucn by NO, suggests the existence of an endogenous autocrine/paracrine chondroprotective mechanism against stimuli inducing chondrocyte apoptosis via the intrinsic/mitochondrial pathway.
Keywords: chondrocyte, apoptosis, nitric oxide, urocortin
Articular cartilage of the joints forms a smooth, shock-absorbing surface covering opposing subchondral bone and is essential for the normal function of the joint.1 This connective tissue is produced by only one type of specialized cell, the chondrocyte. The extracellular matrix produced and recycled by these cells is rich in collagen fibers embedded in proteoglycans and provides high mechanical strength. These cells are therefore crucial for normal joint integrity.2, 3 During the aging process and in pathologies of the aged, such as osteoarthritis (OA), there is a loss of joint mobility and pain especially in the knees, hips, hands and vertebrae. In OA, these changes have been attributed to a reduction in the number of active chondrocytes in articular cartilage, occurring primarily in the superficial zone of the cartilage without distinct changes in the middle or deep zones4 and the severity of cartilage damage correlates very closely with the number of remaining active cells.4, 5 In these conditions, chondrocyte death appears to be essentially apoptotic, but with subtle differences, for example, membrane ‘blebbing' is rarely seen. This has led to the phrase ‘chondroptosis' being suggested to describe the unique form of apoptosis/programmed cell death observed in these cells.6
Although it has now become established that chondrocyte cell death contributes to progressive articular cartilage damage and a loss of joint function, the stimuli involved are unclear. Several locally occurring factors have, however, been implicated including nitric oxide (NO),7 oxygen-free radicals,7 tumor necrosis factor α (TNF-α),8 interleukin 1β (IL-1β)9 and Fas ligand.10 The effects of these stimuli on chondrocytes have been shown to contribute to accelerated damage of articular cartilage in vivo.3 Of these, NO and the pro-inflammatory cytokines TNF-α and IL-1β are likely to be significant contributors to the apparent changes in chondrocyte function and viability observed during cartilage degradation. NO, in particular, is present in significant quantities within OA joints in vivo11 and elevated levels of NO in the cartilage and synovial fluid may be a result of excess production in response to a variety of mechanical and chemical stresses. These increased levels of NO may be a result of the action of a neuronal NO synthetase-like enzyme, which has been detected in OA cartilage but not in normal articular cartilage.12 NO may also mediate cartilage matrix loss by upregulation of matrix metalloproteinases (MMP-9 and MMP-3) causing further degradation of the cartilage matrix.13 It has also been shown in vitro that the addition of a NOS inhibitor can reduce the level of observed apoptotic cell death.14 Certain experimental models of OA in various species also indicate a correlation between the level of NO production and prevalence of apoptotic cells in cartilage tissue.15 It is clear that imbalances in cartilage homeostasis observed in both OA and the aging process need to be redressed and that key to this is the protection of chondrocytes from apoptotic death.
Recently, the neuropeptide urocortin (Ucn) has been found to be elevated in the synovial fluid of patients with rheumatoid arthritis.16 It also reduces inflammation and bone erosion in a mouse model of the disease.17 Beyond this, little is known of the role of Ucn in the pathobiology of OA. This small peptide and its paralogs UcnII (human stresscopin-related peptide) and UcnIII (human stresscopin) are members of the corticotrophin releasing factor (CRF) family. These peptides have been demonstrated to have pleiotrophic effects on numerous cell systems including anti-apoptotic actions in heart18 and the regulation of skeletal osteoclast differentiation and resorption,19 acting in an autocrine or paracrine manner.20 These agonists bind to two classes of receptor CRF receptor 1 (CRFR1) and CRFR2 (which are expressed as multiple isoforms due to alternate RNA splicing21). Signaling complexity is increased further by receptor promiscuity, enabling the activation of different G proteins by the same receptor subtype.22 Studies have demonstrated that Ucn can bind to both CRFR1 and CRFR2, whereas Ucn II and Ucn III bind exclusively to CRFR2.23 The system is completed by a high-affinity binding protein (CRF-BP), which acts as a decoy receptor and regulates functional peptide availability.24
Here we report that Ucn is expressed in the chondrocyte cell line C-20/A4, and that this cell line expresses both CRFR1 and R2 receptor subtypes. Furthermore, Ucn is essential for C-20/A4 cell survival, and is also a potent chondroprotective agent against cell death induced by pro-apoptotic stimuli.
Results
The effects of pro-apoptotic stimuli on C-20/A4 chondrocytes
C-20/A4 cell death was analyzed in the presence of ascending concentrations of the pro-apoptotic stimuli S-nitroso-N-acetyl-𝒟-ℒ-penicillamine (SNAP), a nitric oxide (NO) donor and TNF-α. C-20/A4 cultures were analyzed for apoptotic cell death by Annexin V and terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) assays with the use of cytoplasmic lactate dehydrogenase (LDH) release as a measure of necrosis. The effects of SNAP treatment are shown in Figure 1a and TNF-α treatment in Figure 1b.
