

Abstract
Numerous studies have confirmed that cancer stem cells (CSCs) are more resistant to chemotherapy; however, there is a paucity of data exploring the effect of long-term drug treatment on the CSC sub-population. The purpose of this study was to investigate whether long-term doxorubicin treatment could expand the neuroblastoma cells with CSC characteristics and histone acetylation could affect stemness gene expression during the development of drug resistance. Using n-myc amplified SK-N-Be(2)C and non-n-myc amplified SK-N-SH human neuroblastoma cells, our laboratory generated doxorubicin-resistant cell lines in parallel over 1 year; one cell line intermittently treated with the histone deacetylase inhibitor (HDACi) vorinostat and the other without exposure to HDACi. Cells' sensitivity to chemotherapeutic drugs, the ability to form tumorspheres, and capacity for in vitro invasion were examined. Cell-surface markers and side populations (SPs) were analyzed using flow cytometry. Differentially expressed stemness genes were identified through whole genome analysis and confirmed with real-time PCR. Our results indicated that vorinostat increased the sensitivity of only SK-N-Be(2)C-resistant cells to chemotherapy, made cells lose the ability to form tumorspheres, and reduced in vitro invasion and the SP percentage. CD133 was not enriched in doxorubicin-resistant or vorinostat-treated doxorubicin-resistant cells. Nine stemness-linked genes (ABCB1, ABCC4, LMO2, SOX2, ERCC5, S100A10, IGFBP3, TCF3, and VIM) were downregulated in vorinostat-treated doxorubicin-resistant SK-N-Be(2)C cells relative to doxorubicin-resistant cells. A sub-population of cells with CSC characteristics is enriched during prolonged drug selection of n-myc amplified SK-N-Be(2)C neuroblastoma cells. Vorinostat treatment affects the reversal of drug resistance in SK-N-Be(2)C cells and may be associated with downregulation of stemness gene expression. This work may be valuable for clinicians to design treatment protocols specific for different neuroblastoma patients.
Keywords: neuroblastoma, cancer stem cell, drug resistance, histone deacetylase inhibitor
Neuroblastoma is a heterogeneous pediatric extra-cranial solid tumor, which arises from the neural crest elements of the sympathetic nervous system. It accounts for 15% of pediatric cancer deaths.1 The high mortality associated with high-risk neuroblastoma is partially attributable to therapy-resistant relapse, due to either inherited or acquired drug resistance allowing a subset of tumor cells to survive treatment. Cancer stem cell (CSC) theory posits that there is a small therapy-resistant sub-population of cells within a tumor that possess the capacity to initiate a primary tumor, bear an inherit resistance to chemotherapeutic drugs, and whose survival through treatment leads to persistent and recurrent disease.2, 3 The CSC hypothesis predicts that destruction of a tumor's CSC population is required for tumor elimination.4, 5 Novel therapies targeting CSCs may lead to effective treatment strategies that can overcome drug resistance. Therefore, understanding the role of CSCs in drug resistance and how to target CSCs may lead to critical therapeutic advances in cancer treatment.
The origin of CSCs is uncertain. CSCs have been isolated from bone marrow,6 primary tumor tissues,3, 7, 8, 9 and cultured cancer cell lines.10, 11 They can be identified through detection of specific cell-surface markers and measurement of stem cell-specific metabolic activities.12 CSCs have been identified in both hematopoietic cancers,13, 14 and, more recently, solid malignancies, including brain,15 breast,16 liver,17 prostate,18 lung,19 colon,8 and ovarian cancer.7 Numerous studies have confirmed that CSCs are more resistant to chemotherapy and radiotherapy.2, 3, 20, 21 CD133+ lung cancer cells displayed stem-like features and were spared by cisplatin treatment.21 CD133+ glioblastoma cells demonstrated resistance to temozolomide, carboplatin, paclitaxel, and etoposide compared with CD133− glioblastoma cells.3 There is a paucity of data exploring the effect of long-term drug treatment on the CSC sub-population. A recent study reported that putative breast CSCs were increased after prolonged continuous selection in doxorubicin.22 Another study on colorectal cancer demonstrated that drug resistance cells had a higher percentage of cells expressing CSC-surface markers.23 These data suggest that the CSC population may be expanded in drug-resistant tumors and strategies that can affect stemness gene expression may have an effect in reversing drug resistance.
Histone deacetylase inhibitors (HDACi) have been developed as sensitizers for chemotherapy or radiation therapy.24 They interfere with HDAC activity, inhibiting deacetylation of histone and non-histone proteins leading to varied modulation of gene expression.25 The effect of HDACi on stemness gene expression is varied. HDACi treatment of CD133− ovarian cells leads to increased CD133 expression, a CSC marker of ovarian cells, and promotes stem cell self-renewal.26 HDACi has also been shown to decrease proliferation potential and differentiation capability of human mesenchymal stem cells.27 In combination with imatinib mesylate, HDACi can induce apoptosis in quiescent myelogenous leukemia stem cells and inhibit CML progenitor proliferation.28
In the present study, we sought to determine whether neuroblastoma cells with CSC characteristics are enriched during long-term chemotherapeutic drug selection and whether, during the development of drug resistance, histone acetylation affects stemness gene expression and drug resistance. We found that there was a sub-population of cells with CSC characteristics that is enriched during prolonged drug selection for resistant neuroblastoma cells in n-myc amplified SK-N-Be(2)C cells but not in the non-n-myc amplified SK-N-SH cell line. Treatment with the HDACi vorinostat resulted in differential expression of stemness genes in drug-resistant cells.
Results
Vorinostat cytotoxicity
The cytotoxicity of vorinostat was measured using the MTT assay (Figure 1a). Experimental vorinostat treatment concentrations were chosen to maximize vorinostat treatment levels while minimizing cytotoxicity. On the basis of these MTT results, vorinostat concentrations of 0.5 and 1 μM were used for the SK-N-Be(2)C and SK-N-SH cell lines, respectively. Modulation of histone H3 acetylation for each cell line at chosen vorinostat concentrations was verified with western blot (data not shown).
Figure 1.
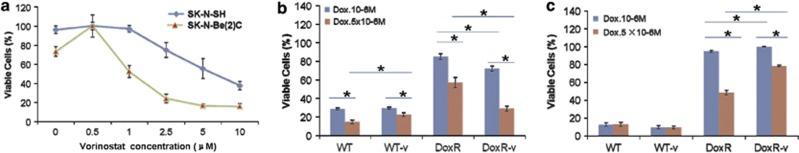
(a) Cytotoxicity of vorinostat was assessed in the wild-type SK-N-Be(2)C and SK-N-SH cell lines using the MTT cell proliferation assay. SK-N-Be(2)C (b) and SK-N-SH (c) cells were treated with doxorubicin at 10−6 M and 5 × 10−6 M for 96 h after which cell survival was assessed using MTT. Error bar=95% confidence intervals. *P<0.0001. Experiments were repeated at least three times
Drug resistance
Wild-type (WT), vorinostat-treated wild-type (WT-v), doxorubicin-resistant (DoxR), and vorinostat treatment doxorubicin resistant (DoxR-v) cells were treated with doxorubicin at 10−6 M and 5 × 10−6 M for 96 h. MTT was then used to assay doxorubicin cytotoxicity. These data are presented in Figures 1b and c. SK-N-Be(2)C and SK-N-SH DoxR and DoxR-v cell lines displayed resistance to doxorubicin at 10−6 M. When treated with doxorubicin at 5 × 10−6 M, the response of DoxR-v cells relative to DoxR cells differed between the SK-N-Be(2)C and SK-N-SH cell lines. The SK-N-Be(2)C-DoxR-v cell line was more sensitive to doxorubicin at 5 × 10−6 M relative to DoxR. The inverse was true in the SK-N-SH cell lines for which doxorubicin had diminished cytotoxicity in the DoxR-v cell line relative to DoxR at 5 × 10−6 M.
Tumorsphere formation
One of the important phenotypic characteristics of CSCs is the ability to form non-adherent spheres in culture medium. WT, WT-v, DoxR, and DoxR-v cells were cultured in tumorsphere culture medium to evaluate the capacity to form tumorspheres. After 4–5 days in culture, SK-N-Be(2)C-DoxR cells were able to form tumorspheres of varying sizes; the tumorspheres were tight and difficult to separate into single-cell suspension. Instead of forming tumorspheres, SK-N-Be(2)C WT, WT-v, and DoxR-v cells adhered to the culture plate (Figure 2a). All SK-N-SH cell lines remained suspended in stem-cell culture medium. SK-N-SH WT, WT-v, and DoxR cells formed loose, grape-like clusters; cells were separated easily. SK-N-SH DoxR-v cells formed tumorspheres, but they were not as tight as SK-N-Be(2)C-DoxR tumorspheres (Figure 2a).The limited dilution assay was performed for SK-N-Be2C-DoxR and SK-N-SH-DoxR-v cells to compare tumorsphere formation frequency. About 1 in 32 SK-N-Be2C-DoxR cells was able to form tumorspheres (Figure 2b). SK-N-SH-DoxR-v cells formed tumorspheres, however, they were difficult to quantify as the tumorspheres were loose and ill defined.
Figure 2.

(a) SK-N-Be(2)C and SK-N-SH WT, WT-v, DoxR, and DoxR-v cell lines were cultured in tumorsphere culture media. SK-N-Be(2)C-DoxR cells formed tumorspheres of varying sizes after 4–5 days in culture. SK-N-Be(2)C WT, WT-v, and DoxR-v cells did not form tumorspheres, adhering to the plate. SK-N-SH-DoxR-v cells formed loose tumorspheres. SK-N-SH-WT, WT-v, and DoxR cell lines failed to form tumorspheres instead forming loose grape-like clusters suspended in culture media. Experiments were repeated at least three times. (b)The sphere formation efficiency experiment for SK-N-Be(2)C-DoxR was repeated four times and the average number of tumorsphere was presented
Colony formation efficiency
Clonogenic ability was evaluated using a colony formation assay (Figures 3a–d). SK-N-Be(2)C-DoxR cells displayed the highest colony formation efficiency among all four SK-N-Be(2)C groups. SK-N-Be(2)C-DoxR-v cells demonstrated low colony formation efficiency, lower than WT and WT-v cells. For SK-N-SH cell lines, the DoxR line exhibited the highest clonogenic ability. The SK-N-SH DoxR-v cell line displayed similar efficiency in colony formation to the SK-N-SH WT cell line.
Figure 3.

One hundred fifty of WT, WT-v, DoxR, and DoxR-v SK-N-Be2C (a) and SK-N-SH (b) cells were seeded into each well of a six-well plate and grown for 10–12 days to assess clonogenic ability. The colonies were counted (only 50 cells or more were counted as a colony) under the microscope. The colony formation efficiency was calculated as the number of colonies formed/the number of the cells seeded × 100. Experiments were repeated at least three times. The number of colonies per plate for the SK-N-Be(2)C cell lines (c) and SK-N-SH cell lines (d) are displayed graphically. Error bar=95% confidence interval. *P=0.03, **P=0.01, ***P=0.005, ****P=0.002, *****P<0.001
Invasive ability
Previous studies have established that CSCs are not only associated with tumor initiation and growth, but also have a crucial role in tumor metastasis.29 In the present study, the invasive properties of all cell lines were assessed using the Matrigel assay (Figures 4a and b). Among the SK-N-Be(2)C cell lines, the DoxR line demonstrated the greatest invasive ability. DoxR-v cells, in contrast, had diminished invasiveness relative to DoxR cells (P<0.001). Both SK-N-SH DoxR and DoxR-v cell lines demonstrated greater invasiveness compared with WT and WT-v cell lines. There was no significant difference between SK-N-SH DoxR and DoxR-v or WT and WT-v cell lines.
Figure 4.

BD Biocoat Matrigel in vitro invasion assay was used to compare the WT, WT-v, DoxR, and DoxR-v (a) SK-N-Be(2)C and (b) SK-N-SH cell lines. Invasion was calculated as the percentage of cells able to invade through a membrane coated with a defined matrix (collagen IV, laminin, and gelatin) during a 24-h period as a fraction of the control inserts. Bars represent the normalized invasion indices. Error bar=95% confidence interval. *P=0.01, **P=0.02, ***P=0.004, ****P<0.001. Experiments were repeated at least five times
Expression of the putative neuroblastoma CSC marker CD133
CD133 has been identified as a CSC marker in some solid tumors.30 In addition, drug-resistant colon cancer and Ewing's sarcoma manifest enriched CD133 expression.31, 32 CD133 has been used as a putative stem cell marker for neuroblastoma.33, 34, 35 Previous work has shown that CD133+ neuroblastoma cells are more resistant to chemotherapy drugs compared with CD133− cells. These CD133+ cells also demonstrate increased tumorsphere formation and greater propensity to form tumors in vivo.34 In contrast, CD133 was minimally detected in neuroblastoma tumor-initiating cells (TIC) and in side population (SP) cells.2, 6 We wanted to determine whether CD133 expression is enriched in drug-resistant neuroblastoma cells and whether vorinostat treatment leads to further upregulation of CD133 as has previously been shown in ovarian cancer.26
CD133 expression was quantified using both reverse transcription-PCR (RT-PCR) and flow cytometry. As shown in Figures 5a and b, CD133 expression, at both the mRNA and protein level, was not increased in any of the SK-N-Be(2)C or SK-N-SH cell lines. We also evaluated whether short-term doxorubicin and/or vorinostat treatment upregulated CD133 expression. SK-N-SH and SK-N-Be(2)C WT cells were treated with doxorubicin alone at 10−9 M, 10−8 M, or 10−7 M for 24 h, vorinostat alone at 0.5 μM (SK-N-Be2C) or 1 μM (SK-N-SH) for 24 h, or combination treatment with doxorubicin and vorinostat (SK-N-Be2C with doxorubicin 10−7 M+vorinostat 0.5 μM and SK-N-SH with doxorubicin 5 × 10−8 M+vorinostat 1 μM) for 24 h. CD133 expression did not vary in any of these treatment groups (data not shown).
Figure 5.

CD133 was not enriched in any of the neuroblastoma cell lines, as confirmed with RT-PCR, the CD133 DNA fragment size is 534 bp (a) and flow cytometry (b), using WERI-Rb-1 retinoblastoma cells as a positive control (c). Experiments were repeated at least four times
SP analysis
SP cells have been identified in 65% of primary neuroblastoma tumors, and neuroblastoma SP cells were reported to be less sensitive to mitoxantrone.2 In this study, SK-N-Be(2)C-DoxR-v cells were found to be more sensitive to doxorubicin relative to DoxR cells, and the inverse was true in SK-N-SH DoxR-v and DoxR cells. We therefore evaluated the percent SP in each cell line to determine whether this variation in drug resistance was associated with changes in the SP (Figure 6a). Flow cytometry using Hoescht 33342 dye revealed that the SP constituted 15.0% of SK-N-Be(2)C-WT cells, 65.6% of DoxR cells, and 26.8% of DoxR-v cells. These data indicate that long-term doxorubicin selection enriches the SP in SK-N-Be(2)C cells, but less so when co-treated with vorinostat. These data suggest that vorinostat treatment may interfere with the expression of drug transporter genes, such as MDR and ABCG2, or stemness-related genes, in this cell line. In contrast to the SK-N-Be(2)C cell lines, the SP was 0.4% in the SK-N-SH-WT cell line, 53.7% in the SK-N-SH-DoxR cell line, and 64.8% in the SK-N-SH-DoxR-v cell line (Figure 6b). In contrast to the SK-N Be(2)C-DoxR-v cell lines, these data suggest that vorinostat may not affect the expression of drug transporter genes and/or stemness genes in SK-N-SH-DoxR-v cells.