Figure 1.

Apoptotic and necrotic chondrocyte cell death assessed as the percentage of Annexin V- and TUNEL-positive cells (apoptosis) and cellular LDH release (necrosis) following treatment of C-20/A4 cells with increasing concentrations of SNAP (a) and TNF-α (b). *P<0.05. **P<0.01 compared with control (untreated)
SNAP treatment exhibited a dose-dependent increase in apoptotic cell death with no significant increase in necrosis (P>0.05 versus control) at all concentrations tested. SNAP (0.1 mM) showed minimal apoptotic death (15% Annexin V- and 8% TUNEL-positive cells) but as the dose of SNAP increased, apoptotic levels increased with 23% Annexin V- and 18% TUNEL-positive cells (P<0.01 versus control) at 1 mM and 35% Annexin V and 33% TUNEL-positive cells (P<0.01 versus control) at 10 mM. Based on these data a concentration of 1 mM SNAP was used for all subsequent experiments.
TNF-α treatment similarly showed a dose-dependent increase in apoptotic cell death, again with no significant increase in necrosis (P>0.05 versus control) at all concentrations tested. Minimal apoptotic cell death was apparent at concentrations up to 40 pg/ml (P>0.05 versus control) but apoptotic cell death was observed at concentrations of 60 pg/ml and above with a significant (P<0.01 versus control) increase to 24% Annexin V-positive cells. A small increase in TUNEL-positive cells was evident, but this was not statistically significant (P>0.05 versus control). 80 pg/ml TNF-α treatment resulted in a significant (P<0.001 versus control) increase in both Annexin V and TUNEL positivity (29% and 33%, respectively). Based on these data a concentration of 70 pg/ml TNF-α was used for all subsequent experiments.
The endogenous expression of Ucn and its receptors by C-20/A4 chondrocytes
Optimum annealing temperature and amplification cycle number (linear part of the amplification curve) were determined for reverse transcription-PCR (RT-PCR) of Ucn and glyceraldehyde 3-phosphate dehydrogenase (GAPDH) in C-20/A4 chondrocytes (full data not shown, optimum conditions described in Table 1). Ucn and GAPDH expression were analyzed by agarose gel electrophoresis (Figure 2a) and quantified by densitometric analysis of the resulting PCR amplicons with Ucn expression ‘normalized' to GAPDH expression (Figure 2b). Basal expression of Ucn and GAPDH was determined in control/untreated cells and the ratio of Ucn/GAPDH expression compared with that in cells treated with SNAP and TNF-α. Figures 2a and b clearly indicate that SNAP treatment results in an increased expression of Ucn (P<0.01 versus control), whereas TNF-α treatment does not appreciably induce Ucn expression (P>0.05 versus control).
Table 1. Sequences of primers used for PCR amplification of cDNA and predicted amplicon sizes.
| Gene | Primer sequence (5′→3′) | Expected product size (bp) | PCR protocol | Cycles |
|---|---|---|---|---|
| UCN | CAGGCGAGCGGCCGCG | 146 | 94°C/57°C/72°C | 33 |
| (Human) | CTTGCCCACCGAGTCGAAT | |||
| GAPDH | CCTGCTTCACCACCTTCTTG | 437 | 94°C/58°C/72°C | 30 |
| (Human) | CATCATCTCTGCCCCCTCTG | |||
| CRH-R1 | ACAAACAATGGCTACCGGGA | 280 (R1α) | 94°C/58°C/72°C | 45 |
| (Human) | GGACCACGAACCAGGTGGCG | 367 (R1β) | ||
| CRH-R2 | AGCCCATTTTGGATGACAAG | 180 | 94°C/58°C/72°C | 45 |
| (Human) | AGGTGGTGATGAGGTTCCAG | |||
| CRH-R2α | TCCACAGCCTGCTGGAGGCC | 239 | 94°C/58°C/72°C | 45 |
| (Human) | CTCCAAGCATTCTCGATAGGC | |||
| CRH-R2β | CCTCCTCTACGTCCCACACC | 310 | 94°C/58°C/72°C | 45 |
| (Human) | CTCCAAGCATTCTCGATAGGC | |||
| CRH-R2γ | TGGGAAGAGAGCCTTGGCC | 212 | 94°C/58°C/72°C | 45 |
| (Human) | CTCCAAGCATTCTCGATAGGC |
Figure 2.