Figure 6.

The side population of SK-N-Be(2)C (a) and SK-N-SH (b) cell lines was analyzed. A representative chart of a total of three experiments was shown. Left: Hoechst 33342 dye treatment only. Middle: 50 μM verapamil treatment. Right: 100 μM verapamil treatment. All cells were incubated with Hoechst 3342 dye, excited at 357 nm. Its fluorescence was analyzed at two wavelengths (blue: 670–700 nm and violet: 450–500 nm), both in linear mode. Incubation with propidium iodide (1 μg/ml) was performed to exclude dead cells immediately before analysis. Singlets were gated for side-population analysis. Side-population cells are shown by the outlined area
Whole genome expression in SK-N-Be(2)C lines
Whole genome expression analysis was performed using a 47 231 probe genechip to identify differentially expressed genes (DEGs) in the SK-N-Be(2)C DoxR and DoxR-v cell lines relative to WT. There were 2197 DEGs in the DoxR versus WT comparison and 1489 DEGs in the DoxR-v versus WT comparison with 696 DEG common to both comparisons. The DEGs in SK-N-Be(2)C DoxR and DoxR-v cells were interrogated for a significant change in the expression of stemness-related genes. The profile of DEGs was compared with previous microarray-based profiling of so-called ‘stemness genes', which are expressed in embryonic stem cells (ESCs), hematopoietic stem cells (HSCs), and neural stem cells (NSCs).36, 37 IGFBP3, S100A10, TCF3, SOX2, LMO2, ERCC5, and ABCB1 were concordant with the expression profiling of stemness genes previously reported.36 Expression of ATP binding-cassette family genes and the putative neuroblastoma stem cell markers used in previous studies, including VIM, CD133, OCT4, CD117, CD34, GD2, CD56, CD71, and CD271, were all evaluated. A total of nine stemness-linked genes, IGFBP3 (4.55-fold), S100A10 (13.10-fold), TCF3 (2.56-fold), SOX2 (2.75-fold), LMO2 (4.07-fold), ERCC5 (2.12-fold), ABCB1 (4.23-fold), VIM (24.3-fold), and ABCC4 (2.12-fold), were found to be significantly upregulated in the SK-N-BE(2)C-DoxR cell line (Table 1). These genes were variably upregulated, but to a lesser fold, in the vorinostat-treated SK-N-Be(2)C-DoxR-v cell line.
Table 1. Stemness-linked genes differentially expressed on whole genome microarray analysis (fold change >2.0, adjusted P<0.1) in the SK-N-BE(2)C DoxR and DoxR-v cell lines relative to their parental (WT) lines.
| Gene symbol | Gene Entrez ID | Gene name | DoxR/WT fold | DoxR-v/WT fold |
|---|---|---|---|---|
| ABCB1 | 5243 | ATP-binding cassette, sub-family B (MDR/TAP), member 1 | 4.23 | 2.04 |
| ABCC4 | 10257 | ATP-binding cassette, sub-family C (CFTR/MRP), member 4 | 2.12 | NS |
| LMO2 | 4005 | LIM domain only 2 (rhombotin-like 1) | 4.07 | 2.28 |
| SOX2 | 6657 | SRY (sex determining region Y)-box 2 | 2.75 | NS |
| ERCC5 | 2073 | Excision repair cross-complementing rodent repair deficiency, complementation group | 2.12 | NS |
| S100A10 | 6281 | S100 calcium binding protein A10 | 13.1 | N.S. |
| IGFBP3 | 3486 | Insulin-like growth factor binding protein 3 | 4.55 | 1.84 |
| TCF3 | 6929 | Transcription factor 3 | 2.56 | 1.78 |
| VIM | 7431 | Vimentin | 24.3 | 5.19 |
NS, No significant fold change
Stemness gene expression
Real-time PCR was used to validate the microarray findings in the SK-N-Be(2)C cell lines (Figure 7a). The results confirmed that the fold change of S100A10, TCF3, Sox2, LMO2, ERCC5, ABCB1, IGFBP3, VIM, and ABCC4 expression in DoxR versus WT comparison was significantly higher than DoxR-v versus WT comparison (P=0.05). Real-time PCR for these same genes in the SK-N-SH cell lines revealed that vorinostat treatment did not have a significant effect on stemness gene expression in SK-N-SH cells. The expression of ABCB1 and VIM in SK-N-SH-DoxR was significantly higher than DoxR-v (P=0.05) and the expression of TCF3 and S100A10 was significantly lower in DoxR than in DoxR-v (P=0.05) (Figure 7b).
Figure 7.

Real-time quantitative reverse transcription-PCR was used to measure mRNA expression levels, confirming nine differentially expressed stemness genes in SK-N-Be(2)C (a) and SK-N-SH (b) DoxR and DoxR-v cell lines relative to their parental (WT) cell lines. Data are the average fold change in DoxR or DoxR-v cells compared with WT cells with error bars representing 95% confidence intervals. Each experiment was performed at least in triplicate
Discussion
Only one previous study has found that the CSC population may be expanded during the acquisition of drug resistance.22 The effect of HDACi on the CSCs sub-population is debated. In the present study, we found that there was a sub-population of cells with CSC characteristics which is enriched during prolonged drug selection for resistant neuroblastoma cells in n-myc amplified SK-N-Be(2)C cells but not in the non-n-myc amplified SK-N-SH cell line. Treatment with the HDACi vorinostat resulted in differential expression of stemness genes in drug-resistant cells.
CSCs are known to have an innate resistance to chemotherapy drugs.2, 3, 20 This innate resistance is attributable to an enhanced capacity for DNA repair, decreased apoptosis, and higher expression of ATP-binding cassette transporters.38 Knowing if CSCs are enriched in drug-resistant cell lines may allow for the development of new treatment strategies targeted against CSCs with the goal of reversing drug resistance. Putative CSCs have been identified in neuroblastoma patients using three different approaches: identifying the TICs through bone marrow analysis, analyzing SPs, and sorting the cells based on specific CSC-surface markers. Kaplan and colleagues isolated putative TICs (also known as CSCs) from the bone marrow of high-stage neuroblastoma patients on the basis of their immature neuronal phenotype.6 These cells have been widely used in neuroblastoma CSC studies. It was however recently reported that the TIC cultures were contaminated by EBV-infected B-lymphoblastoid cells.39 SP analysis is another approach used to identify CSCs. SP is thought to harbor the CSC niche and has been identified in over half of neuroblastoma, breast cancer, lung cancer, and glioblastoma cell lines.2 SP cells are capable of sustained expansion ex vivo and are able to generate both SP and non-SP progeny. SP cells have the capacity to expel cytotoxic drugs leading to increased survival in the face of chemotherapeutics. The proportion of SP in cancer cell lines derived from patients in relapse was significantly higher relative to paired pretreatment cell lines, and these SPs demonstrated high clonogenic ability.40, 41 In addition, other studies have shown that a large fraction of tissue stem cells are of the SP fraction, and most of the cells in the SP fraction are stem cells.42, 43, 44 The third approach used for isolating CSCs is selection using putative CSC markers. CD133 has been used as a putative stem cell marker for neuroblastoma.33, 35, 45, 46, 47 However, CD133 has not been detected within TIC populations or in SPs of neuroblastoma patients in relapse.2, 6 In the present study, the n-myc amplified SK-N-Be(2)C doxorubicin-resistant cells were found to be more invasive, had higher colony formation efficiency, possessed the unique ability to form tumorspheres, had a higher SP percentage and overexpressed ATP binding cassette transporter genes and stemness-related genes (ABCB1, ABCC4, LMO2, SOX2, TCF3, S100A10, IGFBP3, ERCC5, and VIM), relative to their parental wild-type (WT) cells. Unlike breast cancer cells that have definitively identified CSC markers, a marker for the neuroblastoma CSC has yet to be defined, and, thus, neuroblastoma CSCs cannot be separated from the remaining cell population using a marker. The data presented here suggest that there is a CSC-like sub-population in SK-N-Be(2)C drug-resistant cells. Although drug-resistant SK-N-SH cells exhibited increased invasiveness and colony formation capability relative to the parental WT cell, it was unable to form tumorspheres. Therefore, whether the SK-N-SH cell lines harbors a CSC-like population needs to be studied further.
Vorinostat is a broad-spectrum HDACi targeting both class I and II HDACs and was the first HDACi approved by the Food and Drug Administration for the treatment of cutaneous T cell Lymphoma (http://www.cancer.gov/cancertopics/druginfo/fda-vorinostat). Clinical trials studying the effects of vorinostat on solid organ tumors, including neuroblastoma, are in progress and have demonstrated promising early results. There are over 60 clinical trials currently under way to explore additional uses for vorinostat as an anticancer drug using combination therapies. Previous work has shown that high HDAC1 mRNA expression correlated with multidrug resistance in neuroblastoma cell lines and inhibition of HDAC1 expression or activity enhanced the cytotoxicity of chemotherapeutic agents.48 These data indicate that HDACi may have a role in drug resistance and HDACs represent a potential therapeutic target in multidrug-resistant neuroblastoma. In our study, we intermittently treated neuroblastoma cells with low doses of vorinostat during the development of drug resistance to investigate whether vorinostat would interfere with the development of drug resistance. Our results demonstrated that vorinostat had a varied effect in different neuroblastoma cell lines. Vorinostat increased the sensitivity of SK-N-Be(2)C-resistant cells to chemotherapy, caused the loss of the ability to form tumorspheres, reduced in vitro invasive capacity, and reduced the percentage of SP cells. In contrast, vorinostat decreased the sensitivity of SK-N-SH doxorubicin-resistant cells to doxorubicin, enhanced the cells' ability to form tumorspheres, and had minimal effect on the cells' invasion and SP percentage. Previous studies have shown that vorinostat, even at low effective doses, can alter the biology of human mesenchymal stem cells.27 In vitro treatment of high-risk ependymoma stem cells with vorinostat induced neuronal differentiation associated with loss of stem cell-specific properties.49 We further queried whether there was a change in stemness gene expression associated with the effect of vorinostat on neuroblastoma drug resistance. Our microarray analysis for SK-N-Be(2)C WT, WT-v, DoxR, and DoxR-v cells demonstrated that nine stemness-linked genes (ABCB1, ABCC4, LMO2, SOX2, TCF3, S100A10, IGFBP3, ERCC5, and VIM) were upregulated in both SK-N-Be(2)C DoxR cells and SK-N-Be(2)C DoxR-v cells. However, a greater fold increase in stemness gene expression was seen for the DoxR versus WT comparison relative to the DoxR-v versus WT comparison. Of these nine genes, ABCB1 and ABCC4 belong to ATP-binding cassette gene family and are linked with drug resistance. The ABCB1 gene is enriched in ESCs, HSC, and neuronal stem cell (NSC),36, 37 ABCC4 with ABCC1,3 is associated with poor prognosis and affects neuroblastoma biology independent of their role in drug efflux.50 We did not see the overexpression of ABCG2, a gene that is expressed in a wide variety of stem cells and is a molecular determinant of the SP phenotype.44 Sox2, a marker of ESCs, is an essential transcription factor for embryonic development. A recent report showed that induced NSCs (iNSCs) from mouse and human fibroblasts can be generated by direct reprogramming with a single factor, Sox2.51TCF3 is highly expressed in ESCs52 and HSC,53 and is a key regulating factor in the OCT4-Sox2-Nanog self-renewal circuit.54 Although Sox2 and TCF3 were overexpressed in SK-N-Be(2)C-DoxR and DoxR-v cells, we did not see a change in OCT4 and Nanog expression. LMO2, a cysteine-rich protein, also known as Rhombotin-2 and T-cell translocation protein-2, is a transcriptional cofactor that is required for hematopoietic and endothelial development and is implicated in HSC maintenance.55, 56 ERCC5 is a gene enriched in all ESC, HSC, and NSC,36, 37 like ABCB1, and is involved in DNA repair. IGFBP3 and S100A10 are transcription factors that are related to NSC.36 S100A10 is also associated with drug resistance. VIM is a type III intermediate filament protein,57 and is expressed in non-epithelial cells, especially mesenchymal stem cells. Besides these nine genes, we also checked for the other stemness genes that have been studied in other neuroblastoma CSC studies (e.g., CD 133, CD34, CD117, and CD271); none of these genes were upregulated in either SK-N-Be(2)C-DoxR cells or SK-N-Be(2)C-DoxR-v cells. It appears, in this study, that vorinostat was able to reverse drug resistance in N-MYC amplified neuroblastoma cells by altering the expression of certain stemness genes. In contrast, vorinostat did not have a similar effect on stemness gene expression in non-N-MYC amplified SK-N-SH cells. ABCB1 and ABCC4 were downregulated in SK-N-SH-DoxR-v cells relative to DoxR cells, and TCF3 was upregulated <1.5-fold in SK-N-SH-DoxR cells.
There are a number of limitations inherent in this study. Neuroblastoma is a heterogeneous tumor. Even though we used two cell types with different phenotypes (n-myc amplified SK-N-Be(2)C versus non-n-myc amplified SK-N-SH), the results we obtained may not fully represent all neuroblastoma cell lines. In addition, we cannot isolate the pure neuroblastoma CSC population for further analysis because there are no identified CSC markers available for neuroblastoma.
In conclusion, our data demonstrated that a CSC-like sub-population may be enriched in n-myc amplified neuroblastoma cells after long-term drug selection with doxorubicin. Intermittent, low dose treatment of neuroblastoma cells with vorinostat can sensitize drug-resistant cells to chemotherapy, and may be due to downregulation of stemness gene expression. These data suggest that development of CSC targeted therapies may be of particular value in patients with n-myc amplified neuroblastoma tumors.