(a) RT-PCR analysis of Ucn (upper panel) and GAPDH (lower panel) expression in control (untreated) C-20/A4 chondrocytes and cells treated with 1 mM SNAP and 70 pg/ml TNF-α. Ucn and GAPDH amplicon sizes are indicated. (b) Ratio of Ucn/GAPDH expression in control (untreated) C-20/A4 chondrocytes and cells treated with 1 mM SNAP and 70 pg/ml TNF-α as determined by densitometric analysis of PCR products shown in a. (c) RT-PCR analysis of CRFR subtype expression in C-20/A4 chondrocytes. Lanes 1 and 2=CRFR1 product, lane 3=CRFR1-negative control, lanes 5 and 6=CRFR2 product, lane 7=CRFR2-negative control. CRFR amplicon sizes are indicated. (d) RT-PCR analysis of CRFR2 splice variant expression in C-20/A4 chondrocytes. Lanes 1 and 2=CRFR2α PCR – no product, lane 3=CRFR2α-negative control, lanes 5 and 6=CRFR2β PCR product, lane 7=CRFR2β-negative control, lanes 9 and 10=CRFR2γ PCR – no product, lane 11=CRFR2γ-negative control. CRFRβ amplicon size is indicated
Again, following the identification of optimum annealing temperatures and amplification cycle number for RT-PCR of the CRF receptors in C-20/A4 chondrocytes (full data not shown, optimum conditions described in Table 1), we have identified the expression of both CRFR1 and CRFR2 receptor subtypes in these cells. Specifically, we demonstrate the expression of CRFR1α and CRFR2β subtypes, and the identities of the PCR products were confirmed by direct sequencing. Protein expression of both classes of receptor was confirmed by western blotting with antibodies specific for CRFR1 and CRFR2 (Figure 3).
Figure 3.
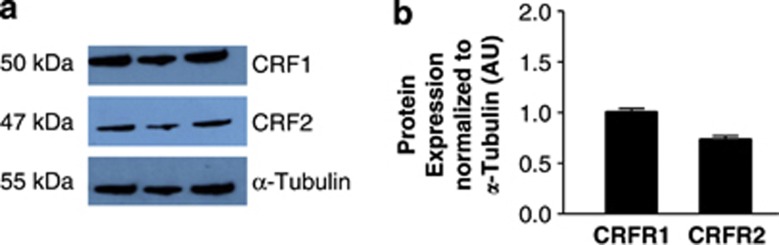
(a) Western-blot analysis of CRFR expression. CRFR1 and CRFR2 protein levels were determined using rabbit anti-CRFR1 and rabbit anti-CRFR2 monoclonal antibodies with a mouse anti-α-tubulin monoclonal antibody used as a loading control. Bands with sizes corresponding to 50 kDa (CRFR1), 47 kDa (CRFR2) and 55 kDa (α-tubulin) were detected. (b) Observed bands were quantified by densitometry and CRFR expression normalized to that of α-tubulin. Data are presented as mean±S.E.M. of three independent experiments
Endogenous Ucn production is required for chondrocyte survival
In order to assess the importance of the endogenous, inducible Ucn expression demonstrated above, we performed receptor blocking and ligand depletion studies and determined the effects of these actions on C-20/A4 chondrocyte death in the absence of pro-apoptotic stimuli (Figure 4). Treatment with the non-selective CRFR antagonist α-helical CRH(9-41) alone (10−8M) resulted in a significant (P<0.01 versus control) increase in apoptotic cell death (23% Annexin V, 23% TUNEL) with no significant increase in necrotic cell death as determined by LDH release. Ligand depletion studies with a specific anti-human Ucn antibody resulted in even greater increases in apoptotic cell death (37% Annexin V, 33% TUNEL; P<0.01 versus control), whereas treatment with an isotype control antibody directed against human albumin resulted in no significant increase in apoptosis as compared with the control. There was no significant increase in necrotic cell death following treatment with either antibody.
Figure 4.

Cytoprotective effects of endogenous Ucn. Apoptotic and necrotic chondrocyte cell death assessed as the percentage of Annexin V- and TUNEL-positive cells (apoptosis) and cellular LDH release (necrosis) following the treatment of C-20/A4 cells, with the CRH receptor antagonist α-helical CRH, anti-UCN antibody and anti-albumin antibody, **P<0.01 compared with control
The addition of exogenous Ucn is chondroprotective against pro-apoptotic stimuli
Using the optimum concentrations of SNAP (1 mM) and TNF-α (70 pg/ml) as determined in Figure 1, C-20/A4 chondrocytes were treated with these stimuli, in conjunction with the addition of exogenous Ucn (10−8M). Figure 5 demonstrates that the addition of concomitant Ucn ameliorates the apoptotic cell death induced by both SNAP and TNF-α but appears more effective against that induced by SNAP. Treatment with both Ucn and SNAP resulted in a significant reduction of apoptosis (14% Annexin V and 10% TUNEL) when compared with SNAP alone (23% Annexin V and 19% TUNEL; P<0.01). Treatment with Ucn and TNF-α resulted in a significant reduction in TUNEL positivity when compared with TNF-α alone (from 19% to 12%, P<0.05) but no significant reduction in Annexin V-positive cells. In both the cases, the chondroprotective effect of Ucn is blocked by the addition of α-helical CRH(9-41) resulting in levels of cell death that are not significantly different to treatment with the pro-apoptotic stimuli alone.
Figure 5.

Cytoprotective effects of exogenous Ucn. Apoptotic and necrotic chondrocyte cell death assessed as the percentage of Annexin V- and TUNEL-positive cells (apoptosis) and cellular LDH release (necrosis) following treatment of C-20/A4 cells with Ucn (U) administered concurrently with SNAP (S) or TNF-α (T) in the presence or absence of α-helical CRH (A) *P<0.05, **P<0.01 compared with control (C).