Materials and Methods
Cell lines and reagents
Human neuroblastoma SK-N-SH and SK-N-Be(2)C cell lines were purchased from American Type Culture Collection (Rockville, MA, USA). SK-N-SH cells were grown at 5% CO2 in Dulbecco's Modified Eagle's Medium (DMEM) supplemented with 10% heat inactivated fetal bovine serum, penicillin 100 units/ml, and streptomycin 100 μg/ml. SK-N-Be(2)C cells were cultured in DMEM/F12 1 : 1 with the same supplement as an SK-N-SH culture medium. For each cell line, four groups were generated: (1) parental or WT, (2) DoxR (DoxR cell lines were generated by continuous incubation of parental cell lines with stepwise increases in doxorubicin concentrations, ranging from 10−9 to 10−6 M, over a period of 6 months to 1 year.), (3) DoxR-v (A parallel group of cells were treated with intermittent doses of vorinostat during the development of drug resistance. Cells were treated with vorinostat (1 μM for SK-N-SH and 0.5 μM for SK-N-Be(2)C) for 48 h at the time of each logarithmic increase in the dose of doxorubicin.), and (4) WT-v (A control group of parental cells received only vorinostat at the same time as the treatment in the doxorubicin cells.).
The following culture media, drugs, and antibodies were obtained from the companies cited: DMEM and FBS (Mediatech, Herndon, VA, USA); DMEM/F12, Hank's Balanced Salt Solution (HBSS) (Hyclone Laboratories, Logan, Utah, USA); vorinostat, verapamil, Hoechst 33342, doxorubicin, heparin, β-mercaptoethanol and antibody against human β-Actin (Sigma, St. Louis, MO, USA); 1% B27 and N2 supplements (Invitrogen, Grand Island, NY, USA); anti-human CD133 (CD133/2-293C3) conjugated with phycoerythrin and mouse IgG2b – phycoerythrin (MiltenyiBiotec, Auburn, CA, USA); recombinant human EGF and recombinant human FGF basic (R&D System, Minneapolis, MN, USA); GeneAmp RNA PCR kit and Power SYBR Green RNA-to-CtTM 1-Step kit (Applied Biosystems, Foster City, CA, USA). BD Biocoat growth factor reduced Matrigel invasion chamber (BD Biosciences, Bedford, MA, USA).
Cytotoxicity assay
The cytotoxicity of drugs to the cells was assessed by MTT assay. The cells were either treated with doxorubicin at 10−6 or 5 × 10−6 M or with vorinostat at concentrations varying between 0.5 and 10 μM for 96 h. Viable cells were quantitatively determined using an MTT colorimetric assay. Briefly, MTT (10 μl of 5 mg/ml solution) was added to each well of the 96-well plate and incubated for 4 h at 37°C. The cells were then solubilized by the addition of 100 μl of 10% SDS/0.01 M HCl and incubated for 15 h at 37°C. The optical density of each well was determined in an ELISA plate reader using an activation wavelength of 570 nm and reference wavelength of 650 nm. The percentage of viable cells was determined by comparison with untreated control cells.
Invasion assay
BD Biocoat growth factor reduced Matrigel invasion chambers were used to assess the invasive properties of the cells. An equal number of Matrigel inserts and Control Inserts (no Matrigel coating) were transferred into a 24-well plate and were rehydrated by adding 0.5 ml warm serum-free culture medium onto the interior of the inserts and incubating in a tissue culture incubator at 37°C for 2 h. After rehydration, the medium was carefully removed without disturbing the layer of Matrigel Matrix on the membrane. The inserts were then transferred into the wells containing 0.75 ml culture medium with 10% fetal calf serum. In all, 25 000 cells were added onto the apical side of the insert. The plate was incubated at 37°C, 5% CO2 atmosphere for 22 h. Non-invasive cells were removed from the upper surface of the membrane with a cotton swab before staining. The cells on the lower surface of the membrane were stained following the procedure of Diff-Quick (Siemens Healthcare Diagnostics, Tarrytown, NY, USA). After staining, the inserts were air dried at room temperature. The stained membranes were peeled from the inserts and placed on the slides with coverslips. For each slide, cells in 12 different fields were counted at a magnification of × 20, and the values were averaged. The invasion percentage was determined by the mean number of cells invading through GFR Matrigel insert membrane/mean number of cells migrating through control insert membrane × 100.
Colony formation assay
Cells were trypsinized to produce a single-cell suspension. The cell suspension was diluted into the desired seeding concentration and 150 cells were plated into each well of a six-well plate for 10–12 days. The cells were washed with cold phosphate-buffered saline (PBS) twice on ice and fixed with ice-cold 100% methanol for 10 min. Colonies were stained with 0.5% crystal violet for 10 min at room temperature and rinsed with tap water. After the plate was air dried at room temperature, the colonies were counted (only 50 cells or more were counted as a colony) under the microscope. The colony formation efficiency was calculated by the number of colonies formed/the number of the cells seeded × 100.
Tumorsphere formation
Cells were plated at clonal density (1 × 104/ml) and cultured in tumorsphere media as described by Mahller et al.34 The medium consisted of DMEM/F12, supplemented with 20 ng/ml EGF, 40 ng/ml bFGF, 1% B27 and N2 supplements, 2 μg/ml heparin, 0.1 mM β-mercaptoethanol and 1 × antibiotic/antimycotic. The medium was changed every 3 days.
Limiting dilution assay
For determining the tumorsphere formation frequency of each cell line, the cells were prepared as a single-cell suspension in tumorsphere medium as described above and seeded in 96 ultra-low attachment flat bottom plates (Costar, Corning, NY, USA) with different cell numbers varying from 500 to 1 cell/per well. The number of tumorspheres was counted after culturing the cells for 8–10 days.
Surface marker analysis by flow cytometry
CD133 expression on the cell surface was detected by flow cytometry. Cells were trypsinized, pelleted in a flow tube, washed with cold PBS twice and then resuspended in FcR Blocking Reagent (MiltenyiBiotec) with 0.5% fetal calf serum in PBS. Mouse anti-human CD133/2-PE monoclonal antibody was diluted at 1 : 11 and added into the tube. The tube was incubated at 4°C for 15 min in the dark. After incubation, the labeled cells were washed with PBS (2 × ) resuspended in 400μl and then analyzed by a FACSCaliber flow cytometer (Becton Dickinson, Downers Grove, IL, USA). The cells were acquired using the CellQuest software (Becton Dickinson) and 10 000 events were collected for analysis. Analysis was done using the BD CellQuest (Becton Dickinson) software. Mouse IgG2b-phycoerythrin was used as an isotype control and was set up for each sample. Weri-Rb-1, a cell line with high CD133 expression, was used as a positive control in each experiment.
Analysis of the SP cell fraction by flow cytometry
SP assay was used for identifying stem cells with overexpression of ABC transporters. Cells were cultured to 80–90% confluence and trypsinized. About 1 × 106/ml cells were resuspended in pre-warmed DMEM or DMEM/F12 media with 10% FBS and 10 mM HEPES buffer. In all, 1 ml of the cell suspension was aliquoted into 2 ml tubes and Hoechst 33342 dye (final concentration 5 μg/ml) was added into each tube, mixed well and then verapamil (final concentration at 0, 50, and 100 μM) was added to the tube. The tubes were incubated at 37°C for 90 min. After incubation, the cells were placed on ice and spun down in a centrifuge (4°C) at 1500 r.p.m. for 5 min. The supernatant was removed and the pellet was resuspended in 1 ml of cold HBSS containing 10% FBS and 10 mM HEPES buffer, 1 μl propidium iodide (1 mg/ml, final concentration 1 μg/ml) was added to the tube. Hoechst 33342-labeled cells were analyzed on a Becton Dickinson LSRFortessa (LSRII) flow cytometer (BD Biosciences). Red cells stained by propidium iodide were excluded from the analysis. Singlets were gated for SP analysis. SP cells were visualized by using Indo-1 (Blue, wavelength 670/30 band pass) versus Indo-1 (Violet, wavelength 450/50 band pass) off the UV laser (355 nm) and analyzed with BD FACSDiva software.
Reverse transcription-PCR
Total RNA was extracted from ∼1 million of SK-N-SH and SK-N-Be(2)C cells using the RNeasy Mini Kit (Qiagen, Valencia, CA, USA) according to the manufacturer's protocol. The RNA concentration was measured using the NanoDrop micro-volume spectrophotometer (Thermo Scientific, Wilmington, DE, USA). One microgram of RNA was used for cDNA synthesis and cDNA was amplified following the protocol from Applied BiosystemsGeneAmp RNA PCR Kit (Applied Biosystems). The sequences for the primers CD133 and glyceraldehyde 3-phosphate dehydrogenase (GAPDH) are listed in Supplementary Table 1. The cycling conditions for PCR were 95°C for 105 s, followed by 35 cycles at 95°C for 15 s, 60°C for 30 s, and then 72°C for 7 min.
Real-time quantitative PCR (qPCR)
Real-time quantitative RT-PCR was used for validating the genes that showed statistically significant changes in the microarray analysis below. Total RNA was extracted as described above. Specific PCR primer sequences for all validated genes and the reference gene were taken either from publications or Primer Bank (pga.mgh.harvard.edu/primerbank). Expression of these genes was determined using Power SYBR Green RNA-to-CtTM 1-Step Kit. Reverse transcription was performed using 100 ng of total RNA in a 20-μl reaction volume and assays were performed in triplicate on an Applied Biosystems 7500 system. Expression of genes was measured using threshold cycle values (CT). The ΔCT was calculated by subtracting the CT of GAPDH mRNA from the CT of target gene mRNA. Fold change in gene expression for DoxR and DoxR-v cell lines compared with parental cells (WT) was determined from the same amount of total RNA by use of the delta, delta threshold cycle (Ct) method. The thermal cycling conditions were as follows: holding at 48°C for 30 min and 95°C for 10 min, 40 cycles at 95°C for 15 s followed by 60°C for 1 min.
Whole genome expression
RNA was isolated from triplicate specimens of each cell line using the RNeasy Mini Kit (Qiagen, Venlo, Netherlands). Samples with RNA integrity numbers greater than 9.5 were used in subsequent expression analyses. All RNA samples were stored at −80°C before labeling and hybridization.
Amplification and labeling of the RNA was performed with the illumine TotalPrep RNA Amplification Kit from Life Technologies (Ambion, Austin, TX) using 150 ng of RNA per samples. In all, 750 ng of labeled cRNA was hybridized to Human HT-12 v4 Expression BeadChips for 18 h and processed (Illumina, San Diego, CA, USA) according to the manufacturer's instruction. Slides were scanned using IlluminaiScan System Human HT-12 v4 beadchip containing 47 231 probes.
Image and data analysis
Quality check and probe level processing of the Illumina microarray data was further made with Rbioconductor package, Lumi.58 The data processing includes a normalization method to reduce the obscuring variation between microarrays, which might be introduced during the processes of sample preparation, manufacturing, fluorescence labeling, hybridization, and/or scanning.59 Hierarchical clustering and principal component analysis was performed on the normalized signal data to assess the sample relationship and variability. Probes that were expressed in none of the samples as judged by the criteria of Illumina detection P-value >0.01 were filtered out. Differential gene expression between the different conditions was assessed by statistical linear model analysis using bioconductor package, limma, in which an empirical Bayes method is used to moderate the standard errors of the estimated log-fold changes of gene expression (http://www.bioconductor.org/packages/release/bioc/html/limma.html). It results in a more stable inference and improved power, especially for experiments with small numbers of microarrays. The moderated T statistic P-value derived from the limma analysis above was further adjusted for multiple testing by Benjamini and Hochberg's method to control false discovery rate (FDR). The list of DEGs was obtained by the FDR criteria of <10% and fold change cutoff of >1.5, and visualized by volcanoplots. The gene ontology analysis of the list of DEGs was performed with Rbioconductor package, topG (http://www.bioconductor.org/packages/release/bioc/html/topGO.html.). All bioconductor packages are available at http://bioconductor.org and all computation was performed under Renvironment (http://www.r-project.org)
Statistical analysis
Statistical analyses were performed using SAS 9.3 (SAS institute Inc., Cary, NC, USA). Comparisons between two groups are made either by Student's t-test or by Wilcoxon Rank Sum test. If not otherwise stated, then all tests are two-sided. P-values of ≤0.05 were considered as significant.
Acknowledgments
This work was supported by generous grants from the John W Anderson Foundation and from the Children's Faculty Practice Plan Faculty Development Program Fund. A special thanks to Dr. Gang Zhang at the Children's Hospital of Chicago Research Center for performing the statistical analyses for this paper. Thanks to Dr. Junzhong Wang and Dr. Shunmei Huang at Union Hospital for artwork. Thank you for the Northwestern University flow cytometry core facility for facilitating flow cytometric experiments and data analysis.
Glossary
- CSC
cancer stem cell
- HDACi
histone deacetylase inhibitor
- WT
wild type
- WT-v
vorinostat-treated wild type
- DoxR
doxorubicin resistant
- DoxR-v
vorinostat treatment doxorubicin resistant
- SP
side population
The authors declare no conflict of interest.
Footnotes
Supplementary Information accompanies this paper on Cell Death and Disease website (http://www.nature.com/cddis)
Edited by G Raschellà
Supplementary Material
References
- Olshan AF, Bunin GR.In: Brodeur GM, Sawada T, Tsuchida Y, Voute PA (ed).Neuroblastoma Elsevier Science: Amsterdam; 2000 [Google Scholar]
- Hirschmann-Jax C, Foster AE, Wulf GG, Nuchtern JG, Jax TW, Gobel U, et al. A distinct "side population" of cells with high drug efflux capacity in human tumor cells. Proc Natl Acad Sci USA. 2004;101:14228–14233. doi: 10.1073/pnas.0400067101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu G, Yuan X, Zeng Z, Tunici P, Ng H, Abdulkadir IR, et al. Analysis of gene expression and chemoresistance of CD133+ cancer stem cells in glioblastoma. Mol Cancer. 2006;5:67. doi: 10.1186/1476-4598-5-67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Besancon R, Valsesia-Wittmann S, Puisieux A. Caron de Fromentel C, Maguer-Satta V. Cancer stem cells: the emerging challenge of drug targeting. Curr Med Chem. 2009;16:394–416. doi: 10.2174/092986709787315531. [DOI] [PubMed] [Google Scholar]
- Reya T, Morrison SJ, Clarke MF, Weissman IL. Stem cells, cancer, and cancer stem cells. Nature. 2001;414:105–111. doi: 10.1038/35102167. [DOI] [PubMed] [Google Scholar]