Discussion
Given that chondrocyte apoptosis or ‘chondroptosis' is thought to be one of the major mechanisms involved in the pathogenesis of OA, and that we have previously shown Ucn to be cytoprotective in cardiac myocytes25 and neuronal cells,26 we sought to investigate a potential role for this peptide in chondrocyte responses to pro-apoptotic stimuli implicated in OA.
NO and the pro-inflammatory cytokine TNF-α have both been detected at elevated levels in osteoarthritic cartilage and both been implicated as possible contributors to chondrocyte death and cartilage damage.11, 8 TNF-α is also involved in the amplification of the local inflammatory response within the joint.27 Initial data gathered in this study (Figures 1a and b) confirm a dose-dependent increase in apoptotic cell death following treatment of C-20/A4 cells with these stimuli as determined by both Annexin V binding and TUNEL assays. No significant increase in necrotic cell death (as determined by cytoplasmic LDH release) was noted at all concentrations tested. Concentrations of 1 mM SNAP (which releases approximately 60 nM of free NO28) and 70 pg/ml TNF-α were selected for all further studies as these concentrations induced a consistent and easily quantifiable level of chondrocyte apoptosis with minimal necrosis. Furthermore, these concentrations are similar to those found within OA joints and similar to those used by others in studies with primary human chondrocytes.11, 29, 30
In order to establish the potential of the Ucn system as a chondroprotective mechanism, expression of Ucn and CRFR mRNA in C-20/A4 cells was first investigated in untreated, control cells where RT-PCR demonstrated constitutive expression of both Ucn and CRFRs (Figure 2a, lane 1 and Figure 2c respectively) with the identity of the resulting amplicons confirmed by sequence analysis. Not only is the demonstration of Ucn expression in chondrocytes novel, these studies and the accompanying western blotting studies (Figure 3) also intriguingly demonstrated the expression of both CRFR1 and CRFR2 receptor subtypes in these cells (CRFR1α and CRFR2β), a phenomenon that, as far as we are aware, has not previously been reported. The identity of these receptor subtype variants further confirms that both subtypes are represented by an active splice-variant with CRFR1α being the only active CRFR1 isoform31 and CRFR2β being one of the three active forms of CRFR2 (CRFR2α, CRFR2β and CRFR2γ).32 Furthermore, although CRFR2α is the predominant CRFR2 isoform expressed in the human periphery, it has been shown through adenylate cyclase activation studies that Ucn and the other CRF-related peptides may preferentially activate the larger CRFR2β isoform over CRFR2α and CRFR2γ,33 despite there being no pharmacological differences in ligand binding.32 This may be explained by the fact that the CRFR2α and CRFR2γ isoforms lack the N-terminal domain encoded by exons 1 and 2 of the CRFR2 gene, an extracellular region thought to be important in ligand recognition and interaction.33 Having confirmed the expression of both Ucn and its receptors, we then studied the effects of pro-apoptotic insults on cellular expression of these components of the Ucn system. Although there was no significant variation in receptor expression following treatment with pro-apoptotic stimuli (data not shown), ligand expression was, however, significantly induced by SNAP, but not TNF-α treatment (Figure 2a, lanes 3 and 5, respectively, and Figure 2b). Following normalization of the PCR data by expressing the data as a ratio of Ucn/GAPDH densitometry readings, it becomes readily apparent that Ucn expression increases nearly twofold following exposure to SNAP (P<0.01 versus control) yet there is no significant change following TNF-α exposure (Figure 2b). These data would suggest that the Ucn system is therefore likely to represent a constitutive chondroprotective mechanism but moreover that it may represent a mechanism specifically targeted toward protection against certain stimuli.
Following the demonstration of Ucn mRNA production by C-20/A4 cells the presence of functional endogenous Ucn production was confirmed by CRFR receptor blockade (by α-helical CRH(9-41), a competitive inhibitor of both CRFR1 and CRFR2 receptor subclasses) and the specific depletion of Ucn ligand released into the culture medium (using an Ucn-specific antibody). These experiments not only demonstrate the presence of functional Ucn peptide in these cells, but also suggest that the Ucn system represents an essential endogenous mediator of chondrocyte survival as both CRFR blockade and selective ligand depletion result in high levels of chondrocyte death, even in the absence of pro-apoptotic stimuli. The addition of isotype control antibody (anti-human albumin) did not result in any significant increase in chondrocyte apoptosis when compared with control cells (Figure 4).
In order to further characterize Ucn-mediated chondroprotection, and its possible selective nature, cytoprotection experiments were performed with the addition of exogenous Ucn, concomitant with SNAP and TNF-α treatment. As shown in Figure 5, co-treatment with SNAP and exogenous Ucn results in a significant decrease in the level of C-20/A4 apoptosis induced by SNAP treatment alone, protection that is again reversed by the addition of α-helical CRH(9-41). When added in conjunction with TNF-α, however, the chondroprotective effects of Ucn are much less evident. The TUNEL assay data reveal a small reduction in cell death but the Annexin V assay, however, does not show any significant difference when compared with TNF-α treatment alone. Taken together, these data would suggest that Ucn appears to principally confer cytoprotection against pro-apototic stimuli that induce cell death via the intrinsic/mitochondrial apoptotic pathway (e.g., NO), but is less effective against stimuli acting through the extrinsic/death receptor-mediated pathway (e.g., TNF-α). These findings are in agreement with cardiac myocyte-derived data where Ucn was shown to prevent mitochondrial damage and inhibit lipid peroxidation resulting from oxidative stress.34, 35, 36
Using a stable, well-established chondrocyte cell line, this study has demonstrated critical roles for Ucn as both an essential chondrocyte survival signal and as a chondroprotective agent in the presence of pro-apoptotic stimuli. As with all studies involving cell lines, these observations should, and will be, confirmed in primary chondrocytes but these two characteristics ultimately represent a putative role for the Ucn system in preserving cartilage tissue. Furthermore, when coupled with our recently published data demonstrating a potent anti-resorbtive role for Ucn in osteoclasts,19 a wider role in bone metabolism and maintenance is suggested. Together, these data would suggest that Ucn, by preserving the ability to lay down new cartilage as well as reducing bone resorption, may have therapeutic potential in both osteoporosis and OA.
Materials and Methods
Cell culture
In vitro experiments were performed with the C-20/A4 human chondrocyte cell line37 cultured in a medium consisting of Dulbecco's modified Eagle's medium containing 1 g/l glucose (Lonza, Wokingham, UK), supplemented with 10% (v/v) fetal calf serum (FCS; Labtech International, Uckfield, UK), 1% (v/v) ℒ-glutamine and 1% (v/v) penicillin/streptomycin (Lonza) at 37 °C, 5% CO2. When confluent, cells were passaged by treatment with Ca2+ and Mg2+-free phosphate-buffered saline (Lonza), prewarmed to 37 °C and used as required. Following passage, the chondrocyte cell suspension (1 × 106 cells/ml) was transferred to six-well tissue culture plates and incubated for 24–48 h in the above medium to allow cells to reach ∼80% confluency. The cells were then ‘serum starved' for 24 h by replacing the normal, 10% FCS growth medium with medium containing 1% FCS (all other components unchanged). The cultured cells were then re-supplied with fresh 1% FCS medium (control) or treated with fresh 1% medium containing the stimuli/reagents detailed below for 6 h, after which levels of cell death were determined.
Six-well plate cultures were treated with individual or combination treatments of: the NO donor SNAP (Calbiochem, Nottingham, UK), TNF-α (R&D Systems, Abingdon, UK), the CRH antagonist α helical CRH(9-41) (10−8 M; Sigma-Aldrich, Gillingham, UK), rabbit anti-human Ucn antibody (100 μg/ml) and an isotype control (anti-human albumin; 100 μg/ml; both Sigma-Aldrich) and the level of chondrocyte cell death compared.
Measurement of cell death
Control and treated C-20/A4 cultures were assessed for both apoptotic and necrotic cell death. Apoptosis was assessed by fluorescence microscopy by Annexin V binding (TACS Annexin V-FITC kit, R&D systems) and by terminal deoxynucleotidyl transferase-mediated dUTP nick end labeling (TUNEL; in situ cell death detection kit, Roche diagnostics, Lewes, UK). Necrotic cell death was assessed by cytoplasmic LDH release (TOX 7 In Vitro Toxicology assay kit, Sigma, Poole, UK). All assays were performed according to the manufacturer's standard protocol.
RNA isolation and RT-PCR
Total RNA was isolated using Qiagen RNeasy Plus micro kit with gDNA eliminator columns to prevent genomic DNA contamination (Qiagen, Crawley, UK) according to the manufacturer's standard protocol. RNA purity and concentration were determined spectrophotometrically at 260 and 280 nm in an Eppendorf Biophotometer (Eppendorf, Cambridge UK) and RNA integrity verified by 1% agarose gel electrophoresis in tris(hydroxymethyl)aminomethane (Tris)-acetate-EDTA buffer followed by ethidium bromide (200 ng/ml) staining and visualization on a UV transilluminator.
cDNA was synthesized from 2 μg of each RNA sample using the Omniscipt reverse transcriptase kit in the presence of an RNAse inhibitor, oligo dt primer and mixed dNTPs (all from Qiagen). RNA and oligo dt mixes were initially heated to 70 °C for 10 min (to denature secondary RNA structures) after which the other components were added and the final mix incubated at 37 °C for 1 h.