- Hansford LM, McKee AE, Zhang L, George RE, Gerstle JT, Thorner PS, et al. Neuroblastoma cells isolated from bone marrow metastases contain a naturally enriched tumor-initiating cell. Cancer Res. 2007;67:11234–11243. doi: 10.1158/0008-5472.CAN-07-0718. [DOI] [PubMed] [Google Scholar]
- Zhang S, Balch C, Chan MW, Lai HC, Matei D, Schilder JM, et al. Identification and characterization of ovarian cancer-initiating cells from primary human tumors. Cancer Res. 2008;68:4311–4320. doi: 10.1158/0008-5472.CAN-08-0364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ricci-Vitiani L, Lombardi DG, Pilozzi E, Biffoni M, Todaro M, Peschle C, et al. Identification and expansion of human colon-cancer-initiating cells. Nature. 2007;445:111–115. doi: 10.1038/nature05384. [DOI] [PubMed] [Google Scholar]
- O'Brien CA, Pollett A, Gallinger S, Dick JE. A human colon cancer cell capable of initiating tumour growth in immunodeficient mice. Nature. 2007;445:106–110. doi: 10.1038/nature05372. [DOI] [PubMed] [Google Scholar]
- Wang J, Zhou JY, Wu GS. ERK-dependent MKP-1-mediated cisplatin resistance in human ovarian cancer cells. Cancer Res. 2007;67:11933–11941. doi: 10.1158/0008-5472.CAN-07-5185. [DOI] [PubMed] [Google Scholar]
- Chiba T, Kita K, Zheng YW, Yokosuka O, Saisho H, Iwama A, et al. Side population purified from hepatocellular carcinoma cells harbors cancer stem cell-like properties. Hepatology. 2006;44:240–251. doi: 10.1002/hep.21227. [DOI] [PubMed] [Google Scholar]
- Haraguchi N, Ishii H, Mimori K, Tanaka F, Ohkuma M, Kim HM, et al. CD13 is a therapeutic target in human liver cancer stem cells. J Clin Invest. 2010;120:3326–3339. doi: 10.1172/JCI42550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lapidot T, Sirard C, Vormoor J, Murdoch B, Hoang T, Caceres-Cortes J, et al. A cell initiating human acute myeloid leukaemia after transplantation into SCID mice. Nature. 1994;367:645–648. doi: 10.1038/367645a0. [DOI] [PubMed] [Google Scholar]
- Bhatia M, Wang JC, Kapp U, Bonnet D, Dick JE. Purification of primitive human hematopoietic cells capable of repopulating immune-deficient mice. Proc Natl Acad Sci USA. 1997;94:5320–5325. doi: 10.1073/pnas.94.10.5320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh SK, Hawkins C, Clarke ID, Squire JA, Bayani J, Hide T, et al. Identification of human brain tumour initiating cells. Nature. 2004;432:396–401. doi: 10.1038/nature03128. [DOI] [PubMed] [Google Scholar]
- Wright MH, Calcagno AM, Salcido CD, Carlson MD, Ambudkar SV, Varticovski L. Brca1 breast tumors contain distinct CD44+/CD24− and CD133+ cells with cancer stem cell characteristics. Breast Cancer Res. 2008;10:R10. doi: 10.1186/bcr1855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suetsugu A, Nagaki M, Aoki H, Motohashi T, Kunisada T, Moriwaki H. Characterization of CD133+ hepatocellular carcinoma cells as cancer stem/progenitor cells. Biochem Biophys Res Commun. 2006;351:820–824. doi: 10.1016/j.bbrc.2006.10.128. [DOI] [PubMed] [Google Scholar]
- Lang SH, Frame FM, Collins AT. Prostate cancer stem cells. J Pathol. 2009;217:299–306. doi: 10.1002/path.2478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eramo A, Lotti F, Sette G, Pilozzi E, Biffoni M, Di Virgilio A, et al. Identification and expansion of the tumorigenic lung cancer stem cell population. Cell Death Differ. 2008;15:504–514. doi: 10.1038/sj.cdd.4402283. [DOI] [PubMed] [Google Scholar]
- Bao S, Wu Q, McLendon RE, Hao Y, Shi Q, Hjelmeland AB, et al. Glioma stem cells promote radioresistance by preferential activation of the DNA damage response. Nature. 2006;444:756–760. doi: 10.1038/nature05236. [DOI] [PubMed] [Google Scholar]
- Bertolini G, Roz L, Perego P, Tortoreto M, Fontanella E, Gatti L, et al. Highly tumorigenic lung cancer CD133+ cells display stem-like features and are spared by cisplatin treatment. Proc Natl Acad Sci USA. 2009;106:16281–16286. doi: 10.1073/pnas.0905653106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calcagno AM, Salcido CD, Gillet JP, Wu CP, Fostel JM, Mumau MD, et al. Prolonged drug selection of breast cancer cells and enrichment of cancer stem cell characteristics. J Natl Cancer Inst. 2010;102:1637–1652. doi: 10.1093/jnci/djq361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dallas NA, Xia L, Fan F, Gray MJ, Gaur P, van Buren G, 2nd, et al. Chemoresistant colorectal cancer cells, the cancer stem cell phenotype, and increased sensitivity to insulin-like growth factor-I receptor inhibition. Cancer Res. 2009;69:1951–1957. doi: 10.1158/0008-5472.CAN-08-2023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Batty N, Malouf GG, Issa JP. Histone deacetylase inhibitors as anti-neoplastic agents. Cancer Lett. 2009;280:192–200. doi: 10.1016/j.canlet.2009.03.013. [DOI] [PubMed] [Google Scholar]
- Bolden JE, Peart MJ, Johnstone RW. Anticancer activities of histone deacetylase inhibitors. Nat Rev Drug Discov. 2006;5:769–784. doi: 10.1038/nrd2133. [DOI] [PubMed] [Google Scholar]
- Baba T, Convery PA, Matsumura N, Whitaker RS, Kondoh E, Perry T, et al. Epigenetic regulation of CD133 and tumorigenicity of CD133+ ovarian cancer cells. Oncogene. 2009;28:209–218. doi: 10.1038/onc.2008.374. [DOI] [PubMed] [Google Scholar]
- Di Bernardo G, Alessio N, Dell'Aversana C, Casale F, Teti D, Cipollaro M, et al. Impact of histone deacetylase inhibitors SAHA and MS-275 on DNA repair pathways in human mesenchymal stem cells. J Cell Physiol. 2010;225:537–544. doi: 10.1002/jcp.22236. [DOI] [PubMed] [Google Scholar]
- Zhang B, Strauss AC, Chu S, Li M, Ho Y, Shiang KD, et al. Effective targeting of quiescent chronic myelogenous leukemia stem cells by histone deacetylase inhibitors in combination with imatinib mesylate. Cancer Cell. 2010;17:427–442. doi: 10.1016/j.ccr.2010.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hermann PC, Huber SL, Heeschen C. Metastatic cancer stem cells: a new target for anti-cancer therapy. Cell Cycle. 2008;7:188–193. doi: 10.4161/cc.7.2.5326. [DOI] [PubMed] [Google Scholar]
- Grosse-Gehling P, Fargeas CA, Dittfeld C, Garbe Y, Alison MR, Corbeil D, et al. CD133 as a biomarker for putative cancer stem cells in solid tumours: limitations, problems and challenges. J Pathol. 2012;229:355–378. doi: 10.1002/path.4086. [DOI] [PubMed] [Google Scholar]
- Jiang X, Gwye Y, Russell D, Cao C, Douglas D, Hung L, et al. CD133 expression in chemo-resistant Ewing sarcoma cells. BMC Cancer. 2010;10:116. doi: 10.1186/1471-2407-10-116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suva ML, Riggi N, Stehle JC, Baumer K, Tercier S, Joseph JM, et al. Identification of cancer stem cells in Ewing's sarcoma. Cancer Res. 2009;69:1776–1781. doi: 10.1158/0008-5472.CAN-08-2242. [DOI] [PubMed] [Google Scholar]