PCR was performed to assess the expression of Ucn system components (Ucn, CRF-BP and CRFR) in C-20/A4 chondrocyte cells, using Qiagen Taq PCR Master Mix (Qiagen). The target sequences were amplified using sequence-specific oligonucleotide primers designed to amplify the transcript of interest along with GAPDH as an internal reference. Primer sequences and PCR conditions are given in Table 1. Temperature gradient PCR was performed on an Eppendorf Mastercycler gradient PCR machine (Eppendorf, Cambridge, UK) to determine optimum annealing temperatures for each of the primer pairs and the same machine used for all subsequent amplifications. Products were visualized on 2% (w/v) agarose gel electrophoresis in Tris-acetate-EDTA buffer followed by ethidium bromide (200 ng/ml) staining and visualization on a UV transilluminator. Where appropriate, densitometric analysis was employed to qualify gene expression relevant to the GAPDH housekeeping gene. Amplicon identity was confirmed by sequence analysis performed by GACT Biotech Ltd. (London, UK).
Western blotting
C-20/A4 expression of CRFR1 and CRFR2 receptors was determined by western blotting as previously described.38, 39 Following electrophoresis in a 10% SDS-polyacrylamide gel, proteins were transferred onto nylon membrane by electroblotting, blocked for 1 h in 5% non-fat milk solution in Tris-hydrochloric acid-buffered saline, pH 7.5 (TBS) containing 0.1% (v/v) Tween-20 and then incubated with either specific anti-CRFR1 or anti-CRFR2 (1 : 1000 dilution SAB4500465 and SAB4500466, Sigma-Aldrich, Dorset, UK) rabbit antibodies in blocking solution. Blots were washed in TBS before the addition of a secondary goat anti-rabbit HRP-conjugated antibody (1 : 2000 dilution) and specific antibody binding was detected by enhanced chemiluminescence (Pierce Biotechnology, Rockford, IL, USA). Following detection, bound antibodies were removed by incubating the membranes in 100 mM glycine-hydrochloric acid, pH 2.5, for 30 min and the blot re-probed to detect α-tubulin as described previously.38, 39 Densitometry analysis was performed using Image J software (NIH, Bethesda, MD, USA).
Statistical analysis
Data values from the figures are expressed as mean±S.D. of n-independent experiments. All experiments were repeated at least nine times with the exception of the western blots, which were repeated three times. All data were subjected to ANOVA followed by Bonferroni's correction for post hoc t-tests where appropriate. Probabilities of P≤0.05 were considered statistically significant. Statistical tests were performed using Microsoft Excel or Prism GraphPad software as applicable.
Glossary
- NO
nitric oxide
- OA
osteoarthritis
- TNF-α
tumor necrosis factor-α
- CRF
corticotrophin releasing factor
- Ucn
urocortin
- SNAP
S-nitroso-N-Acetyl-𝒟,ℒ-penicillamine
- CRFR
corticotrophin releasing factor receptor
- RT-PCR
reverse transcription-PCR
- IL-1β
interleukin 1β
- nNOS
neuronal nitric oxide synthetase
- CRF-BP
corticotrophin releasing factor-binding protein
- TUNEL
terminal deoxynucleotidyl transferase dUTP nick end labeling
- LDH
lactate dehydrogenase
- GAPDH
glyceraldehyde 3-phosphate dehydrogenase
- DMEM
Dulbecco's modified Eagle's medium
- FCS
fetal calf serum
- Tris
tris(hydroxymethyl)aminomethane
The authors declare no conflict of interest.
Footnotes
Edited by A Stephanou
References
- Buckwalter JA, Mankin HJ. Articular cartilage: tissue design and chondrocyte-matrix interactions. Instr Course Lect. 1998;47:477–486. [PubMed] [Google Scholar]
- Muir H. The chondrocyte: architect of cartilage. Biomechanics, structure, function and molecular biology of cartilage matrix macromolecules. Bioesssay. 1995;17:1039–1048. doi: 10.1002/bies.950171208. [DOI] [PubMed] [Google Scholar]
- Buckwalter JA, Mankin HJ. Articular cartilage. J Bone Joint Surg Am. 1997;79A:600–632. [Google Scholar]
- Kobayashi K, Healey RM, Coutts RD, Amiel D. Changes in chondrocyte density with age in human knees. Trans Orth Res Soc. 2003;49:566. [Google Scholar]
- Loeser RF. Aging and osteoarthritis: the role of chondrocyte senescence and aging changes in the cartilage matrix. Osteoarthr Cartil. 2009;17:971–979. doi: 10.1016/j.joca.2009.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roach HI, Aigner T, Kouri JB. Chondroptosis: a variant of apoptotic cell death in chondrocytes. Apoptosis. 2004;15:365–773. doi: 10.1023/b:appt.0000025803.17498.26. [DOI] [PubMed] [Google Scholar]
- Blanco FJ, Ochs RL, Schwartz H, Lotz M. Chondrocyte apoptosis induced by nitric oxide. Am J Pathol. 1995;146:75–85. [PMC free article] [PubMed] [Google Scholar]
- Fernandes JC, Martel-Pelletier J, Pelletier JP. The role of cytokines in osteoarthritis pathophysiology. Biorheology. 2002;39:237–246. [PubMed] [Google Scholar]
- Kim J, Xu M, Xo R, Mates A, Wilson GL, Pearsall AW, et al. Mitochondrial DNA damage is involved in apoptosis caused by pro-inflammatory cytokines in Human OA chondrocytes. Osteoarthr Cartil. 2010;18:424–432. doi: 10.1016/j.joca.2009.09.008. [DOI] [PubMed] [Google Scholar]
- Lotz M, Hashimoto S, Kühn K. Mechanisms of chondrocyte apoptosis. Osteoarthr Cartil. 1999;7:389–391. doi: 10.1053/joca.1998.0220. [DOI] [PubMed] [Google Scholar]
- Farrell AJ, Blake DR, Palmer RMJ, Moncada S. Increased concentrations of nitrite in synovial fluid and serum samples suggest increased nitric oxide synthesis in rheumatic diseases. Ann Rheum Dis. 1992;51:1219–1222. doi: 10.1136/ard.51.11.1219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amin AR, Di Cesare PE, Vyas P, Attur M, Tzeng E, Billiar TR, et al. The expression and regulation of nitric oxide synthase in human osteoarthritis-affected chondrocytes: evidence for up-regulated neuronal nitric oxide synthase. J Exp Med. 1995;182:2097–2102. doi: 10.1084/jem.182.6.2097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murrell GAC, Jang D, Williams RJ. Nitric Oxide activates metalloproteinase enzymes in articular cartilage. Biochem Biophys Res Commun. 1995;206:15–21. doi: 10.1006/bbrc.1995.1003. [DOI] [PubMed] [Google Scholar]
- Pelletier J, Jovanovic D, Fernandes JC, Manning P, Connor JR, Currie MG, et al. Reduction in the structural changes of experimental osteoarthritis by a nitric oxide inhibitor. Osteoarthr Cartil. 1999;7:416–418. doi: 10.1053/joca.1998.0229. [DOI] [PubMed] [Google Scholar]
- Allen RT, Robertson CM, Harwood FL, Sasho T, Williams SK, Pomerleau AC, et al. Characterization of mature versus aged rabbit articular cartilage: analysis of cell density, apoptosis-related gene expression and mechanisms controlling chondrocyte apoptosis. Osteoarthr Cartil. 2004;12:917–923. doi: 10.1016/j.joca.2004.08.003. [DOI] [PubMed] [Google Scholar]
- Kohno M, Kawahito Y, Tsubouchi Y, Hanshiramoto A, Yamada R, Inoue KI, et al. Urocortin expression in synovium of patients with rheumatoid arthritis and osteoarthritis: relation to inflammatory activity. J Clin Endocrinol Metab. 2001;86:4344–4352. doi: 10.1210/jcem.86.9.7827. [DOI] [PubMed] [Google Scholar]
- Gonzalez-Rey E, Chorny A, Varela N, O'Valle F, Delgado M. Therapeutic effect of Urocortin on collagen–induced Arthritis by down-regulation of Inflammatory and Th1 responses and induction of regulatory T cells. Arthritis Rheum. 2007;56:531–543. doi: 10.1002/art.22394. [DOI] [PubMed] [Google Scholar]
- Lawrence KM, Latchman DS. The Urocortins: mechanism of cardioprotection and therapeutic potential. Mini Rev Med Chem. 2006;6:1119–1126. doi: 10.2174/138955706778560111. [DOI] [PubMed] [Google Scholar]
- Combs CE, Fuller K, Kumar H, Albert AP, Pirianov G, McCormick J, et al. Urocortin is a novel regulator of osteoclast differentiation and function through inhibition of a canonical transient receptor potential 1-like cation channel. J Endocrinol. 2012;212:187–197. doi: 10.1530/JOE-11-0254. [DOI] [PubMed] [Google Scholar]
- Florio P, Rossi M, Sigurdarottir M, Ciarmela P, Luisi S, Vigano P, et al. Paracrine regulation of myometrial function: interaction between progesterone and corticotropin-releasing factor (CRF) and Activin A. Steroids. 2003;68:801–807. doi: 10.1016/s0039-128x(03)00137-5. [DOI] [PubMed] [Google Scholar]
- Perrin MH, Vale WW. Corticotropin releasing factor receptors and their ligand family. Ann NY Acad Sci. 1999;885:312–328. doi: 10.1111/j.1749-6632.1999.tb08687.x. [DOI] [PubMed] [Google Scholar]
- Grammatopoulos DK, Randeva HS, Levine JD, Katsanou ES, Hillhouse EW. Urocortin, but not corticotropin-releasing hormone (CRH), activates the mitogen-activated protein kinase signal transduction pathway in human pregnant myometrium: an affect mediated via R1α and R2β CRH receptor subtypes and stimulation of Gq-proteins. Mol Endocrinol. 2000;14:2076–2091. doi: 10.1210/mend.14.12.0574. [DOI] [PubMed] [Google Scholar]