- Liang Y, Zhong Z, Huang Y, Deng W, Cao J, Tsao G, et al. Stem-like cancer cells are inducible by increasing genomic instability in cancer cells. J Biol Chem. 2010;285:4931–4940. doi: 10.1074/jbc.M109.048397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahller YY, Williams JP, Baird WH, Mitton B, Grossheim J, Saeki Y, et al. Neuroblastoma cell lines contain pluripotent tumor initiating cells that are susceptible to a targeted oncolytic virus. PLoS One. 2009;4:e4235. doi: 10.1371/journal.pone.0004235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vangipuram SD, Wang ZJ, Lyman WD. Resistance of stem-like cells from neuroblastoma cell lines to commonly used chemotherapeutic agents. Pediatr Blood Cancer. 2010;54:361–368. doi: 10.1002/pbc.22351. [DOI] [PubMed] [Google Scholar]
- Ramalho-Santos M, Yoon S, Matsuzaki Y, Mulligan RC, Melton DA. "Stemness": transcriptional profiling of embryonic and adult stem cells. Science. 2002;298:597–600. doi: 10.1126/science.1072530. [DOI] [PubMed] [Google Scholar]
- Geschwind DH, Ou J, Easterday MC, Dougherty JD, Jackson RL, Chen Z, et al. A genetic analysis of neural progenitor differentiation. Neuron. 2001;29:325–339. doi: 10.1016/s0896-6273(01)00209-4. [DOI] [PubMed] [Google Scholar]
- Dean M. ABC transporters, drug resistance, and cancer stem cells. J Mammary Gland Biol Neoplasia. 2009;14:3–9. doi: 10.1007/s10911-009-9109-9. [DOI] [PubMed] [Google Scholar]
- Mohlin S, Pietras A, Wigerup C, Ora I, Andang M, Nilsson K, et al. Tumor-initiating cells in childhood neuroblastoma--letter Cancer Res 201272821–822.author reply 823. [DOI] [PubMed] [Google Scholar]
- Steiniger SC, Coppinger JA, Kruger JA, Yates J, 3rd, Janda KD. Quantitative mass spectrometry identifies drug targets in cancer stem cell-containing side population. Stem Cells. 2008;26:3037–3046. doi: 10.1634/stemcells.2008-0397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newton TC, Wolcott K, Roberts SS. Comparison of the side populations in pretreatment and postrelapse neuroblastoma cell lines. Transl Oncol. 2010;3:246–251. doi: 10.1593/tlo.09301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alvi AJ, Clayton H, Joshi C, Enver T, Ashworth A, Vivanco MM, et al. Functional and molecular characterisation of mammary side population cells. Breast Cancer Res. 2003;5:R1–R8. doi: 10.1186/bcr547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lassalle B, Bastos H, Louis JP, Riou L, Testart J, Dutrillaux B, et al. 'Side Population' cells in adult mouse testis express Bcrp1 gene and are enriched in spermatogonia and germinal stem cells. Development. 2004;131:479–487. doi: 10.1242/dev.00918. [DOI] [PubMed] [Google Scholar]
- Zhou S, Schuetz JD, Bunting KD, Colapietro AM, Sampath J, Morris JJ, et al. The ABC transporter Bcrp1/ABCG2 is expressed in a wide variety of stem cells and is a molecular determinant of the side-population phenotype. Nat Med. 2001;7:1028–1034. doi: 10.1038/nm0901-1028. [DOI] [PubMed] [Google Scholar]
- Cournoyer S, Nyalendo C, Addioui A, Belounis A, Beaunoyer M, Aumont A, et al. Genotype analysis of tumor-initiating cells expressing CD133 in neuroblastoma. Genes Chromosomes Cancer. 2012;51:792–804. doi: 10.1002/gcc.21964. [DOI] [PubMed] [Google Scholar]
- Sartelet H, Imbriglio T, Nyalendo C, Haddad E, Annabi B, Duval M, et al. CD133 expression is associated with poor outcome in neuroblastoma via chemoresistance mediated by the AKT pathway. Histopathology. 2012;60:1144–1155. doi: 10.1111/j.1365-2559.2012.04191.x. [DOI] [PubMed] [Google Scholar]
- Takenobu H, Shimozato O, Nakamura T, Ochiai H, Yamaguchi Y, Ohira M, et al. CD133 suppresses neuroblastoma cell differentiation via signal pathway modification. Oncogene. 2011;30:97–105. doi: 10.1038/onc.2010.383. [DOI] [PubMed] [Google Scholar]
- Keshelava N, Davicioni E, Wan Z, Ji L, Sposto R, Triche TJ, et al. Histone deacetylase 1 gene expression and sensitization of multidrug-resistant neuroblastoma cell lines to cytotoxic agents by depsipeptide. J Natl Cancer Inst. 2007;99:1107–1119. doi: 10.1093/jnci/djm044. [DOI] [PubMed] [Google Scholar]
- Milde T, Kleber S, Korshunov A, Witt H, Hielscher T, Koch P, et al. A novel human high-risk ependymoma stem cell model reveals the differentiation-inducing potential of the histone deacetylase inhibitor Vorinostat. Acta Neuropathol. 2011;122:637–650. doi: 10.1007/s00401-011-0866-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henderson MJ, Haber M, Porro A, Munoz MA, Iraci N, Xue C, et al. ABCC multidrug transporters in childhood neuroblastoma: clinical and biological effects independent of cytotoxic drug efflux. J Natl Cancer Inst. 2011;103:1236–1251. doi: 10.1093/jnci/djr256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ring KL, Tong LM, Balestra ME, Javier R, Andrews-Zwilling Y, Li G, et al. Direct reprogramming of mouse and human fibroblasts into multipotent neural stem cells with a single factor. Cell Stem Cell. 2012;11:100–109. doi: 10.1016/j.stem.2012.05.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lluis F, Ombrato L, Pedone E, Pepe S, Merrill BJ, Cosma MP. T-cell factor 3 (Tcf3) deletion increases somatic cell reprogramming by inducing epigenome modifications. Proc Natl Acad Sci USA. 2011;108:11912–11917. doi: 10.1073/pnas.1017402108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ivanova NB, Dimos JT, Schaniel C, Hackney JA, Moore KA, Lemischka IR. A stem cell molecular signature. Science. 2002;298:601–604. doi: 10.1126/science.1073823. [DOI] [PubMed] [Google Scholar]
- Marson A, Foreman R, Chevalier B, Bilodeau S, Kahn M, Young RA, et al. Wnt signaling promotes reprogramming of somatic cells to pluripotency. Cell Stem Cell. 2008;3:132–135. doi: 10.1016/j.stem.2008.06.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamada Y, Warren AJ, Dobson C, Forster A, Pannell R, Rabbitts TH. The T cell leukemia LIM protein Lmo2 is necessary for adult mouse hematopoiesis. Proc Natl Acad Sci USA. 1998;95:3890–3895. doi: 10.1073/pnas.95.7.3890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warren AJ, Colledge WH, Carlton MB, Evans MJ, Smith AJ, Rabbitts TH. The oncogenic cysteine-rich LIM domain protein rbtn2 is essential for erythroid development. Cell. 1994;78:45–57. doi: 10.1016/0092-8674(94)90571-1. [DOI] [PubMed] [Google Scholar]
- Dang H, Lin AL, Zhang B, Zhang HM, Katz MS, Yeh CK. Role for Notch signaling in salivary acinar cell growth and differentiation. Dev Dyn. 2009;238:724–731. doi: 10.1002/dvdy.21875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du P, Kibbe WA, Lin SM. lumi: a pipeline for processing Illumina microarray. Bioinformatics. 2008;24:1547–1548. doi: 10.1093/bioinformatics/btn224. [DOI] [PubMed] [Google Scholar]
- Bolstad BM, Irizarry RA, Astrand M, Speed TP. A comparison of normalization methods for high density oligonucleotide array data based on variance and bias. Bioinformatics. 2003;19:185–193. doi: 10.1093/bioinformatics/19.2.185. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.