- Fekete EM, Zorrilla EP. Physiology, pharmacology and therapeutic relevance of urocortins in mammals: Ancient CRF paralogs. Front Neuroendocrinol. 2007;28:1–27. doi: 10.1016/j.yfrne.2006.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seasholtz AF, Valverde RA, Denver RJ. Corticotropin-releasing hormone-binding protein: biochemistry and function from fishes to mammals. J Endocrinol. 2002;175:89–97. doi: 10.1677/joe.0.1750089. [DOI] [PubMed] [Google Scholar]
- Brar BK, Stephanou A, Okosi A, Lawrence KM, Knight RA, Marber MS, et al. CRH-like peptides protect cardiac myocytes from lethal ischaemic injury. Mol Cell Endocrinol. 1999;158:55–63. doi: 10.1016/s0303-7207(99)00183-5. [DOI] [PubMed] [Google Scholar]
- Abuirmeileh A, Lever R, Kingsbury AE, Lees AJ, Locke IC, Knight RA, et al. The corticotrophin releasing factor like peptide urocortin reverses key deficits in two rodent models of Parkinson's disease. Eur J Neurosci. 2007;26:417–423. doi: 10.1111/j.1460-9568.2007.05653.x. [DOI] [PubMed] [Google Scholar]
- Horton WE, Jr., Feng L, Adams C. Chondrocyte apoptosis in development, aging and disease. Matrix Biol. 1998;17:107–115. doi: 10.1016/s0945-053x(98)90024-5. [DOI] [PubMed] [Google Scholar]
- Matthews JR, Botting CH, Panico M, Morris HR, Hay RT. Inhibition of NF-kappaB DNA binding by nitric oxide. Nucleic Acids Res. 1996;24:2236–2242. doi: 10.1093/nar/24.12.2236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hulejová H, Barešová V, Klézl Z, Polanská M, Adam M, Šenolt L. Increased level of cytokines and matrix metalloproteinases in osteoarthritic subchondral bone. Cytokine. 2007;38:151–156. doi: 10.1016/j.cyto.2007.06.001. [DOI] [PubMed] [Google Scholar]
- Kühn K, Shikhman AR, Lotz M. Role of NO, reactive oxygen species and P38 MAP kinase in the regulation of human chondrocyte apoptosis. J Cell Physiol. 2003;197:379–387. doi: 10.1002/jcp.10372. [DOI] [PubMed] [Google Scholar]
- Zmijewski MA, Slominski AT. Emerging role of alternative splicing of CRF1 receptor in CRF signaling. Acta Biochem Pol. 2010;57:1–13. [PMC free article] [PubMed] [Google Scholar]
- Dautzenberg FM, Hauger RL. The CRF peptide family and their receptors: yet more partners discovered. Trends Pharmacol Sci. 2002;23:71–77. doi: 10.1016/s0165-6147(02)01946-6. [DOI] [PubMed] [Google Scholar]
- Grammatopoulos DK, Chrousos GP. Functional characteristics of CRH receptors and potential clinical applications of CRH-receptor antagonists. Trends Endocrin Met. 2002;13:436–444. doi: 10.1016/s1043-2760(02)00670-7. [DOI] [PubMed] [Google Scholar]
- Lawrence KM, Townsend PA, Davidson SM, Carroll CJ, Eaton S, Hubank M, et al. The cardioprotective effect of urocortin during ischaemia/ reperfusion involves the prevention of mitochondrial damage. Biochem Biophys Res Commun. 2004;321:479–486. doi: 10.1016/j.bbrc.2004.06.170. [DOI] [PubMed] [Google Scholar]
- Townsend PA, Davidson SM, Clarke SJ, Khaliulin I, Carroll CJ, Scarabelli TM, et al. Urocortin prevents mitochondrial permeability transition in response to reperfusion injury indirectly by reducing oxidative stress. Am J Physiol Heart Circ Physiol. 2007;293:928–938. doi: 10.1152/ajpheart.01135.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barry SP, Lawrence KM, McCormick J, Soond SM, Hubank M, Eaton S, et al. New targets of urocortin-mediated cardioprotection. J Mol Endocrinol. 2010;45:69–85. doi: 10.1677/JME-09-0148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldring MB, Birkhead JR, Suen LF, Yamin R. Use of immortalized human chondrocytes in studies of IL-1-mediated gene expression. Arthritis Rheum. 1993;36:S49. [Google Scholar]
- Getting SJ, Riffo-Vasquez Y, Pitchford S, Kaneva M, Grieco P, Page CP, et al. A role for MC3R in modulating lung inflammation. Pulm Pharmacol Ther. 2008;21:866–873. doi: 10.1016/j.pupt.2008.09.004. [DOI] [PubMed] [Google Scholar]
- Kaneva MK, Kerrigan MJ, Grieco P, Curley GP, Locke IC, Getting SJ. Chondroprotective and anti-inflammatory role of melanocortin peptides in TNF-α activated human C-20/A4 chondrocytes. Br J Pharmacol. 2012;167:67–79. doi: 10.1111/j.1476-5381.2012.01968.x. [DOI] [PMC free article] [PubMed] [Google Scholar]